
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlock copolymer compatibilization driven frustrated crystallization in electrospun nanofibers of polystyrene/poly(ethylene oxide) blends†
Pratick Samanta,
Rajiv Srivastava and
Bhanu Nandan*
and
Bhanu Nandan*
Department of Textile Technology, Indian Institute of Technology Delhi, Hauz Khas, New Delhi 110016, India. E-mail: nandan@textile.iitd.ac.in
First published on 16th May 2018
Abstract
The confined crystallization behaviour of poly(ethylene oxide) (PEO) has been studied in electrospun nanofibers of the phase-separated blends of polystyrene (PS) and PEO compatibilized with polystyrene-block-poly(ethylene oxide) (PS-b-PEO) block copolymer. The PS was present as the majority component such that the electrospun nanofibers consisted of PEO domains dispersed in the PS matrix. The phase separation in the blend occurred under the radial constraint of the nanofibers which led to the formation of small-sized fibrillar PEO domains. The use of block copolymer compatibilizer resulted in a noticeable decrease in the PEO domain size in the as-spun nanofibers. Moreover, the decrease in the domain size and domain connectivity was more substantial in the thermally annealed blend nanofibers due to the suppression of the domain coalescence mechanism resulting from the localization of the PS-b-PEO block copolymer at the interface. Consequently, the fraction of PEO domains crystallizing via homogeneous nucleation increased in the compatibilized blend nanofibers due to the presence of higher number of heterogeneity free PEO domains and disruption in their spatial connectivity. Interestingly, in the compatibilized blend nanofibers consisting of low molecular weight PEO, additional crystallization event attributed to surface nucleation was observed. The surface nucleation, plausibly, resulted from the formation of wet-brush structures where the PEO homopolymers homogeneously wet the PEO blocks present at the interface. In such a scenario, the PEO crystallization occurred via surface nucleation at the domain interface. The surface nucleated crystallization was absent in the compatibilized blend nanofibers composed of high molecular weight PEO presumably due to the formation of morphology with dry-brush structures.
Introduction
In recent past, there has been an increased focus on understanding the fundamental properties of materials when present as nanosized structures. The research carried out so far has shown that material properties of nanosized materials may differ significantly in comparison to its bulk counterparts. Polymers, with long chain molecules, also depicts interesting properties when present as nanosized domains. One of the fascinating aspect of polymer is its crystallization behavior which differs considerably from the way the small molecules crystallizes. The many studies done in past decades have made our understanding on the various aspects of polymer crystallization, in bulk, more or less very clear. However, when the polymer is allowed to crystallize in confined environment, where the confinement length-scale is only few times that of the long period of polymer crystal lamellae, the situation is expected to be more complex. In such a scenario, the confinement may frustrate the polymer crystallization resulting in significant deviation from the crystallization occurring in the bulk conditions.1–17 With the advent of nanotechnology, study of confinement induced frustration in polymer crystallization is of significant fundamental and practical importance considering the focus on miniaturizing of devices.The past studies done on polymer crystallization have shown that the confinement brings about significant changes in the crystallization and melting behaviour of polymers. Most notably, under well-partitioned nano-confined conditions, the number of domains per unit volume is much higher than the number density of heterogeneities present; hence, a significant fraction of the polymer chains have to crystallize through self-nucleation or homogeneous nucleation mechanism which occurs at very large degree of supercooling. The confinement also was found to influence the melting behaviour as well as development of total degree of crystallinity in the polymer.9–15 The past studies on confined crystallization behaviour of polymers has been done mostly using block copolymers,18–31 porous anodic alumina (AAO) membrane32–42 and multilayer thin films.43–52 It must be noted that, in block copolymers, chain connectivity between the two homopolymer blocks additionally influence the crystallization behaviour. Furthermore, in case of AAO membranes, the polymer film present on the surface is somewhat tedious to remove and even trace amount of polymers on the surface may leave an imprint of bulk crystallization on the expected confined crystallization behaviour. However, AAO membrane provide tailored and uniform confinement sizes which is useful for the quantitative evaluation of the resulting crystallization behaviour.
The electrospun nanofibers fabricated from an immiscible blend of an amorphous and a crystalline polymer, with the latter forming the dispersed phase in the fibers, offers another fascinating and complex system for studying the effect of confinement on the crystallization behaviour.53–59 Here, the limited dimension along the radial direction of the nanofibers restricts the length scale of the phase separation among the immiscible constituents, so as to generate nanosized domains. However, compared to the other commonly studied systems, the size of the domains formed in the nanofibers is highly polydisperse and shape is irregular, which may lead to a more complex crystallization behaviour. Furthermore, the confinement induced crystallization behaviour observed in the electrospun nanofiber based systems provides information under actual non-equilibrium processing conditions. In the past, we investigated the crystallization behaviour of poly(ethylene oxide) (PEO) in the nanofibers electrospun from polystyrene (PS)/PEO and PS/poly(ε-caprolactone) (PCL) immiscible blends, where the PEO or PCL weight faction was 0.4 and less.57,58 It was demonstrated that the PEO or PCL domains size and shape, in the nanofibers, differed considerably from that in the cast films. This was due to the submicron dimensions of the nanofibers in atleast two dimensions and the extensional forces experienced by the polymer solution during electrospinning. The phase-separated morphology in turn significantly influenced the crystallization behaviour of PEO in the blend nanofibers. At PEO weight fraction greater than 0.3, the crystallization was observed to occur through heterogeneous nucleation mechanism similar to that in cast blend films. However, as PEO weight fraction in the blend nanofibers was reduced from 0.3 to 0.2 and less, an abrupt transformation of nucleation mechanism from heterogeneous to predominantly self or homogenous type was revealed.
It is well known that compatibilization of immiscible polymer blends reduces the interfacial energy between the phases and allows for a finer dispersion during mixing.60–65 The process also leads to an improved interfacial adhesion and, hence, has been widely used to improve the mechanical properties of immiscible polymer blends. One of the most frequently proposed compatibilization strategy is the addition of a block copolymer composed of blocks that are each miscible with one of the homopolymers. Hence, an A–B diblock copolymer could be used to compatiblize A/B blend. The logic behind using such a diblock copolymer is that the respective blocks prefers to mix with the corresponding homopolymers of the A/B blend and, hence, this drives the block copolymer to localize at the interface. In the case of electrospun nanofibers prepared from a polymer blend, it is expected that the use of compatibilizer will further reduce the droplet size and also is likely to make them more uniform. Furthermore, the presence of the block copolymer at the interface prevents droplet coalescence and, hence, is expected to make the blend system more stable during post annealing processes. To our knowledge, block copolymer controlled compatibilization of polymer blends in electrospun nanofibers has not been investigated till now. More significantly, such compatibilization induced reduction in droplet size in electrospun nanofibers of immiscible blend could further drastically confine the crystallization behavior if the crystallizable polymer constitutes the minority component. In the present study, we investigate the morphology and crystallization behavior of electrospun nanofibers composed of PS/PEO blends which were compatibilized by a symmetric PS-b-PEO diblock copolymer. It will be demonstrated that compatibilization by PS-b-PEO indeed led to a reduction in the PEO droplet size which were dispersed in the PS matrix and, hence, effectively increased the confinement effect on the PEO crystallization behavior. Moreover, the interfacial localization of the PS-b-PEO block copolymer in the PS/PEO blend prevented the PEO domain coalescence during static annealing above the Tg of PS. Hence, the domain break-up had a dominating influence on the final morphology and crystallization behavior of compatibilized blend nanofibers. This was in contrary to the observed behavior in uncompatibilized blend nanofibers where the domain coalescence phenomena competed with the domain break-up and had a more significant influence on the final morphology. Furthermore, it will also be shown that the crystallization of PEO homopolymer was significantly influenced by its ability to wet the PEO brushes located at the domain interface.
Experimental
Materials
Poly(ethylene oxide) (PEO) (Mv = 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and polystyrene (PS) (Mw = 190000) were procured from Sigma Aldrich. The low molecular weight PEO (Mn = 4000) and polystyrene-block-poly(ethylene oxide) (PS-b-PEO) block copolymer with Mn,PS = 61000, Mn,PEO = 60000 was procured from Polymer Source Inc. Dimethylformamide (DMF), used as solvent for electrospinning, was procured from Merck chemicals and used as it is.
000) and polystyrene (PS) (Mw = 190000) were procured from Sigma Aldrich. The low molecular weight PEO (Mn = 4000) and polystyrene-block-poly(ethylene oxide) (PS-b-PEO) block copolymer with Mn,PS = 61000, Mn,PEO = 60000 was procured from Polymer Source Inc. Dimethylformamide (DMF), used as solvent for electrospinning, was procured from Merck chemicals and used as it is.
Sample preparation
| Sl. no. | Samples name | Code | Electrospinning condition | |||
|---|---|---|---|---|---|---|
| Conc. (wt%) | Voltage (kV) | Distance (cm) | Flow rate (ml h−1) | |||
| a Block copolymer concentration was with respect to the total weight of PS and PEO in the blend. The cast film and nanofiber blend samples were differentiated by adding a prefix of ‘C’ and ‘N’, respectively, to the blend composition codes. | ||||||
PS (Mw ∼ 190![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 000 g mol−1), PEO (Mv ∼ 400000 g mol−1) 000 g mol−1), PEO (Mv ∼ 400000 g mol−1) |
||||||
| 1 | PS-70%, PEO-30%, PS-b-PEO-0% | S7E3B0 | 6 | 20 | 20 | 0.5 |
| 2 | PS-70%, PEO-30%, PS-b-PEO-2% | S7E3B2 | 6 | 20 | 20 | 0.5 |
| 3 | PS-70%, PEO-30%, PS-b-PEO-4% | S7E3B4 | 6 | 20 | 20 | 0.5 |
| 4 | PS-70%, PEO-30%, PS-b-PEO-6% | S7E3B6 | 6 | 20 | 20 | 0.5 |
| 5 | PS-80%, PEO-20%, PS-b-PEO-0% | S8E2B0 | 8 | 20 | 20 | 0.5 |
| 6 | PS-80%, PEO-20%, PS-b-PEO-2% | S8E2B2 | 8 | 20 | 20 | 0.5 |
| 7 | PS-80%, PEO-20%, PS-b-PEO-4% | S8E2B4 | 10 | 25 | 20 | 0.5 |
| 8 | PS-80%, PEO-20%, PS-b-PEO-6% | S8E2B6 | 10 | 25 | 20 | 0.5 |
|
||||||
| PS (Mw ∼ 190000 g mol−1), PEO (Mn ∼ 4000 g mol−1) | ||||||
| 9 | PS-80%, PEO-20%, PS-b-PEO-0% | S8E22B0 | 16 | 25 | 20 | 0.5 |
| 10 | PS-80%, PEO-20%, PS-b-PEO-2% | S8E22B2 | 16 | 25 | 20 | 0.5 |
| 11 | PS-80%, PEO-20%, PS-b-PEO-4% | S8E22B4 | 16 | 25 | 20 | 0.5 |
| 12 | PS-80%, PEO-20%, PS-b-PEO-6% | S8E22B6 | 16 | 25 | 20 | 0.5 |
Characterization techniques
Differential scanning calorimetry (DSC)
A TA Q 2000 DSC instrument was used to measure the crystallization and melting behaviour of PEO. The non-isothermal crystallization studies of the blend samples were carried out by heating them from 30 to 90 °C at the heating rate of 10°C min−1 and further holding for 4 min to destroy the previous thermal history. Subsequently, the samples were cooled to −50 °C at the rate of 5°C min−1 and reheated upto 90 °C again at the rate of 10°C min−1. For thermal annealing study, samples were annealed at different temperature above the glass transition temperature (Tg) of PS for 30 minutes and then cooled to −50 °C at the rate of 5°C min−1 and reheated again upto 90 °C at the rate of 10°C min−1.Scanning electron microscopy (SEM)
The surface morphology of the samples was observed under a scanning electron microscope (SEM), ZEISS EVO 50. Before observation, samples were sputter coated with thin layer of gold to avoid the accumulation of electrons on the samples surface. In order to develop contrast, the samples were washed with water to etch out PEO.Wide angle X-ray diffraction
The crystalline structure of the PEO crystals were ascertained using wide-angle X-ray diffraction (WAXD). The WAXD analysis was carried out using a PANalytical X'Pert Pro powder diffractometer with Ni-filtered Cu Kα radiation with λ = 1.54 Å and operating at 40 kV voltage and 30 mA current. The samples were scanned at a step size of 0.02° s−1 with 2θ ranging from 10° to 40°.Results and discussion
Electrospinning
The morphology of electrospun nanofibers prepared from PS/PEO (70/30) (S7E3) and PS/PEO (80/20) (S8E2) compositions with different weight fraction of PS-b-PEO block copolymer, are shown in Fig. 1 and 2, respectively. The SEM figures showed that uniform and bead-free nanofibers were produced at the electrospinning conditions optimized during the experiments (Table 1). The distribution of nanofiber diameter is shown as inset in each of the SEM micrograph. The average diameter of the blend nanofibers was maintained at 1.1 ± 0.2 μm and 0.6 ± 0.2 μm for S8E2 and S7E3 blends containing different fraction of block copolymers. The nanofiber diameter depends on a complex interplay among various parameters such as the polymer molecular weight, the solution concentration and other various instrument parameters. It must be emphasized here that we did not observe any significant effect of polydispersity in the diameter of electrospun fibers on the crystallization behavior of PEO vis-à-vis compositional variations in the blends since most of the significant observations were a direct result of the change in the polymer concentrations. | ||
| Fig. 1 SEM micrographs of electrospun PS/PEO/PS-b-PEO blend nanofibers (a) N-S7E3B0; (b) N-S7E3B2; (c) N-S7E3B4 and (d) N-S7E3B6. | ||
 | ||
| Fig. 2 SEM micrographs of electrospun PS/PEO/PS-b-PEO blend nanofibers (a) N-S8E2B0; (b) N-S8E2B2; (c) N-S8E2B4 and (d) N-S8E2B6. | ||
Morphology of blend nanofibers
The morphology of PEO domains developed during the phase separation in PS/PEO blends was first investigated in order to further correlate it with the observed crystallization behaviour of PEO which will be discussed later. For imaging the domain morphology using SEM, the PEO fraction in the blends was first selectively etched using deionized water such that the nanofiber morphology was retained.As will be discussed later, the crystallization behaviour of PEO in the PS/PEO blend nanofibers was studied after thermally annealing the samples at three different temperatures (Ta) i.e. 90, 130 and 160 °C. Since PS has a Tg ∼ 100 °C, the morphology of samples annealed at 90 °C was expected to correspond to that of as-spun nanofibers. However, at Ta > 100 °C, the PEO domain morphology was expected to get perturbed due to the softening of the PS matrix phase and which than could affect the crystallization behaviour of the blend samples. Fig. 3 and 4 displays, respectively, the SEM micrographs of the N-S7E3B and N-S8E2B blend nanofibers, annealed at different temperatures. In the uncompatibilized blend nanofibers (N-S7E3B0 and N-S8E2B0), melt annealed at 90 °C, short empty channels were observed on the surface (Fig. 3(a) and 4(a)). These channels were aligned along the long axis of the nanofibers. The average length along the long axis of these channels was ca. 400 nm whereas the thickness was less than 100 nm. It has been reported in the past that the minority component in the nanofibers of immiscible blend may form fibrillar domains in the fibers due to the extensional flow encountered during fiber spinning.53,54 In the present case, the SEM results revealed that similar elongated domains of PEO were formed during the electrospinning of N-S8E2B and N-S7E3B blend nanofibers due to the extension of the phase-separated PEO droplets. The fibril length was seemingly longer and interconnectivity between fibrils was more prevalent in the uncompatibilized as-spun N-S7E3 blend nanofibers.
 | ||
| Fig. 3 SEM micrographs showing the morphology of PEO domains in the electrospun PS/PEO (70/30) with different weight percent of block copolymer in the nanofibers after annealing at different temperature. (a–a′′) N-S7E3B0; (b–b′′) N-S7E3B2; (c–c′′) N-S7E3B4; (d–d′′) N-S7E3B6. | ||
 | ||
| Fig. 4 SEM micrographs showing the morphology of PEO domains in the electrospun PS/PEO (80/20) with different weight fraction of block copolymer in the nanofibers after annealing at different temperatures. (a–a′′) N-S8E2B0; (b–b′′) N-S8E2B2; (c–c′′) N-S8E2B4; (d–d′′) N-S8E2B6. | ||
However, when the N-S7E3B0 blend nanofibers were melt annealed at the higher annealing temperatures (e.g. Ta = 130 and 160 °C) where the PS matrix softened, the elongated fibrils were found to undergo further breakup and/or coalescence resulting in a significant increase in the domain size as could also be observed from Fig. 5(a) which depicts the domain size variation with annealing temperature for the N-S7E3B blend nanofiber system. The ultimate PEO domain size and morphology was controlled by a competition between Plateau–Rayleigh instability induced domain breakup and the coalescence of the resulting domains as shown schematically in Fig. 6(a). At Ta ≫ TPSg, the PS matrix viscosity was low enough to facilitate the coalescence of the PEO domains such that the domain breakup and coalescence process competed against each other. In this case, the ultimate PEO domain morphology was governed by the outcome of the competition between these processes. The significant increase in domain size observed for uncompatibilized N-S7E3B0 nanofiber blend revealed that domain coalescence had a dominating influence on the final morphology.
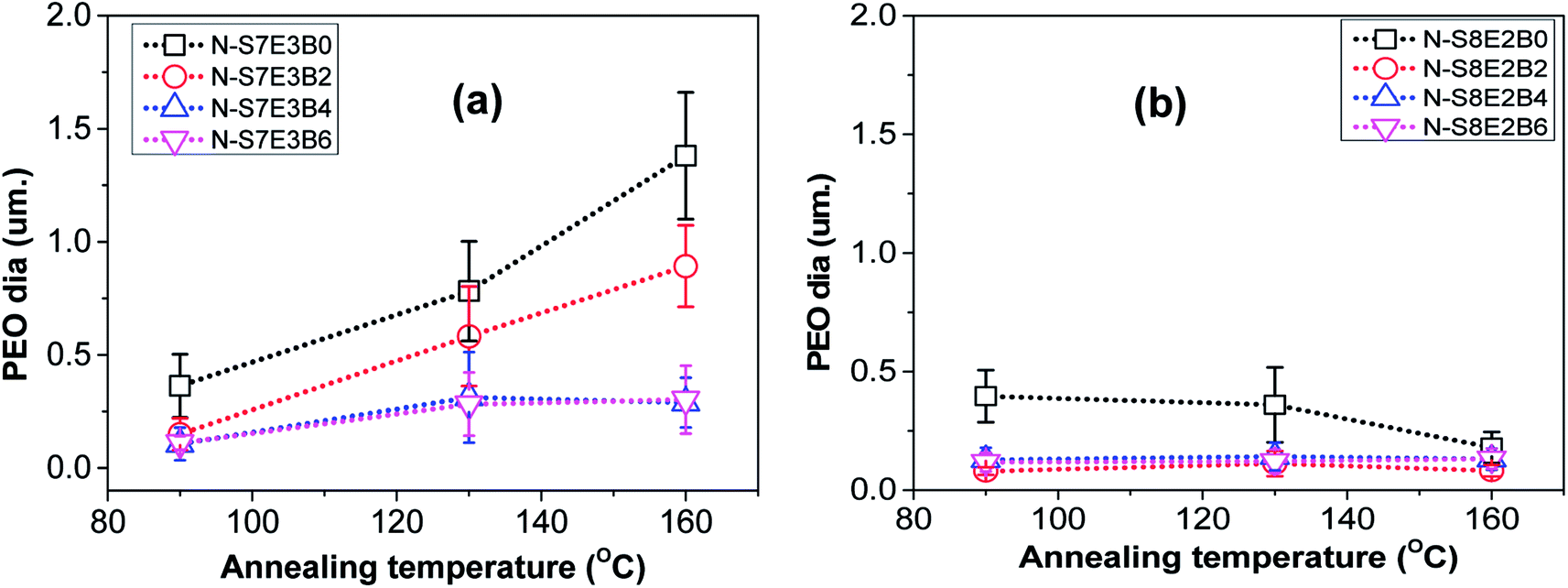 | ||
| Fig. 5 Variation in PEO domain size in nanofibers after thermal annealing at different temperatures. (a) PS/PEO (70/30) and (b) PS/PEO (80/20) nanofibers with different concentration of block copolymer. For irregular PEO domains, the diameter was approximated as the length corresponding to the major axis of the domains. | ||
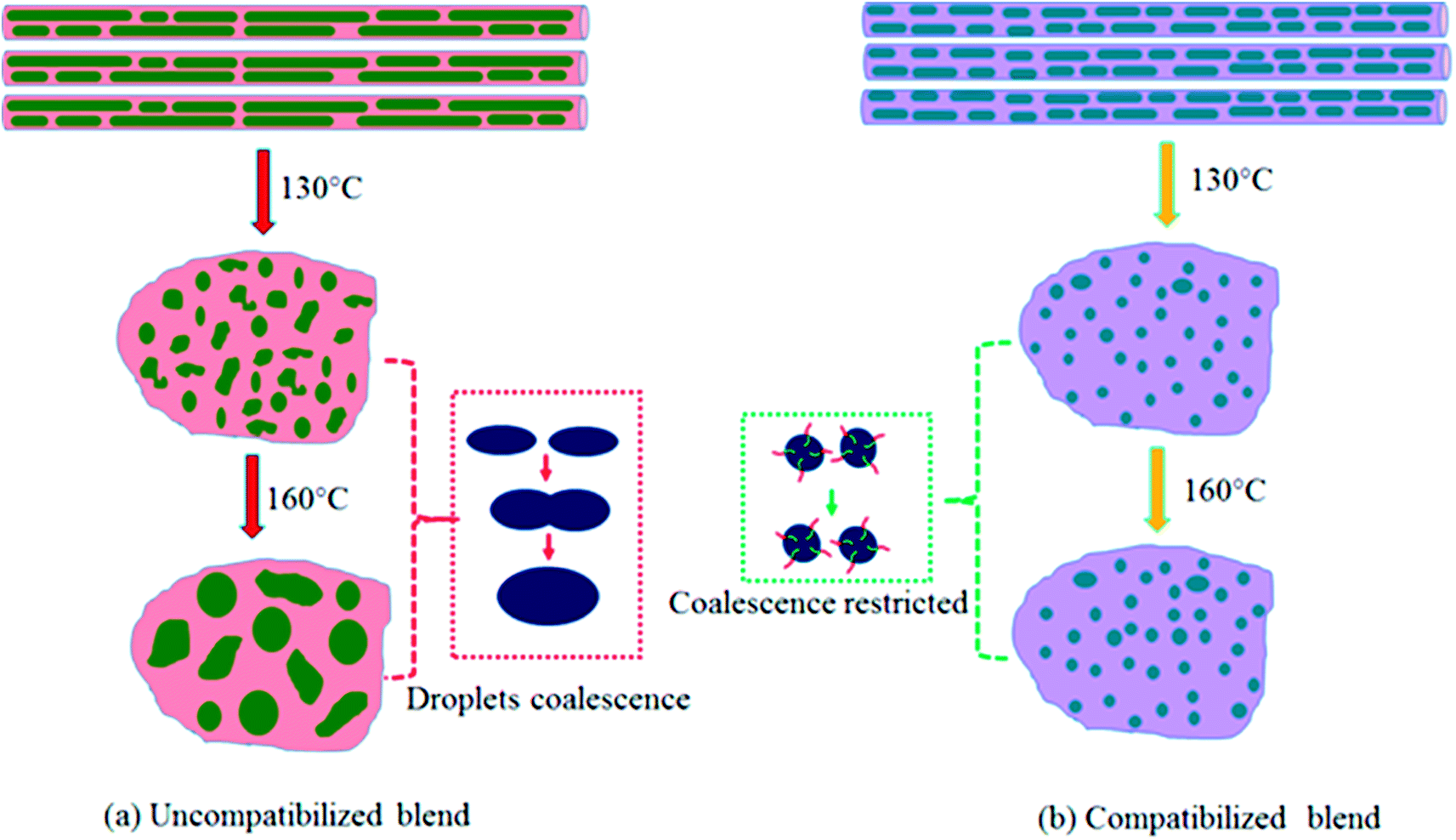 | ||
| Fig. 6 Schematic illustration depicting the morphological changes of nanofibers after treatment at different Ta. (a) Uncompatibilized blend nanofibers, (b) compatibilized blend nanofibers. | ||
Addition of block copolymer compatibilizer was found to have a profound influence on the blend nanofiber morphology. The PEO domains in the as-spun N-S7E3B2 blend nanofibers, with 2 wt% of compatibilizer, had significantly less elongated shape compared to uncompatibilized nanofiber blends. Furthermore, the compatibilization also led to a noticeable reduction in the domain size. Subsequently, further increase in the block copolymer concentration had a negligible influence on the morphology of the as-spun blend nanofibers. However, the effect of block copolymer compatibilizer was much more pronounced for the melt annealed samples. At 2 wt% of added block copolymer, the PEO domain size in the N-S7E3B2 blend nanofibers was found to increase on thermal annealing at 130 °C and 160 °C. This behaviour was similar to that observed for the uncompatibilized blend which revealed that the 2 wt% concentration of block copolymer was not sufficient to prevent the dominance of droplet coalescence during thermal annealing. When the block copolymer concentration was increased to 4 and 6 wt%, the increase in the PEO domain size at 130 °C was not very significant and, more interestingly, the domain size remained stable after further annealing at 160 °C. This revealed that the increased concentration of the block copolymer was sufficient in preventing coalescence of domains during thermal annealing. The stabilization of the PEO domains, at higher weight fraction of block copolymers, was also evident from the comparatively more regular shape of the domains after thermal annealing compared to uncompatibilized blends where the PEO domains were highly irregular in shape. Furthermore, the dispersity in the domain sizes were also markedly reduced in the compatibilized blends as could be noted from the error bars provided in Fig. 5.
The surface morphology as observed from Fig. 3, however, provided information only about the PEO domain shape and size in the N-S7E3B blends. Since the domain connectivity was another important factor influencing the crystallization behaviour, the bulk morphology of the thermally annealed blends was further investigated. Fig. 7(a) and (b) shows the cross-sectional SEM images of cryo-fractured N-S7E3B0 and N-S7E3B4 blend samples thermally annealed at 160 °C. The samples were washed with deionized water to etch out PEO domains. The first striking feature which could be ascertained from these images is that, in the N-S7E3B0 blend, the domains had extensive visible interconnectivity (Fig. 7(a)). However, in the presence of block copolymer compatibilizer, the connectivity between the domains was observed to be reduced and, hence, a higher fraction of PEO domains were present as isolated domains (Fig. 7(b)). The second significant observation was that the PEO domains were visibly more irregular and polydisperse in size in the uncompatibilized blend compared to that in the compatibilized blend. This observation further corroborated the inferences drawn from the surface morphology images as discussed above. The domain connectivity and dispersity in domain sizes was expected to additionally influence the PEO crystallization apart from their domain sizes.
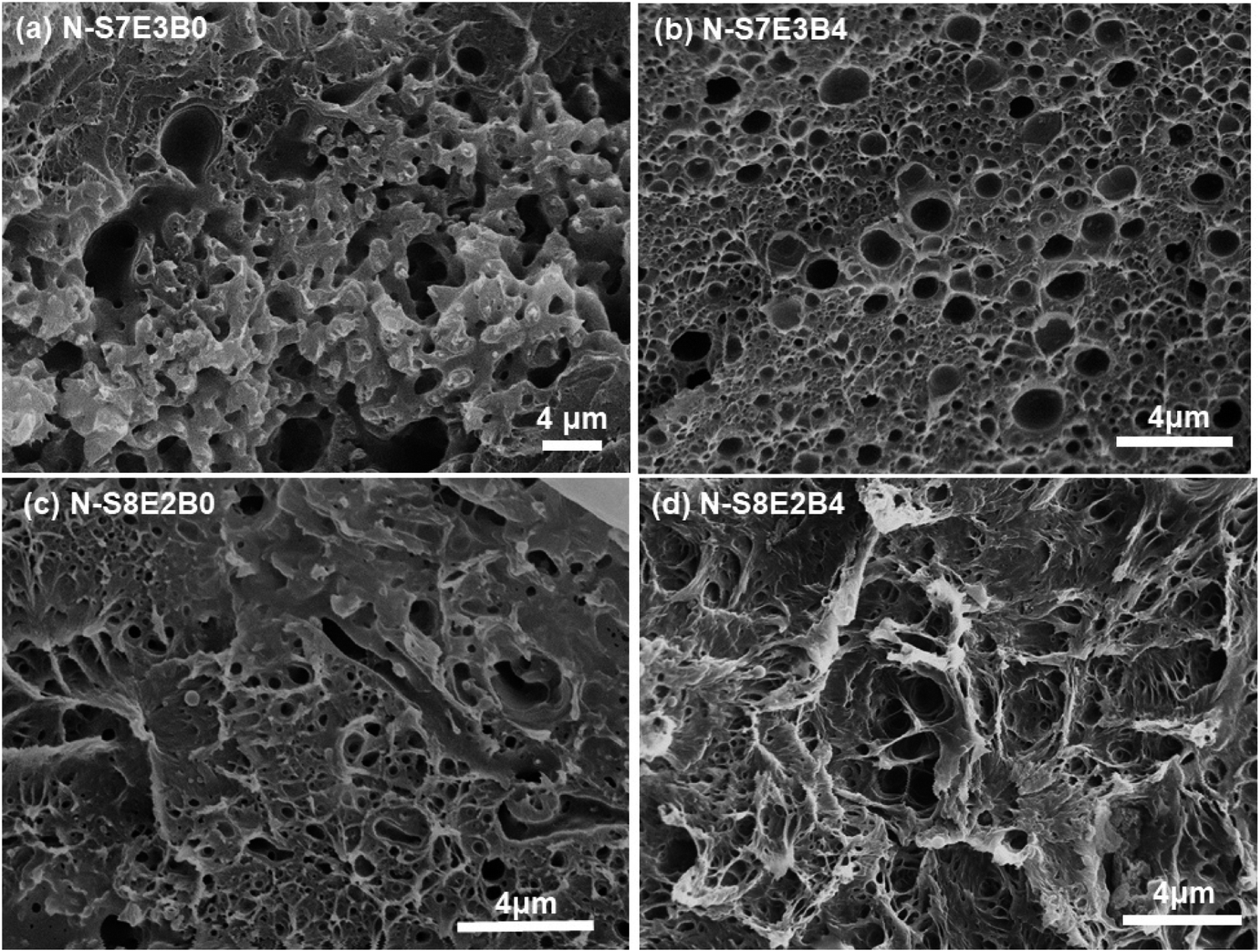 | ||
| Fig. 7 PEO domain's shape and size inside nanofibers after treatment at 160 °C. (a and c) Uncompatibilized blend nanofibers, (b and d) compatibilized blend nanofibers. | ||
In the case of N-S8E2B blend nanofibers, the PEO domain size in the uncompatibilized blend was found to significantly reduce after thermal annealing. This was in sharp contrast to the case of N-S7E3B0 blend nanofibers where the domain size increased significantly on annealing at higher temperatures. The reduction in the size of PEO domains in N-S8E2B0 blend suggested that the Rayleigh instability induced droplet break-up mechanism had a dominating influence on the final morphology. This plausibly was due to the fact that, in the N-S8E2B0 blend nanofibers, the individual PEO domains in as-spun blend nanofibers were distinctly separated from each other. Hence, in this case, the probability of domain coalescence during high temperature annealing was less such that the break-up of these domains controlled the morphology development process. The addition of block copolymer as compatibilizer, even at 2 wt%, was found to significantly reduce the PEO droplet size in the as-spun electrospun nanofibers. Further increase in the block copolymer concentration did not had any additional significant effect on the domain morphology. The domain size also remained unchanged on thermally annealing the compatibilized N-S8E2B blend nanofibers which revealed the stabilization effect of the block copolymer on domain morphology. The bulk morphology of the thermally annealed N-S8E2B blends was revealed from Fig. 7(c) and (d). The SEM images showed that, in the uncompatibilized N-S8E2B0 blends, the PEO domains were present without any connectivity which was different than that observed for the N-S7E3B0 blends (Fig. 7(c)). Hence, the effect of compatibilization in the N-S8E2B blends was seen only in further reduction in the average domain size as also observed from the surface morphology results (Fig. 7(d)). Furthermore, the effect of compatibilization was also visible from the significant difference in the fracture mode observed for the samples. The N-S8E2B4 blends demonstrated higher ductile mode of deformation than the N-S8E2B0 blends due to improved interfacial strength between the PS matrix and PEO droplet phase.
Discussion on the block copolymer compatibilization effect
The compatibilization effect of block copolymer results only if they are localized at the interface. This lowers the interfacial tension (facilitating droplet break-up), prevent the droplet coalescence, and enhance interfacial adhesion. Hence, the role of PS-b-PEO block copolymer in compatibilizing the PS/PEO blend nanofibers will now be discussed in this section in terms of interfacial coverage i.e. how much of them might be at the interface. Distinction will also be made between dynamic and static coalescence of the domains expected during processing (electrospinning) and thermal annealing, respectively. It must also be mentioned that, in the present work, a symmetric PS-b-PEO block copolymer was used as it has been shown in the past that a symmetric diblock copolymer generally is more efficient as a compatibilizer than the asymmetric diblock copolymer of the same molecular weight.66,67According to Macosko and coworkers,60,62,64 if it is assumed that all the added copolymer is at the interface, then the number of block copolymer chains per nm2 (Σ) will be:
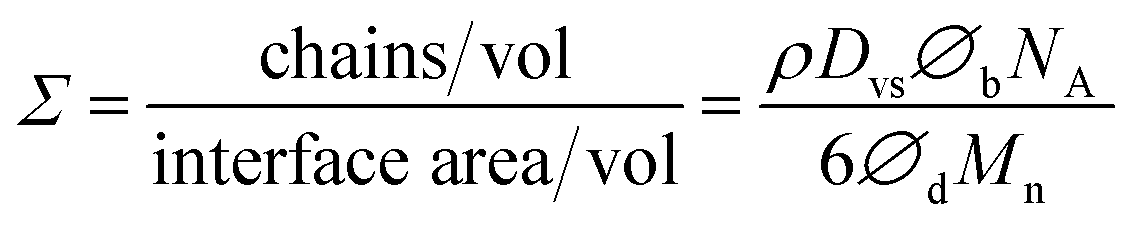 | (1) |
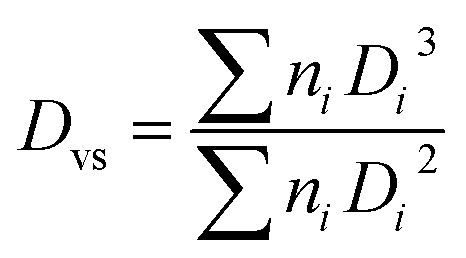 | (2) |
In the case of electrospun nanofiber blends since the dispersed phase domains in the as-spun nanofibers were mostly elongated, the interfacial coverage calculations were done using the domain size values for samples thermally annealed at 130 °C. Values of Σ so obtained for different blend nanofibers is given in Table 2. The maximum block copolymer coverage (Σo) can be estimated by assuming a dense monolayer of the block copolymer at the interface. Hence, considering symmetric diblock copolymer at the PS/PEO interface to be similar to lamellar dry-brush:60,62,64
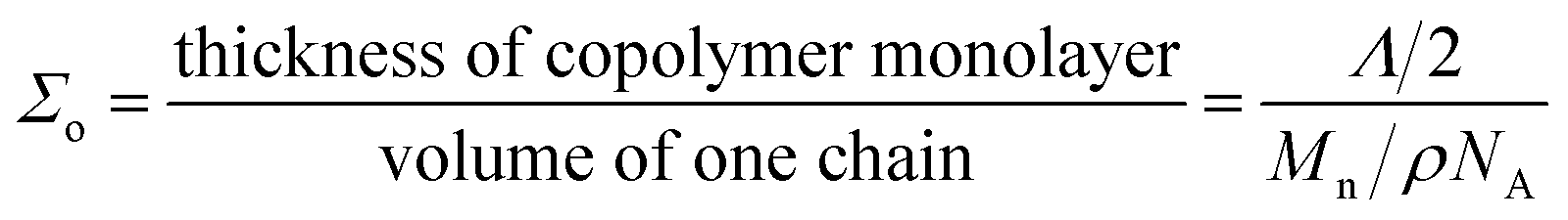 | (3) |
| Composition (w/w) | D (μm) at 130 °C | DA (μm) at 160 °C | Σ (chains per nm2) | Σ/Σo | Stability |
|---|---|---|---|---|---|
| N-S7E3B0 | 0.78 | 1.38 | 0 | 0 | No |
| N-S7E3B2 | 0.62 | 0.99 | 0.044 | 0.40 | No |
| N-S7E3B4 | 0.31 | 0.29 | 0.063 | 0.57 | Yes |
| N-S7E3B6 | 0.28 | 0.30 | 0.068 | 0.62 | Yes |
| N-S8E2B0 | 0.36 | 0.18 | 0 | 0 | No |
| N-S8E2B2 | 0.12 | 0.08 | 0.015 | 0.13 | Yes |
| N-S8E2B4 | 0.14 | 0.13 | 0.031 | 0.28 | Yes |
| N-S8E2B6 | 0.13 | 0.13 | 0.038 | 0.34 | Yes |
Macosko and coworkers60 have shown that, in case of PS/PMMA (70/30) blends, 20% interfacial coverage was enough for the PS-b-PMMA block copolymer to stabilize the droplets against static coalescence. However, in their case, the ratio of the homopolymer molecular weight was lower compared to the corresponding block molecular weight in the diblock copolymer. This situation, according to Leibler's brush theory,69 corresponds to the wet-brush case where the homopolymer solubilizes in the corresponding block of the block copolymer. In this case, the block copolymer present at the interface was stable due to the entanglement of the blocks with the respective homopolymers. However, in the present system, the homopolymer molecular weight (both PS and PEO) was much higher than the corresponding block molecular weight such that it corresponded to the dry-brush case. Here, the homopolymers were not soluble with their respective blocks of the block copolymer. In such a scenario, the block copolymer preferred to localize at the interface. However, the blocks were likely to remain untangled with the corresponding homopolymer such that the block copolymer was free to slide along the interface. Hence, a higher interfacial coverage of the block copolymer was necessary to prevent coalescence of the domains. In the case of blend nanofibers with 4 and 6 wt% block copolymer, annealing at 160 °C did not led to any notable change in the domain size. Hence, an interfacial coverage of 57% at 4 wt% of added block copolymer was optimum for preventing domain coalescence in the N-S7E3B blend nanofibers. In the case of N-S8E2B blends, the interfacial coverage of block copolymers was not very crucial for stabilization of PEO droplet size since even in the absence of compatibilizer the droplet coalescence was not significant as is evident from the reduction in the domain size on thermal annealing. The PEO domains, in this case, were already well separated from each other plausibly due to lower dispersed phase content in the blend. Hence, even at 2 wt% of added block copolymer in the N-S8E2B2 blend nanofibers and with an interfacial coverage of 13%, the PEO droplets were found to be stable against static annealing at 160 °C.
Crystallization behaviour
The crystallization behaviour of the compatibilized PS/PEO blend nanofibers and cast films was investigated using DSC measurements. The PS and PEO were expected to be immiscible in the melt state.70 The crystallization of PEO thus occurred predominantly within the PEO-rich domains. Our previous study had revealed that in the case of uncompatibilized PS/PEO blends, PEO crystallized through heterogeneous nucleation mechanism in bulk samples.57,58 However, in the blend nanofibers, PEO showed composition dependent crystallization behaviour where the nucleation mechanism shifted from heterogeneous to homogenous when the weight fraction of PEO in the blend was lowered from 0.3 to 0.2. In the present study, the immiscible PS/PEO blend was compatibilized using PS-b-PEO diblock copolymer. The weight fraction of the diblock copolymer, with respect to total PS/PEO weight, in the blend was varied from 0 to 0.06. It was expected that the compatibilization will lead to a finer dispersion of PEO in the blend leading to a stronger confinement effect on its crystallization behaviour.Fig. 8 shows the DSC cooling curves for recording the crystallization exotherm of PEO in bulk and nanofiber samples of S8E2B and S7E3B blends containing different fraction of block copolymers. For the bulk, as-casted, uncompatibilized blend sample (C-S7E3B0 and C-S8E2B0), the crystallization exotherm for PEO was observed at 46 °C. The crystallization at such a low supercooling is known to occur via heterogeneous nucleation mechanism which is prevalent for crystallization in the bulk state. The compatibilization using PS-b-PEO block copolymers was not found to have any significant influence on the crystallization behaviour of PEO in the bulk blend samples (Fig. 8(a) and (b)). Hence, the dominance of heterogeneous nucleation mechanism even in the compatibilized bulk blend samples implied that the average size of PEO domains was still sufficiently large, such that the number density of the PEO-rich domains remained less than that of the heterogeneous nuclei present. The reduction of domain size in bulk blend samples due to compatibilization, in absence of strongly interacting block copolymer compatibilizer, was not expected to decrease the domain size below micrometer range. However, in the past63,71,72 it has been shown that a strongly interacting block copolymer compatibilizer can lead to appreciable reduction in domain size where the domain diameter could be in the range of few hundreds of nanometers.
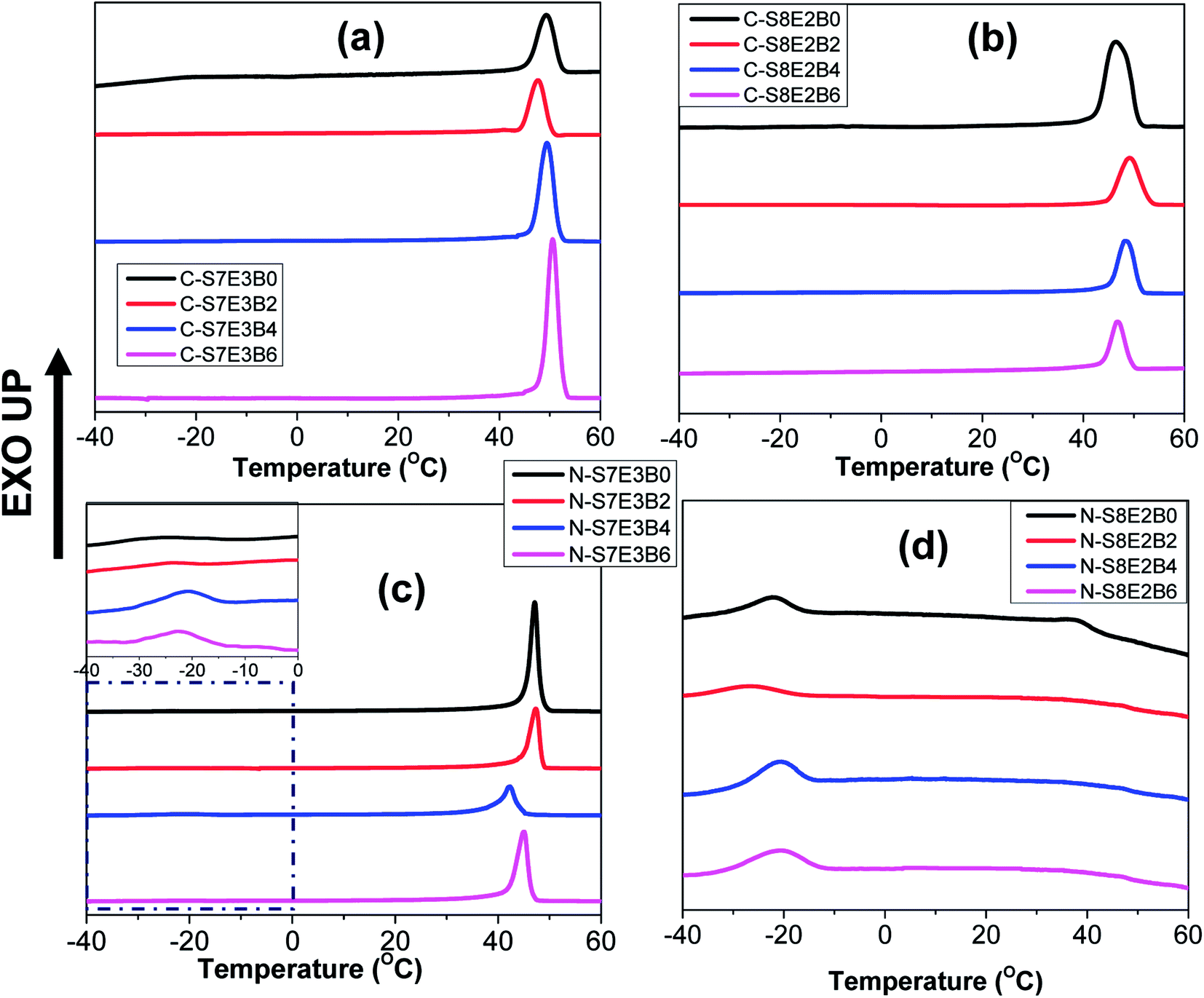 | ||
| Fig. 8 DSC cooling curves of PS/PEO/PS-b-PEO blends, prepared as as-casted films and nanofibers, after first heating upto 90 °C. (a and b) Cast film and (c and d) nanofibers with different weight fraction of block copolymer added samples. | ||
The compatibilization of PS/PEO blends using PS-b-PEO block copolymer, however, noticeably influenced the PEO crystallization behaviour in their electrospun nanofibers. Fig. 8(c) and (d) shows the DSC cooling curves for N-S7E3B and N-S8E2B blend nanofibers. The Tc, in the N-S7E3B blend nanofiber, was observed approximately at the same location as that of bulk PEO samples, signalling that the crystallization in the PEO-rich domains developed in these nanofibers proceeded through heterogeneous nucleation mechanism. However, a very weak second crystallization exotherm was also observed at much higher supercooling (Tc ∼ −23 °C). In the case of N-S7E3B blend nanofibers containing the block copolymer compatibilizer, the Tc observed at lower supercooling, due to heterogeneous nucleated crystallization, was found to shift slightly to further lower temperatures. More significantly, the intensity of the weak crystallization peak, located at higher supercooling, gradually increased as the weight fraction of the block copolymer was increased and a maximum was observed for the N-S7E3B4 blend nanofiber containing 0.04 weight fraction of the PS-b-PEO compatibilizer. In the N-S8E2B0 blend nanofibers containing 20 wt% of PEO homopolymer, the crystallization exotherm at lower undercooling had considerably diminished intensity and the broad peak was centred at ∼37 °C (Fig. 8(d)). However, the second peak observed at much higher supercooling (Tc ∼ −23 °C) was relatively sharp. Significantly, the addition of PS-b-PEO block copolymer resulted in almost complete depression of the crystallization peak observed at lower supercooling. Furthermore, the crystallization exotherm at higher supercooling either shifted further to lower temperature or depicted a marked increase in the intensity in the presence of block copolymer compatibilizer.
The crystallization observed for the blend nanofibers, at higher supercooling, is known to occur due to homogeneous or self-nucleation. A crystalline polymer generally crystallizes via heterogeneous nucleation mechanism where the inherent heterogeneities in the polymer act as nucleating agents. However, if a given polymer volume is partitioned into a number of smaller domains such that it exceeds the number of impurities present, the polymer in a fraction of the domains than can only crystallize via homogenous nucleation at much higher supercoolings. The crystallization of PEO blocks, forming cylindrical or spherical nanodomains, in polybutadiene-block-poly(ethylene oxide) (PB-b-PEO) block copolymers was shown to occur predominantly through homogenous nucleation mechanism.73 Similarly, when PEO was infiltrated in nanopores of AAO membrane with pore diameter less than 20 nm, the crystallization was observed to occur mostly through homogenous nucleation mechanism.35,36 Apart from this, effect of polymer architecture, polydispersity, additives and impurities on the crystallization behaviour of PEO confined in AAO membranes have also been reported.74,75 Considering that PEO in the PS/PEO blends under study was the minor component, it was expected that PEO will form the dispersed phase in the PS matrix. In the solution casted films, the size of PEO dispersed domains was typically of several tens of micrometers, such that heterogeneous nucleation became the dominant nucleation mechanism. However, the smaller diameter of the nanofibers inhibit the coarsening of the PEO domains, thereby restricting the PEO domain size to few tens or hundreds of nanometers. In this case, the number of PEO domains per unit volume may became higher than that of the heterogeneities present which suppressed the heterogeneous nucleation process.
Floudas and coworkers36 have shown that a typical PEO spherulite has a diameter of ∼300 μm. Hence, the resulting volume per impurity per nucleus will be ∼10−2 mm3. The average size of the PEO domains in the uncompatiblized as-spun (or annealed at Ta = 90 °C) N-S7E3B and N-S8E2B blend nanofibers were found to be ca. 400 nm (Fig. 5). The volume of the domain, assuming them to be spherical, with a 400 nm diameter amounts to ∼3.3 × 10−11 mm3. It must be noted that for the, as-spun nanofibers, the domain geometry was more like ellipsoid and the size of the domain measured corresponded to that along their long axis. Hence, the actual volume of such a domain was expected to be less than 3.3 × 10−11 mm3. Since this pore volume was many orders of magnitude smaller than the volume per heterogeneous nucleus in bulk PEO, only a small portion of the PEO domains were expected to contain heterogeneous nuclei. If the domains were isolated, such a scenario would have resulted in only small fraction of PEO crystallizing via heterogeneous nucleation. However, the N-S7E3B0 blend nanofibers crystallized dominantly through heterogeneous nucleation which attested to the fact that the spatial connectivity between the domains was extensive as also revealed from the SEM results. This was further corroborated by the fact that N-S8E2B0 blend nanofibers crystallized dominantly via homogenous nucleation mechanism even though the average domain size was almost same as that in N-S7E3B0 blend nanofibers. As revealed from the SEM images, the PEO domains in the N-S8E2B0 blend nanofibers had limited spatial connectivity thus limiting the crystallization growth initiated via heterogeneities to few domains. It must also be noted that in the as-casted PS/PEO blends, the domain dimensions were several tens of micrometers57 and, hence, a significant fraction of PEO domains were likely to contain heterogeneities such that they crystallized almost exclusively through heterogeneous nucleation mechanism.
The addition of PS-b-PEO block copolymer in the PS/PEO blend nanofibers resulted in the suppression of the crystallization peak due to heterogeneous nucleation, whereas, at the same time, the intensity of the crystallization peak attributed to homogenous nucleation increased. This demonstrated that the size of the PEO domains decreased in the compatibilized PS/PEO blends such that the number density of the PEO domains increased after compatibilization. The increased number density of PEO domains resulted in a higher fraction of them being heterogeneity free and, hence, a higher fraction of PEO crystallized via homogenous nucleation mechanism. During the electrospinning process, as the solvent evaporated and solution jet was stretched towards the collector plate, the PS-b-PEO block copolymers were preferentially adsorbed at the PS/PEO domain interface. Hence, this interfacial localization of the block copolymer prevented domain coarsening through coalescence during the solvent evaporation process resulting in reduced PEO domain size. This indeed was revealed also from the morphological data, extracted from SEM and discussed in previous section, which showed a reduction in the domain size as well as their dispersity signifying that a higher fraction of PEO was heterogeneity free. Furthermore, spatial connectivity between domains has significant effect on the crystallization behaviour as is clear from a comparison of the crystallization behaviour of N-S7E3B4 and N-S8E2B0 blends. The domain size of N-S8E2B0 blend was almost 4 times that of the N-S7E3B4 blends (Fig. 5). However, whereas N-S8E2B0 blend crystallized mostly through homogenous nucleation mechanism, heterogeneous nucleation was dominant in N-S7E3B4 blends. As indeed revealed from the bulk morphology, this was due to more spatial connectivity between PEO domains in the N-S7E3B4 blends in comparison to that in N-S8E2B0 blends. The spatial connectivity between PEO domains enabled the spread of a single heterogeneity driven crystallization event to many neighbouring PEO domains.
The melting behaviour of the as-cast bulk and nanofiber blend samples is shown in Fig. 9. In the case of bulk uncompatibilized bulk blends (C-S7E3B0 and C-S8E2B0) the peak melting temperature (Tm) of PEO was observed at ca. 66 °C. The Tm of uncompatibilized blend nanofibers were found to be significantly depressed as compared to that of the bulk blend samples especially for the N-S8E2B0 nanofibers. The reduction of melting point suggested that the crystalline lamellar thickness of PEO in the nanofibers, particularly at the lower weight fractions, was significantly thinner than that of bulk PEO.38 It must be noted that under confined condition, the c-axis i.e. the thickness direction of the crystalline lamellae normally aligns perpendicularly to the long axis of the confining geometry since this orientation provides maximum opportunity for the kinetic growth of the crystallites. However, in the fibril shaped PEO domains, the crystal growth in the lateral direction normal to the long axis of the PEO fibrillar domains were expected to be restricted by the fibril diameter resulting in the thinner lamellae and, hence, of lower thermal stability.
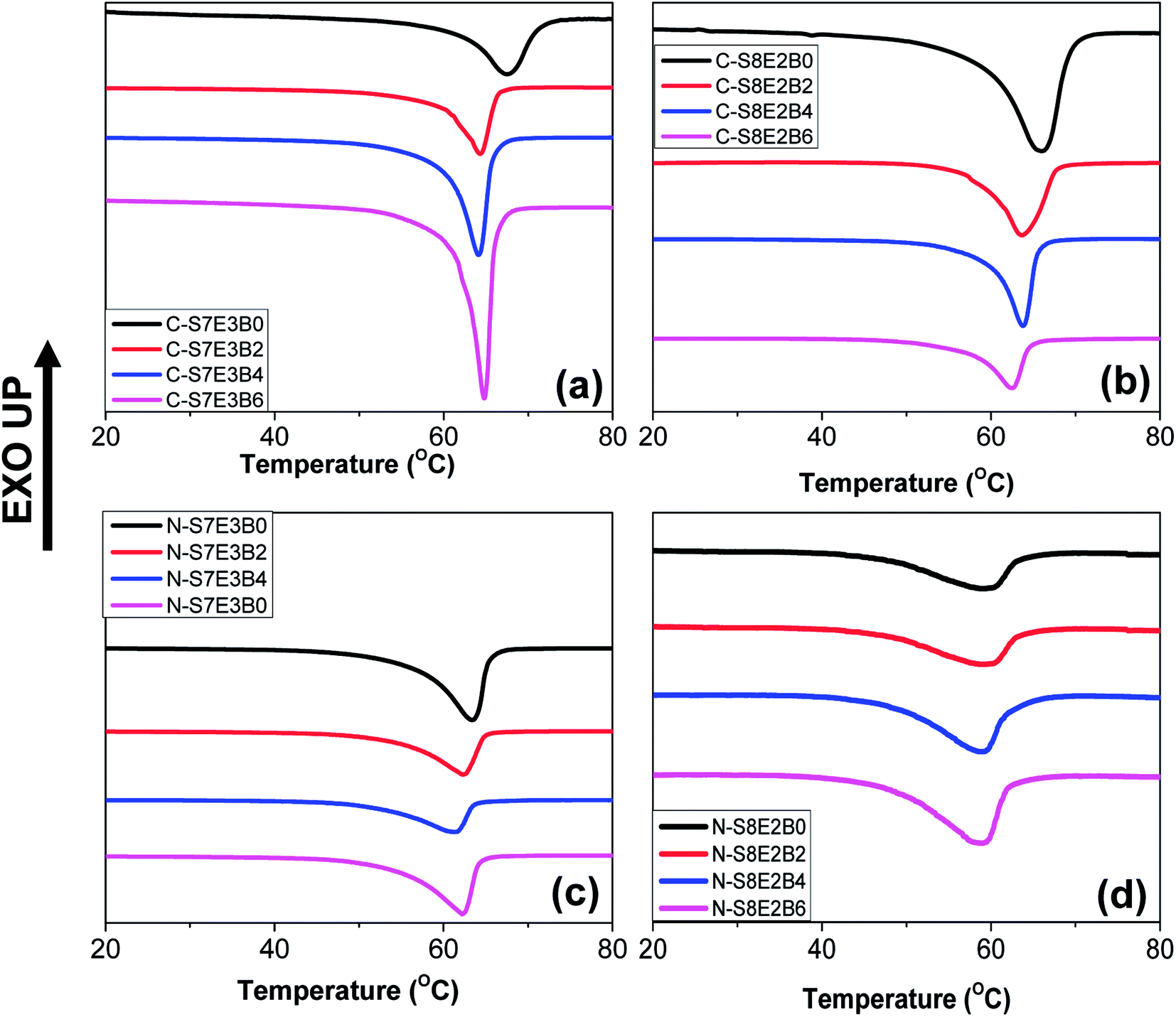 | ||
| Fig. 9 DSC heating curves of PS/PEO/PS-b-PEO blend nanofibers and cast film after first heating up to 90 °C. (a and b) Cast film and (c and d) nanofibers with different weight fraction block copolymer added samples. | ||
The variation of Tm with block copolymer fraction in the PS/PEO blends is shown in Fig. 10(a). The Tm depressed by upto 2–4 °C both in the bulk as well as nanofiber blend samples after addition of the block copolymer compatibilizer. Furthermore, it was also found that this effect nearly saturated after 4 wt% of block copolymer has been added. The depression in the melting plausibly resulted from decreased domain size due to interfacial stabilization effect of the block copolymer. Furthermore, the variation of heat of fusion per unit weight of PEO, which is a measure of the degree of crystallinity of PEO, with varying fraction of PS-b-PEO block copolymer in the blend is shown in Fig. 10(c). The crystallinity in nanofibers was much lower than that in bulk blend samples and was found to be least when the PEO composition was reduced to 10 wt%. As has already been indicated, PEO domains were expected to be small and well partitioned in the PS matrix at lower PEO weight fraction; hence, the suppression of crystallizability could be attributed to the restricted growth of the crystallites in the geometrically mismatched and nanoscopically confined PEO domains. The addition of block copolymer as compatibilizer additionally influenced the degree of crystallinity of the blends. In both the bulk as well as nanofiber PS/PEO blends, the heat of fusion was found to decrease with increase in the weight fraction of block copolymer upto 0.04 weight fraction after which the effect was not significant. Furthermore, the effect was more pronounced in the compatibilized N-S7E3 blend nanofibers as the crystallization was already considerably suppressed in the neat N-S8E2 blend nanofibers. Hence, the crystallization and melting behaviour of the PS/PEO blends clearly demonstrated that addition of PS-b-PEO block copolymer as compatibilizer does significantly influenced the PEO domain stability leading plausibly to reduction in the overall average domain size.
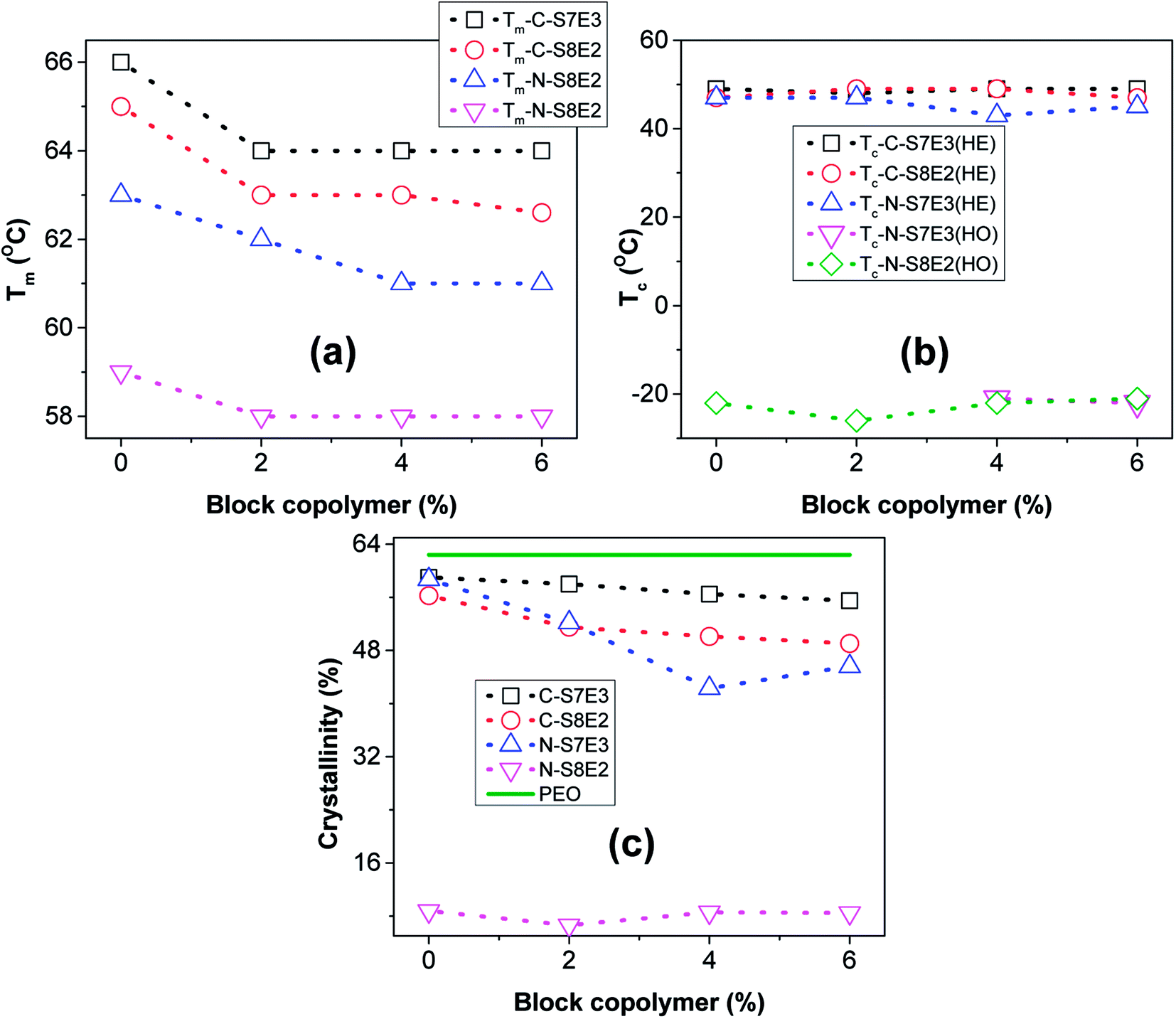 | ||
| Fig. 10 Variations of the thermal properties as a function of PS-b-PEO content, as ascertained from DSC measurements, in as-casted and electrospunned PS/PEO blends (a) melting peak temperature (Tm); (b) crystallization peak temperature (Tc); (c) normalized heat of fusion. The respective values for the neat PEO sample, in each case, has been represented with the dashed line. | ||
Effect of melt annealing temperature
The crystallization behaviour discussed so far was studied by heating the blends upto 90 °C which was below the Tg of PS (TPSg). Hence, in this case, the crystallization of PEO domains occurred under the hard glassy confinement imposed by PS matrix. Since the fibrillar geometry of the PEO domains may be metastable in nature, the domain morphology may get perturbed after annealing the blend nanofibers above the TPSg of PS which would also influence the crystallization behaviour. Furthermore, the use of block copolymer as compatibilizer is known to prevent domain coalescence during static annealing and, hence, the more significant effect of compatibilization of PS/PEO blend nanofibers was expected to be observed via the present melt annealing study. For this study, N-S7E3B and N-S8E2B blend samples were crystallized after annealing at different temperatures (Ta > TPSg) for 30 min. Fig. 11 shows the DSC cooling curves of N-S7E3B blend samples, containing different fraction of block copolymers, obtained after annealing at different temperatures. For the corresponding cast film samples, the Tc and Tm of PEO were found to remain almost unperturbed after the samples had been annealed between 90 and 160 °C (please see ESI†). This showed that the spatial continuity of the PEO domains was not strongly disturbed in the bulk after the melt annealing treatment even in the presence of block copolymer compatibilizer. Hence, even though the compatibilizer may restrict the domain coarsening during annealing, the size of the PEO domain remained relatively large such that the crystallization behaviour of PEO in compatibilized blends was similar to that for uncompatibilized blends even after the thermal annealing for the bulk samples. The melt annealing treatment, however, had profound effect on the crystallization behaviour in the compatibilized PS/PEO blend nanofibers.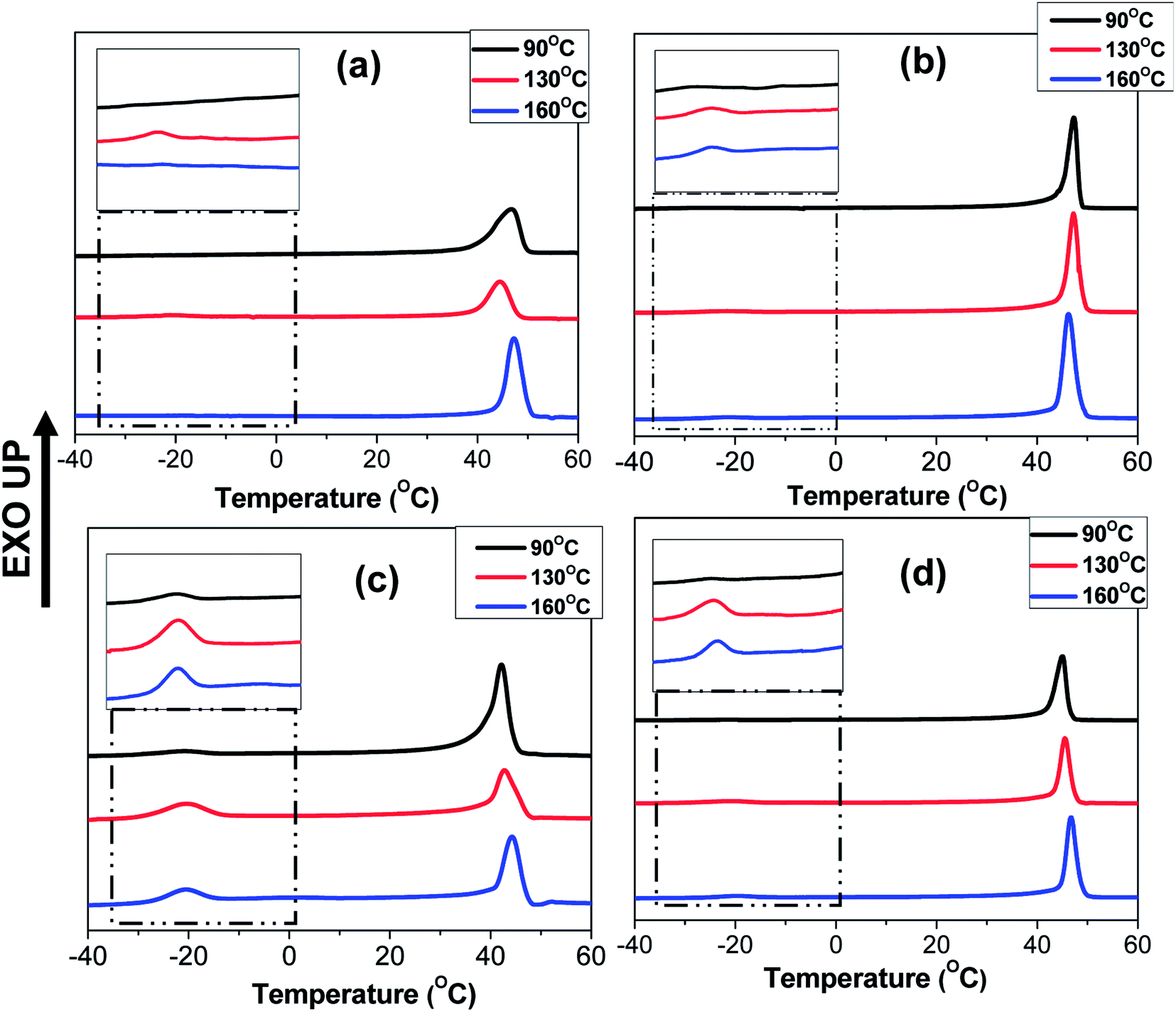 | ||
| Fig. 11 DSC cooling curves of PS/PEO (70/30) with different weight fraction of block copolymers in nanofibers after annealing at different temperature. (a) N-S7E3B0, (b) N-S7E3B2, (c) N-S7E3B4 and (d) N-S7E3B6. | ||
In the case of N-S7E3B0 blend nanofibers, crystallization after annealing above TPSg at 130 °C resulted in a small crystallization exotherm at much higher supercooling apart from the dominant exotherm at lower supercooling, as shown in the enlarged thermogram in the inset of Fig. 11(a). However, the small exotherm at higher supercooling completely disappeared when Ta was further increased to 160 °C. The additional exotherm observed at the exceedingly low Tc was associated with the homogeneously nucleated crystallization. Hence, the melt annealing experiments, in case of uncompatibilized blend nanofibers, demonstrated that with increasing Ta the confinement effect on PEO crystallization first increased but then vanished when Ta was sufficiently high (≥160 °C). This is also evident from Fig. 12 which depicts heat of crystallization occurring via homogenous nucleation at different annealing temperatures. However, in the compatibilized blend nanofibers, the increase in the intensity of the crystallization exotherm observed at higher supercooling was more significant after annealing at 130 °C. Furthermore, the intensity of the exothermic peak was observed to be highest when the weight fraction of the block copolymer in the blend was 0.04 and subsequent increase in the block copolymer concentration did not had any further noticeable effect. However, more significantly, it was observed that the intensity of the crystallization exotherm further increased for samples melt annealed at higher temperature i.e. Ta ∼ 160 °C. This is also evident from the heat of crystallization data in Fig. 12. This strongly indicated that the confinement effect on PEO crystallization increased in the presence of block copolymer compatibilizer plausibly due to the presence of higher fraction of smaller PEO domains at higher annealing temperatures.
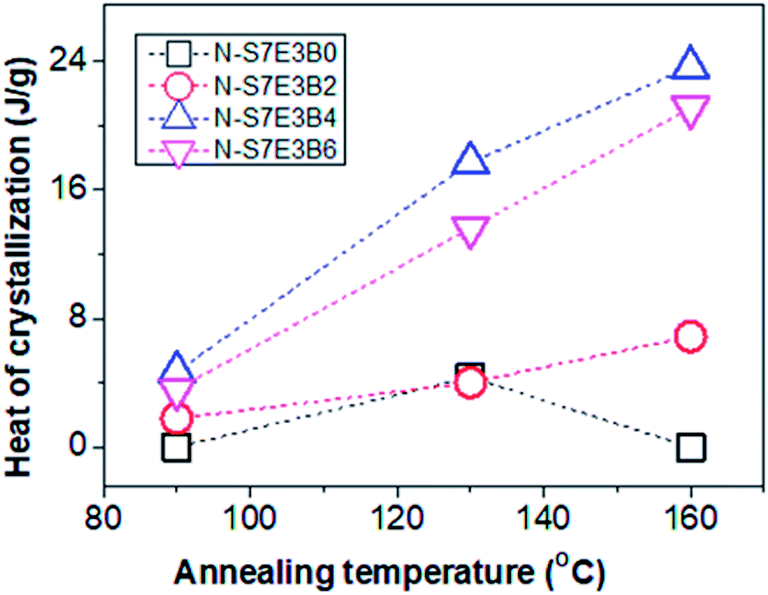 | ||
| Fig. 12 Heat of crystallization of homogeneous nucleated crystallization of PEO, in PS/PEO (70/30) nanofibers with different weight fraction of block copolymer, after annealing at different temperatures. | ||
The crystallization behaviour of melt annealed N-S8E2B blend nanofibers also further demonstrated increased effect of confinement in the presence of block copolymer compatibilizer. In this case, the crystallization behaviour of uncompatibilized N-S8E2B0 blend nanofibers already was dominated by homogenous nucleation due to smaller domain size and poor domain connectivity (please see ESI†). Hence, the effect of block copolymer compatibilizer on crystallization behaviour was comparatively less significant such that the crystallization was dominated by homogenous nucleated crystallization. However, the total heat of crystallization in the compatibilized blend nanofibers was less than that in the uncompatibilized blend nanofibers for samples melt annealed at higher temperatures (please see ESI†). This was plausibly due to much smaller domain volumes created after melt annealing, in the presence of block copolymer, leading to increasingly frustrated crystallization behaviour. Hence, in the presence of PS-b-PEO block copolymer, the melt annealing driven confinement effect on crystallization was much more prominent. Furthermore, the block copolymer also had a strong stabilization effect, on the increased confinement effect, against high temperature melt annealing.
The crystallization behaviour observed for the compatibilized N-S7E3B and N-S8E2B sample after the melt annealing can be correlated with the domain morphologies observed and discussed in the previous sections. In the uncompatibilized N-S7E3B0 blend nanofibers, the large domain size and interdomain connectivity prescribed a high probability of finding heterogeneities in each PEO domain, such that the crystallization was bound to take place through heterogeneous nucleation mechanism. However, at Ta > TPSg, i.e. 130 °C, the Rayleigh–Plateau instability induced breakup of the fibrillar PEO domains occurred, leading to the formation of small sized domains as well as reduction in the interdomain connectivity as shown schematically in Fig. 6(a). In this case, the crystallization within these small fraction of smaller domains occur through homogeneous nucleation. At sufficiently high Ta (160 °C), the coalescence of the domains was more dominant and the merged domains were large enough crystallize through heterogeneous nucleation again. In the compatibilized N-S7E3B blend nanofibers, the PEO domains were relatively small even in the as-spun nanofiber such that several of such domains were heterogeneity free which crystallized through homogeneous nucleation. The thermal annealing of the compatibilized blends led to the formation of a higher fraction of smaller PEO domains such that a higher content of polymer crystallized via homogenous nucleation and, hence, increase in the heat of homogeneous crystallization was observed. It must also be noted that the presence of block copolymer at the interface suppressed the coalescence phenomena such that annealing at further higher temperatures did not led to further increase in the domain diameters (Fig. 6(b)). Furthermore, since the fraction of smaller PEO domains were higher in the blends with 4 and 6 wt% of block copolymer due to enhanced stabilization against coalescence, the PEO fraction crystallizing via homogenous nucleation was also higher. It must also be mentioned that apart from reduction in domain size, decrease in the spatial connectivity between the domains after compatibilization also was a major factor contributing to enhanced homogenous nucleated crystallization. Similarly, in the case of N-S8E2B blend nanofibers, the increase in homogenous nucleated crystallization observed was due to the reduced domain size because of compatibilization effect.
Effect of PEO molecular weight
The compatibilization effect of PS-b-PEO block copolymer on PEO crystallization in the PS/PEO blend nanofibers was further investigated by using a low molecular weight PEO (Mn ∼ 4000). In this case, the ratio of the PEO homopolymer molecular weight with that of PEO block molecular weight was less than one. This corresponded to the wet-brush situation according to the Leibler's brush theory.69 Hence, it was expected that the PEO homopolymers will homogeneously wet the PEO blocks of the block copolymers localized at the interface. This was expected to further influence the morphology and crystallization behaviour of PEO in the compatibilized PS/PEO blend nanofibers.Fig. 13(a–c) shows the DSC curves collected in the cooling cycle for recording the crystallization exotherm of PEO in nanofiber samples of N-S8E22B blends, containing different fraction of block copolymers, after thermal annealing at different temperatures. Fig. 13(a) corresponded to cooling curves after thermal annealing at 90 °C i.e. below the Tg of PS. The crystallization behaviour observed in this case could be related to the blend morphology in the as spun nanofibers. The DSC cooling profile of the uncompatibilized N-S8E22B blend nanofiber showed a sharp crystallization peak situated at ca. 39 °C and two more broad crystallization peaks at higher supercooling (−17 °C and −25.5 °C). Hence, the heterogeneous and homogenous nucleated crystallization were equally dominant for low molecular weight PEO in the blend nanofibers. This behavior was distinct from that observed for high molecular weight PEO where the heterogeneous nucleation was suppressed in the N-S8E2 blend nanofibers. Furthermore, whereas only one crystallization peak was observed at higher supercooling in the N-S8E2 blend nanofibers, the blend nanofibers with low molecular weight PEO depicted two prominent peaks. This plausibly was due to significantly lower viscosity of the low molecular weight PEO which aided the domain break-up as well as coalescence. This was expected to increase the polydispersity in the PEO domain sizes where domains of much larger sizes could also be present owing to dominance of coalescence in the absence of compatibilizer. Increase in the size of domain favoured the heterogeneous nucleation whereas the polydispersity in the domain size led to multiple homogenous nucleated crystallization events. Furthermore, the crystallization peak at −25.5 °C was lower than that observed for the homogenous nucleated crystallization peak in N-S8E2 blends where high molecular weight PEO was used. Floudus and coworkers have shown that the homogenous nucleation temperature decreases as the molecular weight of polymer decreases due to decrease in the liquid to glass temperature for low molecular weight polymers.35,36
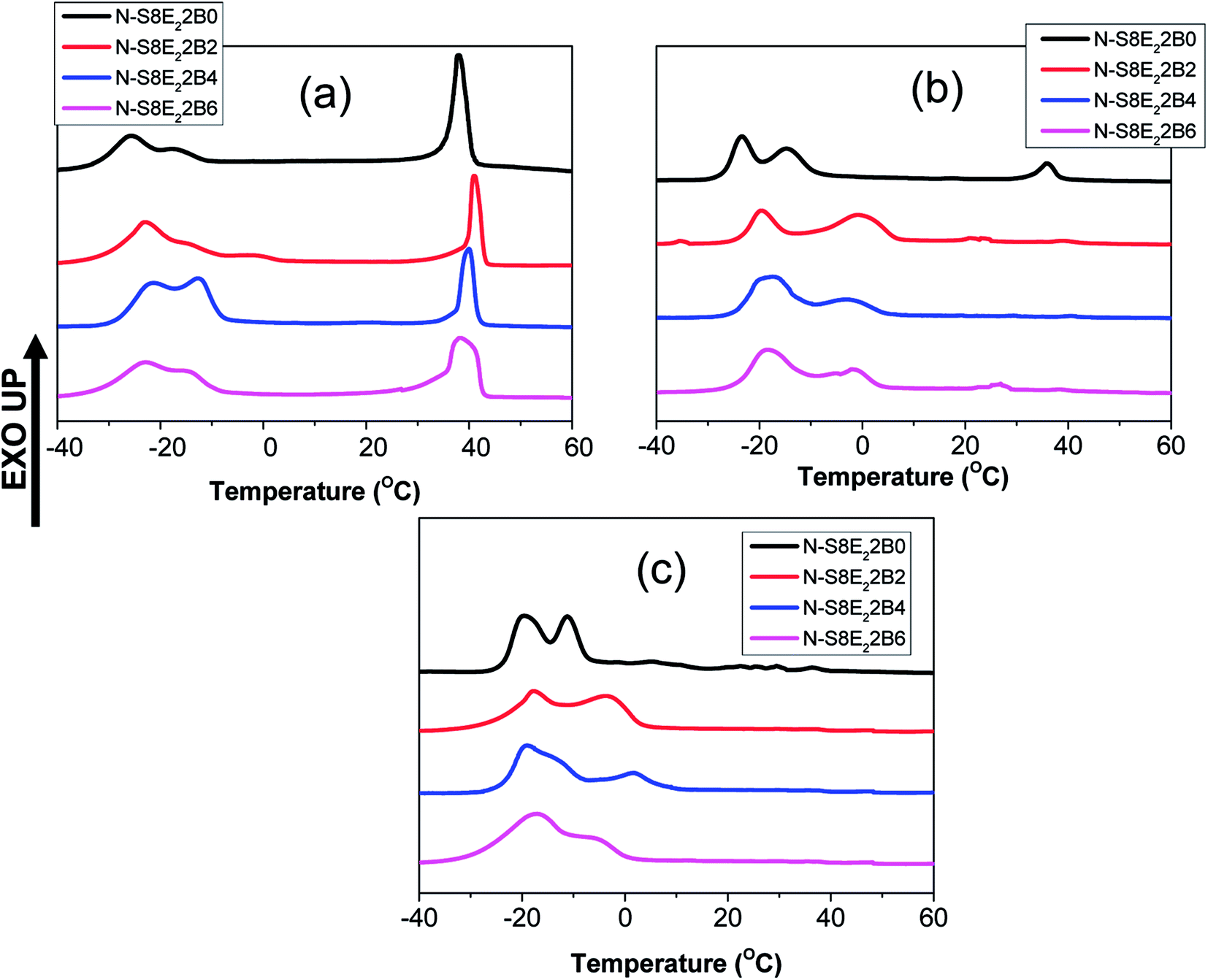 | ||
| Fig. 13 DSC cooling curves of PS/PEO2/PS-b-PEO blend nanofibers obtained after annealing at different temperatures (a) 90 °C (b) 130 °C and (c) 160 °C. | ||
Interestingly, the crystallization behaviour of the N-S8E22B blend nanofibers after compatibilization, using PS-b-PEO block copolymer, was not significantly different than that for the uncompatibilized blend nanofibers. The higher mobility of the shorter PEO homopolymer chains together with the rapid solvent evaporation during the electrospinning process plausibly did not provided enough time for the block copolymer chains to preferentially diffuse to the interface. In the absence of enough block copolymer at the interface, the morphology and, hence, the crystallization behavior of the blend nanofiber was expected to be similar to that of the uncompatibilized blends.
The crystallization behavior of the N-S8E22B blends was subsequently studied after melt annealing at higher temperatures such that the PS matrix phase was softened. Fig. 13(b) and (c) shows the DSC cooling curves obtained after melt annealing at 130 and 160 °C, respectively. The intensity of the exothermic peak observed at lower supercooling was considerably diminished after melt annealing. This suggested that during melt annealing the dimensions of the PEO domains were further reduced due to Plateau–Rayleigh instability induced droplet break-up process. Hence, the fraction of heterogeneity free domains increased after melt annealing such that crystallization was dominated by homogenous nucleation mechanism. In the case of block copolymer compatibilized N-S8E22B blends, after melt annealing at higher temperatures, the heterogeneous nucleated crystallization was more or less completely suppressed as was evident from the absence of exothermic peak at lower undercooling. In this case, during melt annealing at 130 and 160 °C, the block copolymer was able to diffuse to the interface such that it minimized the domain coalescence. Most interestingly, the high temperature melt annealed compatibilized N-S8E22B blends exhibited a crystallization exotherm at ca. −1 °C which was absent in the uncompatibilized blend. The position of the exothermic peak was such that it neither corresponded to homogenous nucleated crystallization (observed at T < −15 °C) nor to the heterogeneity induced heterogeneous nucleated crystallization (observed at T > 30 °C). The crystallization process, in this case, plausibly was the result of surface nucleation which is further elaborated in the discussion below.
Considering that most of the block copolymer localizes at the blend interface, the nature of the interface could be described by Leibler's brush theory.69 When the block copolymer segments are shorter than the corresponding homopolymer segments, the homopolymers are excluded from the respective segments of block copolymer resulting in a dry-brush. However, if the homopolymer segments are shorter than that of the block copolymer, a wet-brush is formed where the homopolymer homogenously mixes with the corresponding segments of block copolymer. In this study, when the high molecular weight PEO was used, the brush was always dry both on the PS as well as PEO side in the blend nanofibers as shown schematically in Fig. 14(a). In this case, the PEO crystallization was not expected to be influenced by the domain interface. However, in the N-S8E22B blend nanofibers, the molecular weight of PEO homopolymer was considerably less than that of the PEO block molecular weight such that a wet-brush was formed on the PEO side even though the brush was dry on the PS side. Hence, the PEO homopolymer was uniformly distributed and mixed with the PEO brushes localized at the interface as shown schematically in Fig. 14(b). In this case, the crystallization of PEO may get induced by the already solidified PS surface. Floudas et al. have shown such a surface induced nucleation during crystallization of PCL confined in nanopores of AAO membrane.35 The surface nucleation attributed to AAO wall was inhibited when the wall was made hydrophobic via chemical modification. Hence, the crystallization exotherm observed at ca. −1 °C, in the N-S8E22B blend nanofibers, may be attributed to such a surface induced nucleation mechanism.
 | ||
| Fig. 14 Schematic illustration depicting the dry brush and wet brush morphology of PEO in the electrospun compatibilized blend nanofibers. (a) Dry-brush morphology (b) wet-brush morphology. | ||
Conclusions
The morphology and crystallization behaviour of electrospun PS/PEO blend nanofibers compatibilized with PS-b-PEO block copolymer has been investigated. The radial constraint imposed by the nanofibers, during the phase separation in the blend, directed the formation of nano-sized PEO domains dispersed in the majority PS matrix. The compatibilization of the blends using PS-b-PEO block copolymer led to a noticeable reduction in the PEO domain size as well as domain connectivity. Furthermore, the reduction in the domain size was more significant when the blend nanofibers were thermally annealed above the glass transition temperature of PS matrix phase. This was attributed to the suppression of the domain coalescence due to interface localization of the block copolymers leading to reduction in the interfacial tension. In such a scenario, the Plateau–Rayleigh instability induced droplet breakup mechanism became dominant reducing the domain size. The reduction in the domain size as well as their spatial connectivity resulted in an increased fraction of PEO domains crystallizing via homogenous nucleation. Furthermore, in the compatibilized blends, with low molecular weight PEO homopolymer, the heterogeneous nucleation was completely suppressed on thermal annealing. Most interestingly, in the blend nanofibers with low molecular weight PEO, additional crystallization event attributed to surface nucleation was observed. The surface nucleation, in the case, plausibly resulted from the formation of wet-brush structures such that PEO homopolymers homogenously wetted the PEO blocks present at the interface. The surface nucleated crystallization was absent in the compatibilized blend nanofibers composed of high molecular weight PEO presumably due to the formation of morphology with dry-brush structures. In conclusion, to the best of our knowledge, the present work provides, for the first time, insight into the compatibilization of binary blends in an electrospun nanofibers using block copolymer. The findings, apart from improving the blend properties, could be further used to construct a more uniformly dispersed system for a systematic study of confined crystallization in electrospun nanofibers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Council for Scientific and Industrial Research (CSIR) under grant no. 22(0707)/15/EMR-II.References
- Z. Liu, X. Fu, L. Jiang, B. Wu, J. Wang and J. Lei, Sol. Energy Mater. Sol. Cells, 2016, 147, 177–184 CrossRef
.
- H.-L. Cheng, J.-W. Lin, J. Ruan, C.-H. Lin, F.-C. Wu, W.-Y. Chou, C.-H. Chen, C.-K. Chang and H.-S. Sheu, ACS Appl. Mater. Interfaces, 2015, 7, 16486–16494 Search PubMed
- H. Lee and J. Lee, J. Ind. Eng. Chem., 2015, 21, 1183–1190 CrossRef
- M. Kim, C. Bae, H. Kim, H. Yoo, J. M. Montero Moreno, H. S. Jung, J. Bachmann, K. Nielsch and H. Shin, J. Mater. Chem. A, 2013, 1, 14080 Search PubMed
- X. Chen, Y. Wang, K. Cai, Y. Bai, S. Bo and D. Guo, J. Appl. Phys., 2014, 116, 66821 CrossRef
- J. M. Carr, D. S. Langhe, M. T. Ponting, A. Hiltner and E. Baer, J. Mater. Res., 2012, 27, 1326–1350 CrossRef
- W.-N. He and J.-T. Xu, Prog. Polym. Sci., 2012, 37, 1350–1400 CrossRef
- J. K. Kim, S. Y. Yang, Y. Lee and Y. Kim, Prog. Polym. Sci., 2010, 35, 1325–1349 CrossRef
- R. V. Castillo and A. J. Müller, Prog. Polym. Sci., 2009, 34, 516–560 CrossRef
- R. M. Michell, I. Blaszczyk-Lezak, C. Mijangos and A. J. Müller, Polymer, 2013, 54, 4059–4077 CrossRef
- R. M. Michell, I. Blaszczyk-Lezak, C. Mijangos and A. J. Müller, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 1179–1194 CrossRef
- R. M. Michell and A. J. Müller, Prog. Polym. Sci., 2016, 54–55, 183–213 CrossRef
- B. Nandan, J. Hsu and H. Chen, J. Macromol. Sci., Polym. Rev., 2006, 46, 143–172 CrossRef
- M.-C. Lin, B. Nandan and H.-L. Chen, Soft Matter, 2012, 8, 7306 RSC
- S. Nakagawa, H. Marubayashi and S. Nojima, Eur. Polym. J., 2015, 70, 262–275 CrossRef
- Q. Jiang and M. D. Ward, Chem. Soc. Rev., 2014, 43, 2066–2079 RSC
- W. Zhao, Y. Su, X. Gao, J. Xu and D. Wang, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 414–423 CrossRef
- Y.-L. Loo, R. A. Register, A. J. Ryan and G. T. Dee, Macromolecules, 2001, 34, 8968–8977 CrossRef
- Y.-Y. Huang, J.-Y. Hsu, H.-L. Chen and T. Hashimoto, Macromolecules, 2007, 40, 3700–3707 CrossRef
- C. De Rosa, R. Di Girolamo, F. Auriemma, M. D'Avino, G. Talarico, C. Cioce, M. Scoti, G. W. Coates and B. Lotz, Macromolecules, 2016, 49, 5576–5586 CrossRef
- C.-Y. Chu, H.-L. Chen, M.-S. Hsiao, J.-H. Chen and B. Nandan, Macromolecules, 2010, 43, 3376–3382 CrossRef
- Y. Cong, H. Liu, D. Wang, B. Zhao, T. Yan, L. Li, W. Chen, Z. Zhong, M.-C. Lin, H.-L. Chen and C. Yang, Macromolecules, 2011, 44, 5878–5882 CrossRef
- H. Takeshita, K. Fukumoto, T. Ohnishi, T. Ohkubo, M. Miya, K. Takenaka and T. Shiomi, Polymer, 2006, 47, 8210–8218 CrossRef
- S. Nojima, Y. Fukagawa and H. Ikeda, Macromolecules, 2009, 42, 9515–9522 CrossRef
- S. Gondo, S. Osawa, T. Sakurai and S. Nojima, Polymer, 2013, 54, 6768–6775 CrossRef
- L. Zhu, B. H. Calhoun, Q. Ge, R. P. Quirk, S. Z. D. Cheng, E. L. Thomas, B. S. Hsiao, F. Yeh, L. Liu and B. Lotz, Macromolecules, 2001, 34, 1244–1251 CrossRef
- S. Nakagawa, K. Kadena, T. Ishizone, S. Nojima, T. Shimizu, K. Yamaguchi and S. Nakahama, Macromolecules, 2012, 45, 1892–1900 CrossRef
- S. Nakagawa, T. Tanaka, T. Ishizone, S. Nojima, Y. Kakiuchi, K. Yamaguchi and S. Nakahama, Macromolecules, 2013, 46, 2199–2205 CrossRef
- S. Nojima, K. Kato, S. Yamamoto and T. Ashida, Macromolecules, 1992, 25, 2237–2242 CrossRef
- G. Floudas, R. Ulrich and U. Wiesner, J. Chem. Phys., 1999, 110, 652–663 CrossRef
- S.-H. Huang, Y.-W. Huang, Y.-W. Chiang, T.-J. Hsiao, Y.-C. Mao, C.-H. Chiang and J.-C. Tsai, Macromolecules, 2016, 49, 9048–9059 CrossRef
- J. Maiz, J. Martin and C. Mijangos, Langmuir, 2012, 28, 12296–12303 CrossRef PubMed
- Y. Suzuki, H. Duran, M. Steinhart, H.-J. Butt and G. Floudas, Macromolecules, 2014, 47, 1793–1800 CrossRef
- A. Houachtia, P. Alcouffe, G. Boiteux, G. Seytre, J.-F. Gérard and A. Serghei, Nano Lett., 2015, 15, 4311–4316 CrossRef PubMed
- Y. Suzuki, H. Duran, W. Akram, M. Steinhart, G. Floudas and H.-J. Butt, Soft Matter, 2013, 9, 9189 RSC
- Y. Suzuki, H. Duran, M. Steinhart, H.-J. Butt and G. Floudas, Soft Matter, 2013, 9, 2621 RSC
- H. Duran, M. Steinhart, H.-J. Butt and G. Floudas, Nano Lett., 2011, 11, 1671–1675 CrossRef PubMed
- K. Shin, E. Woo, Y. G. Jeong, C. Kim, J. Huh and K.-W. Kim, Macromolecules, 2007, 40, 6617–6623 CrossRef
- C.-L. Liu and H.-L. Chen, Macromolecules, 2017, 50, 631–641 CrossRef
- Y. Guan, G. Liu, P. Gao, L. Li, G. Ding and D. Wang, ACS Macro Lett., 2013, 2, 181–184 CrossRef
- H. Masuda and K. Fukuda, Science, 1995, 268, 1466–1468 Search PubMed
- J. L. Lutkenhaus, K. McEnnis, A. Serghei and T. P. Russell, Macromolecules, 2010, 43, 3844–3850 CrossRef
- L. Kailas, C. Vasilev, J.-N. Audinot, H.-N. Migeon and J. K. Hobbs, Macromolecules, 2007, 40, 7223–7230 CrossRef
- H. Wang, J. K. Keum, A. Hiltner and E. Baer, Macromol. Rapid Commun., 2010, 31, 356–361 CrossRef PubMed
- H. Wang, J. K. Keum, A. Hiltner and E. Baer, Macromolecules, 2010, 43, 3359–3364 CrossRef
- S.-W. Hahm, D. Kim and D.-Y. Khang, Polymer, 2014, 55, 175–181 CrossRef
- A. M. Jordan, W. R. Lenart, J. M. Carr, E. Baer and L. T. J. Korley, ACS Appl. Mater. Interfaces, 2014, 6, 3987–3994 Search PubMed
- D. E. Martínez-Tong, B. Vanroy, M. Wübbenhorst, A. Nogales and S. Napolitano, Macromolecules, 2014, 47, 2354–2360 CrossRef
- G. Zhang, P. C. Lee, S. Jenkins, J. Dooley and E. Baer, Polymer, 2014, 55, 663–672 CrossRef
- G. Zhang, P. C. Lee, S. Jenkins, J. Dooley and E. Baer, Polymer, 2014, 55, 4521–4530 CrossRef
- M. Karunakaran, S. P. Nunes, X. Qiu, H. Yu and K.-V. Peinemann, J. Membr. Sci., 2014, 453, 471–477 CrossRef
- Khasanah, K. R. Reddy, S. Ogawa, H. Sato, I. Takahashi and Y. Ozaki, Macromolecules, 2016, 49, 4202–4210 CrossRef
- G. Zhong, K. Wang, L. Zhang, Z.-M. Li, H. Fong and L. Zhu, Polymer, 2011, 52, 5397–5402 CrossRef
- G. Zhong, R. Su, L. Zhang, K. Wang, Z. Li, H. Fong and L. Zhu, Polymer, 2012, 53, 4472–4480 CrossRef
- H. Luo, Y. Huang and D. Wang, Polymer, 2013, 54, 4710–4718 CrossRef
- A. Gradys, Polymer, 2017, 108, 383–394 CrossRef
- P. Samanta, T. V. S. Singh, R. Srivastava, B. Nandan, C.-L. Liu and H.-L. Chen, Soft Matter, 2016, 12, 5110–5120 RSC
- P. Samanta, R. Srivastava, B. Nandan and H.-L. Chen, Soft Matter, 2017, 13, 1569–1582 RSC
- R. Lv, R. Tian, B. Na, P. Zhang and Q. Liu, J. Phys. Chem. B, 2015, 119, 15530–15535 CrossRef PubMed
- C. W. Macosko, P. Guégan, A. K. Khandpur, A. Nakayama, P. Marechal and T. Inoue, Macromolecules, 1996, 29, 5590–5598 CrossRef
- S. H. Anastasiadis, I. Gancarz and J. T. Koberstein, Macromolecules, 1989, 22, 1449–1453 CrossRef
- M. Maric and C. W. Macosko, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 346–357 CrossRef
- J. Kim, R. W. Sandoval, C. M. Dettmer, S. T. Nguyen and J. M. Torkelson, Polymer, 2008, 49, 2686–2697 CrossRef
- J. A. Galloway, H. K. Jeon, J. R. Bell and C. W. Macosko, Polymer, 2005, 46, 183–191 CrossRef
- C. Creton, E. J. Kramer and G. Hadziioannou, Macromolecules, 1991, 24, 1846–1853 CrossRef
- R. Fayt, R. Jérôme and P. Teyssié, J. Polym. Sci., Polym. Phys. Ed., 1982, 20, 2209–2217 CrossRef
- Polymer blends, ed. D. R. Paul and S. Newman, Academic Press, New York, 1978 Search PubMed
- M.-S. Hsiao, W. Y. Chen, J. X. Zheng, R. M. Van Horn, R. P. Quirk, D. A. Ivanov, E. L. Thomas, B. Lotz and S. Z. D. Cheng, Macromolecules, 2008, 41, 4794–4801 CrossRef
- L. Leibler, Macromolecules, 1982, 15, 1283–1290 CrossRef
- L. Zhu, S. Z. D. Cheng, B. H. Calhoun, Q. Ge, R. P. Quirk, E. L. Thomas, B. S. Hsiao, F. Yeh and B. Lotz, Polymer, 2001, 42, 5829–5839 CrossRef
- Z. Lin, Z. Guan, B. Xu, C. Chen, G. Guo, J. Zhou, J. Xian, L. Cao, Y. Wang, M. Li and W. Li, J. Ind. Eng. Chem., 2013, 19, 692–697 CrossRef
- M. F. Díaz, S. E. Barbosa and N. J. Capiati, Polymer, 2005, 46, 6096–6101 CrossRef
- H.-L. Chen, S.-C. Hsiao, T.-L. Lin, K. Yamauchi, H. Hasegawa and T. Hashimoto, Macromolecules, 2001, 34, 671–674 CrossRef
- Y. Yao, T. Sakai, M. Steinhart, H.-J. Butt and G. Floudas, Macromolecules, 2016, 49, 5945–5954 CrossRef
- Y. Suzuki, M. Steinhart, M. Kappl, H.-J. Butt and G. Floudas, Polymer, 2016, 99, 273–280 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02391c |
| This journal is © The Royal Society of Chemistry 2018 |