
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of a new cytarabine-based prodrug for highly efficient oral delivery of cytarabine†
Jing Zhanga,
Di Zhanga,
Xu Hua,
Ruiling Liua,
Zhonghao Lib and
Yuxia Luan *a
*a
aSchool of Pharmaceutical Science, Shandong University, 44 West Wenhua Road, Jinan, Shandong Province 250012, China. E-mail: yuxialuan@sdu.edu.cn
bKey Laboratry of Colloid & Interface Chemistry, Shandong University, Ministry of Education, 250100, China
First published on 9th April 2018
Abstract
Because of the drawbacks of cytarabine (Ara-C) such as poor lipid solubility, deamination inactivation and low oral bioavailability limiting its application by oral administration, herein we propose a novel amphiphilic low molecular weight cytarabine prodrug (PA-Ara) by conjugating palmitic acid (PA) to Ara-C, making it possible to avoid the deamination inactivation by protecting the active 4-amino, as well as improving lipid solubility. Thanks to the rational design, the oil/water partition coefficient (P) of PA-Ara was improved tremendously compared with Ara-C, and the PA-Ara conjugation was stable enough in artificial digestive juice, ensuring that most molecules could be absorbed in the form of the prodrug. Results from an MTT assay conducted to measure the cytotoxicity of Ara-C and PA-Ara to HL60 (acute myeloblastic leukemia cell line) and K562 cells (chronic granulocytic leukemia cell line) showed that PA-Ara had significantly stronger antiproliferation activities than Ara-C. Significantly, we firstly compared the bioavailability of the oral fatty acid chain modified cytarabine prodrug preparation with injection and the relative bioavailability was up to 61.77% for our PA-Ara, which was much superior to that of oral Ara-C solution (3.23%). Overall, these findings make it clear that the PA-Ara suspension has the potential to be a promising new cytarabine oral preparation for leukemia therapy.
Introduction
1-(β-D-Arabinofuranosyl) cytosine (cytarabine, Ara-C) is a clinically widely used anticancer agent, especially for the treatment of both acute and chronic leukemia, which is a dangerous cancer with an estimated 62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 130 new cases and 24500 deaths in the United States in 2017.1–4 As a potent chemotherapeutical anti-metabolic drug, Ara-C will go through activation and inactivation in vivo. On the one hand, after intravenous injection into the blood circulation, part of the cytarabine crosses the cell membrane with the help of nucleoside transporters, and is then converted to a major active metabolite, 1-β-D-arabinofuranoside 5′-triphosphate (Ara-CTP), which forcefully inhibits the production of DNA polymerase and is finally also incorporated into the DNA to exert anticancer therapeutic effects.5,6 On the other hand, the bare active 4-amino of cytarabine is extremely prone to being deaminated to biologically inactive 1-D-arabinofuranosyluracil (Ara-U) by cytidine deaminase,7 resulting in a short plasma half-life and requirements for frequent injections to maintain an effective concentration, which may lead to poor compliance. However, as an acknowledged potent therapeutic drug, there is no clinical alternative oral preparation of cytarabine due to its limitations, such as poor lipid solubility and quite low oral bioavailability caused by the abundance of cytidine deaminase in the gastrointestinal tract and liver. As a result, cytarabine can only be administrated intravenously at present. Therefore, developing an alternative therapy suitable for practical oral administration is of particular importance and in great demand from the viewpoints of both fundamental research and practical applications.
130 new cases and 24500 deaths in the United States in 2017.1–4 As a potent chemotherapeutical anti-metabolic drug, Ara-C will go through activation and inactivation in vivo. On the one hand, after intravenous injection into the blood circulation, part of the cytarabine crosses the cell membrane with the help of nucleoside transporters, and is then converted to a major active metabolite, 1-β-D-arabinofuranoside 5′-triphosphate (Ara-CTP), which forcefully inhibits the production of DNA polymerase and is finally also incorporated into the DNA to exert anticancer therapeutic effects.5,6 On the other hand, the bare active 4-amino of cytarabine is extremely prone to being deaminated to biologically inactive 1-D-arabinofuranosyluracil (Ara-U) by cytidine deaminase,7 resulting in a short plasma half-life and requirements for frequent injections to maintain an effective concentration, which may lead to poor compliance. However, as an acknowledged potent therapeutic drug, there is no clinical alternative oral preparation of cytarabine due to its limitations, such as poor lipid solubility and quite low oral bioavailability caused by the abundance of cytidine deaminase in the gastrointestinal tract and liver. As a result, cytarabine can only be administrated intravenously at present. Therefore, developing an alternative therapy suitable for practical oral administration is of particular importance and in great demand from the viewpoints of both fundamental research and practical applications.
A prodrug is a biologically inactive or low active agent in vitro, and only when it is administrated into the body, can it be metabolized to an active drug to perform therapeutic effects.8 In view of the superiorities of high drug loading and less carrier materials over traditional polymer prodrugs, low molecular weight prodrugs are attracting increasing attention in achieving targeted drug delivery and environmental response.9,10 In particular, small amphiphilic prodrugs are capable of self-assembling into different architectures, like particles,11 capsules,12 rods13 and so on. To date, several cytarabine conjugations have been constructed, and the structural modifications are mostly focused on the free amino of the cytosine base or the 5′-hydroxyl group of the arabinose sugar, obtaining some derivatives such as squalenoyl-cytarabine,14 N4-L-valyl-Ara-C15,16 and intestinal PepT1 targeting 5′-amino acid ester.17 Unfortunately, N4-L-valyl-Ara-C failed to live up to initial expectations with a poor oral bioavailability of 4% in rats, indicating the infeasibility of amino acid modified 4-amino. In addition, although the oral absorption of a 5′-amino acid ester prodrug was promoted, it still suffered from the risk of inactivation due to the exposed amino. Therefore, there is still much room to improve oral bioavailability without the risk of inactivation and to realize a practical oral administration by the rational design of a cytarabine-based prodrug. Inspired by the amphiphilic structure of phospholipid molecules, we came up with the idea of introducing a hydrophobic fatty acid chain into the amino of cytarabine. In past research, some fatty acid conjugated prodrug delivery systems have been investigated intensively,18 but most were mainly about hydrophobic anticancer drugs. For instance, Luo and co-workers constructed a PTX-S-OA prodrug with TPGS as a stabilizer, and the prodrug nanoparticle performed with excellent anticancer activity.19 However, the dual hydrophobic molecule conjugation had poor stability without TPGS. In contrast, we expected the conjugation of a hydrophobic fatty acid with a hydrophilic Ara-C forming a low molecular weight amphiphilic prodrug to be stable enough. As for research into anti-leukemic agents, such as Ara-C, their modifications mostly relied on the 5′-hydroxyl group rather than the free active animo. Elacytarabine, an elaidic acid ester of Ara-C, has been developed using lipid vector technology in an attempt to overcome the low response rate, but the agent was circumventing resistance mechanisms.20 More importantly, the oral delivery of fatty acid modified cytarabine as a convenient and acceptable mode of administration has rarely been investigated. Therefore, developing a fatty acid modified Ara-C prodrug delivery platform was worth trying. Lauric acid (LA), palmitic acid (PA), stearic acid (SA), oleic acid (OA) etc are general fatty acids. Among these fatty acids, PA has mostly participated in the construction of some listed drugs, such as dexamethasone palmitate injections,21 chloramphenicol palmitate suspensions,22 clindamycin hydrochloride palmitate dispersible tablets,23 and vitamin A palmitate eye gel.24 Applications in different dosage forms and administrations proved the biosafety of PA. Consequently, PA was selected as the fatty acid moiety to take part in the design of a cytarabine prodrug in the present work.
In this work, PA was covalently attached to the free 4-amino of cytarabine by a simple amidation reaction. Here, the long fatty acid chain with 16 carbons would endow the conjugation with two charming properties. On the one hand, the added alkyl chain moiety provided amphiphilicity for an absolutely hydrosoluble cytarabine, which endows it with self-assembly and enhances the absorption efficiency by facilitating passive diffusion. On the other hand, the protection of the active amino would decrease the deamination and first-pass effect, so the plasma half-life would be prolonged and oral bioavailability would be greatly enhanced. Moreover, due to the presence of amidase, the as-prepared preparation would achieve the sustained release of free Ara-C in vivo, making it possible to avoid an enormous change in drug concentrations compared with injection and importantly to decrease its toxicity. With a convenient kind of dosage form, PA-Ara assembly suspensions may greatly improve patient compliance. To confirm the successful synthesis of the prodrug, we utilized nuclear magnetic resonance hydrogen (1H-NMR), mass spectrometry (MS), and Fourier transform infrared (FTIR) measurements to characterize the chemical structure of the conjugation, and transmission electron microscopy (TEM) was applied to observe the morphology of its assemblies prepared by a nanoprecipitation method. In addition, the PA-Ara conjugation was stable enough in artificial digestive juice, which ensured enough oral absorption in the form of a prodrug. More importantly, a cytotoxicity assay was conducted to investigate the antiproliferation activities of Ara-C and PA-Ara against HL60 and K562 cells, and the unexpected results showed that PA-Ara had a more excellent performance than Ara-C, proving its high efficiency in inhibiting leukemia cells. What is more, we carried out pharmacokinetic experiments to compare the bioavailability via injection of the oral fatty acid chain modified cytarabine prodrug preparation. To our surprise, the relative bioavailability was as high as 61.77% and the half-life of Ara-C was prolonged, along with a change in the pharmacokinetic behaviors of Ara-C due to the rational design. Last but not the least, the rats administrated intravenously were all dead within 8 h, whereas those which took the preparation orally were all alive after the assay, showing the reduced toxicity of the as-prepared suspension over the present clinically used injection. With one-step synthesis and one-step preparation, our prodrug assemblies not only go a long way to meeting the requirements of industrialization, but also show great potential for the construction of an oral drug delivery platform. Therefore, our newly rational designed PA-Ara prodrug provides a promising alternative oral preparation to injection and broadens the clinical application of Ara-C.
Experimental section
Materials
Cytarabine (Ara-C) was purchased from the Aladdin Industrial Corporation. Palmitic acid (PA), sodium hydroxide, potassium dihydrogen phosphate, sodium dihydrogen phosphate and disodium hydrogen phosphate were bought from Sinopharm Chemical Reagent Co., Ltd. Ethyl chloroformate (EtOCOCl) was obtained from Chengdu Beisite Reagent Co., Ltd. Pepsin (1:3000) and trypsin (1:250) were purchased from Amresco and Sangon Biotech (Shanghai) Co., Ltd, respectively. Acyclovir (HPLC > 98%) was purchased from Dalian Meilun Biotech Co., Ltd. HL60 and K562 cells were kindly provided by the Immunopharmacology Institute of Shandong University and the Shandong Analysis and Test Center. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was purchased from Beijing Solarbio Technology Co., Ltd.
The spectroscopy equipment was: an NMR spectrometer (1H-NMR, Bruker Avance 400), an electrospray tandem mass spectrometer (MS, AB SCIEX API 4000), an FTIR spectrometer (Nicolet 6700), a transmission electron microscope (TEM, JEM-200CX), a high performance liquid chromatograph (HPLC, Agilent Technologies 1200 Series), an HPLC-MS (Agilent 1260 triple quadrupole mass spectrometer equipped with an ESI source), and a microplate reader (ELISA, PerkinElmer).
Synthesis of prodrug PA-Ara
PA-Ara was the product of Ara-C and palmitic acid through an amidation reaction (Scheme 1). Firstly, PA (300 mg, 1.170 mmol) was dissolved in anhydrous N,N-dimethylformamide (DMF, 5 mL) followed by the dropwise addition of triethylamine (TEA, 180 μL) and EtOCOCl (122 μL) under nitrogen at a temperature of 0 °C. After 20 min of stirring, Ara-C (313 mg, 1.287 mmol), which was also dissolved in anhydrous DMF (5 mL), was added to the mixture dropwise and the reaction lasted for 72 h at room temperature. After that, the raw products could be obtained by filtration under diminished pressure to remove the solid byproduct produced in the first step. The solvent was further removed with a rotary evaporator. Finally, the raw products were purified on a silica gel column with a mixed solution of dichloromethane and methanol (from 100:1 to 30:1) as the eluent to obtain the pure PA-Ara prodrug products.
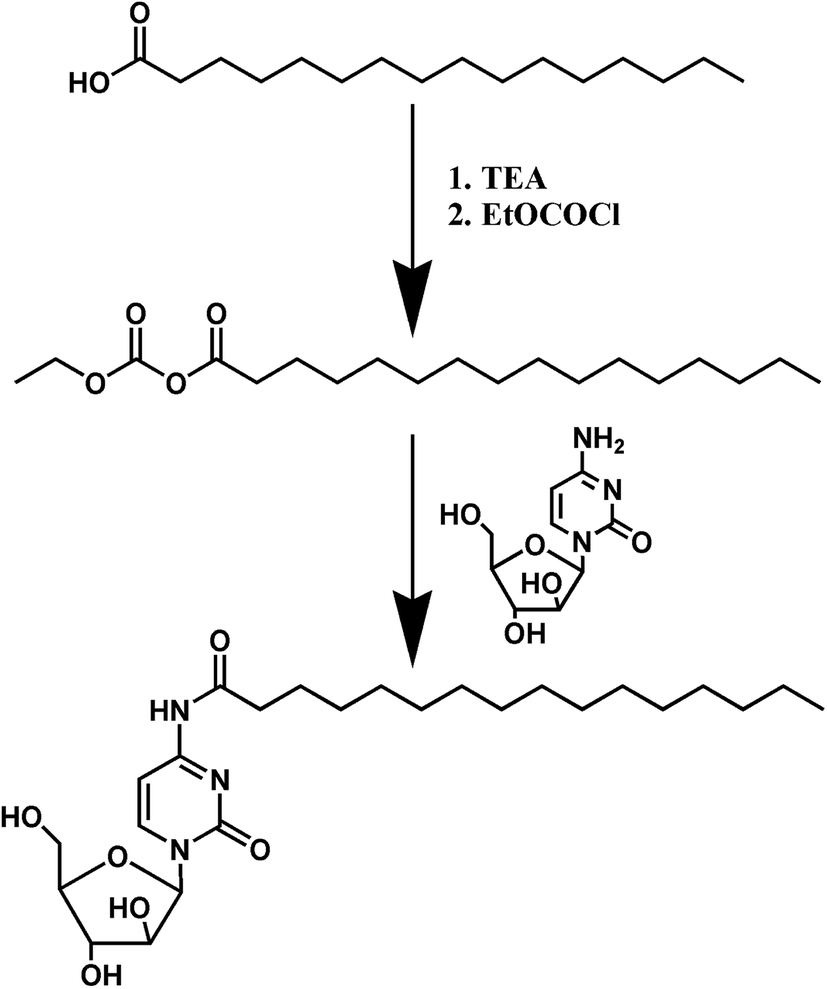 | ||
| Scheme 1 The synthesis route of PA-Ara. | ||
1H-NMR characterization
The newly obtained PA-Ara prodrug was precisely characterized by 1H-NMR. Fresh PA-Ara was dissolved in 0.5 mL of DMSO-d6 in an NMR tube, and then the measurement was taken on a Bruker Avance 400 spectrometer at 400 MHz at room temperature to record chemical shifts reported in ppm in the presence of tetramethylsilane.MS measurement
MS measurement was conducted to obtain the molecular weight information of the prodrug. 2 mg of PA-Ara powder was dissolved in methane and then analyzed by electrospray tandem mass spectrometry (AB SCIEX API 4000).FTIR determinations
Ara-C, PA and PA-Ara were crushed to a fine powder, milled with anhydrous potassium bromide, and finally compressed into thin pellets to record the near infrared absorption peaks from 4000 to 400 cm−1 on a Nicolet 6700 FTIR spectrometer.The preparation of PA-Ara assembly and TEM characterization
A nanoprecipitation method was applied to prepare a PA-Ara prodrug assembly suspension.25 20 mg of PA-Ara products were dissolved in 400 μL of methanol, followed by their dropwise addition into 40 mL of distilled water with stirring at 500 rpm. After stirring for 15 min, the mixture was centrifuged at 14000 rpm for 10 min to obtain the precipitate. In order to remove the undesired methanol as far as possible, the precipitate was washed three times. The final obtained precipitate was resuspended in 4 mL of distilled water under ultrasound until it was dispersed evenly to get a 5 mg mL−1 suspension. TEM is a widely used technique to observe assembled structures, such as nanoparticles,26 polymeric micelles27 and fibers,28 so TEM was applied to observe the morphology of our assemblies. The obtained thick suspension was diluted to 1 mg mL−1, and one drop was deposited on a copper grid and air-dried at room temperature before observation by TEM (JEM-200CX).
Oil/water partition coefficients (lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P)
P)
The oral absorption of a drug requires a proper oil/water partition coefficient for passive transport; herein we measured its lgP by the shaking flask method.29 N-Octanol (60 mL) and water (120 mL) were mixed in a capped conical flask in an incubator shaker at 37 °C for 24 h to achieve equilibration, and thus we obtained an aqueous phase saturated with n-octanol and an n-octanol phase saturated with water. PA-Ara was dissolved in the n-octanol phase in the capped test tubes to obtain a 200 μg mL−1 solution followed by the addition of an equal volume of the aqueous phase, and then the mixture was kept at 37 °C. After 24 h, the concentration of PA-Ara in the water phase was analyzed by high performance liquid chromatography (HPLC, Agilent Technologies 1200 Series). The value of P was calculated according to the following equation:where Coil refers to the concentration of drug in the n-octanol phase and C represents the prepared concentration.
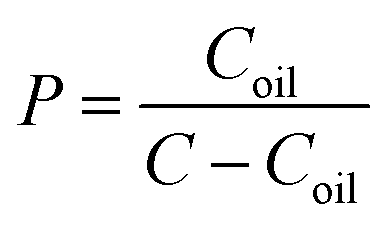
In vitro stability
Unlike drugs administrated intravenously without the process of absorption, the fierce metabolism of oral agents caused by various enzymes in the gastrointestinal tract and the pH value of different positions act as negative factors for sufficient absorption in the form of the original molecular structure.30,31 That is to say, oral drugs should be stable enough in the gastrointestinal tract to guarantee efficient absorption and therapeutic effect. Artificial gastric juice and artificial intestinal fluids were prepared according to Chinese Pharmacopoeia 2015 to estimate the gastrointestinal stability against pepsin and trypsin and pH 2.0, 5.0, 7.4, 8.0 phosphate buffered solution (PBS) to estimate chemical stability. In addition, 0.5% Tween-80 was added to solubilize the PA-Ara prodrug. PA-Ara was dissolved in artificial digestive juice and PBS, and then 200 μL of the sample was taken out to vortex and centrifuge after the addition of 400 μL of cold acetonitrile to stop a possible metabolic reaction at predetermined time point at 37 °C. The supernatant was filtered for HPLC analysis (Agilent Technologies 1200 Series).In vitro cytotoxicity
On account of the application in the treatment of leukemia, the cytotoxicity of Ara-C and PA-Ara prodrug were evaluated on HL60 and K562 cell lines with an MTT assay and were cultured in IMDM and RPMI-1640 medium, respectively both containing 10% fetal bovine serum (GIBCO, Vitrogen Corporation) and antibiotics (100 units mL−1 penicillin, and 100 μg mL−1 streptomycin) at 37 °C under a humidified atmosphere containing 5% CO2.32 Exponentially growing HL60 and K562 in 100 μL of culture medium containing different concentrations of samples were seeded into 96-well plates at a density of 3 × 104 and 1.5 × 104 cells per well, respectively. After 24 h or 48 h of incubation at 37 °C, 10 μL of 5% MTT regent was added to each well and incubated at 37 °C for another 4 h. Then the solution containing unreacted MTT was removed carefully and 150 μL of DMSO was added to dissolve the newly generated blue formazan crystals. The optical density of each well was measured using a microplate reader (ELISA, PerkinElmer) at a wavelength of 490 nm, and the cell inhibition ratio of each sample was calculated according to the following equation:where Apositive control and Ablack refer to the absorbance of the culture without drug treatment and the culture without cells or drug, respectively, whereas the other treatments were the same as for the samples.
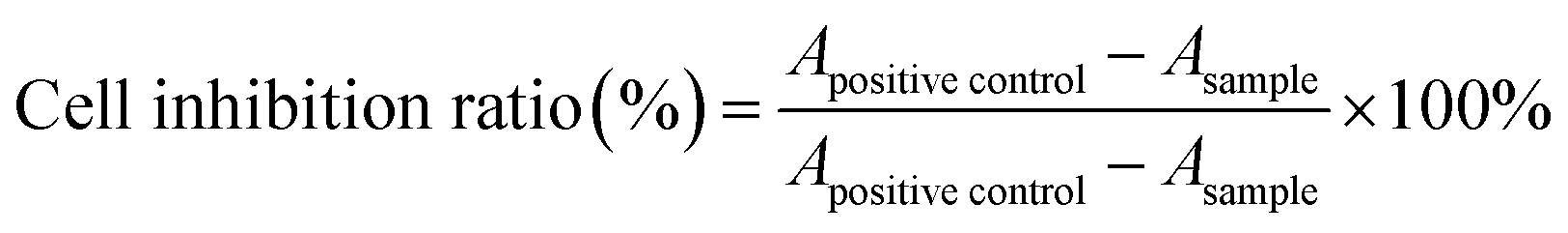
Pharmacokinetics experiments
In view of the difficulties in the separation of Ara-C and its metabolites in HPLC, HPLC-MS/MS was selected as the approach to measure the plasma drug concentrations in Wister rats. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Shandong University and approved by the Animal Ethics Committee of Shandong University.Eight healthy Wistar rats weighting 200 ± 10 g were randomly divided into two groups with administration of intravenous Ara-C saline solution (10 mg kg−1, 2 mg mL−1) and oral PA-Ara suspension (20 mg kg−1, 4 mg mL−1), respectively. Before administration, all the rats were not fed but were given water for 12 h. In this method, 0.5 mL of blood was taken from the jugular vein of rats into 1.5 mL Ep tubes which had previously been washed with heparin (10 mg mL−1) at 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 12, 24, 36, 48, 60 and 72 h after treatment, and then the blood was centrifuged at 10000 rpm for 5 min to obtained a plasma which was kept in a −20 °C fridge.
To measure the concentration of Ara-C and PA-Ara in the samples, standard solutions in plasma with different concentrations of Ara-C or PA-Ara containing acyclovir (800 ng mL−1) were prepared. Briefly, 180 μL of blank plasma and 20 μL of different concentrations of standard solutions of Ara-C or PA-Ara in methanol were added into a 1.5 mL Ep tube, and then 200 μL of acyclovir solution and 400 μL of acetonitrile were added. After vortexing for 60 s and centrifuging for 10 min, the standard solutions of plasma with different concentrations of Ara-C or PA-Ara containing acyclovir were obtained for HPLC-MS/MS measurement. In addition, all the samples were treated in the same manner under the same conditions. A triple quadrupole mass spectrometer (Agilent 1260) equipped with an ESI (electrospray ionization) source, and a C18 column (150 mm × 4.6 mm i.d. 5 mm, Thermo) coupled with a Phenomenex C18 guard column (4 mm × 3.0 mm i.d. 5 mm) was used to conduct the quantitative analysis. For better separation of analytes with endogenous materials and to avoid peak trailing, methanol and 0.1% formic acid were selected as the mobile phase at a flow rate of 0.6 mL min−1 with gradient elution (Table S1 in ESI†). In the positive ionization mode, the Q1 and Q2 masses were 226 to 152 for acyclovir, 244 to 112 for Ara-C and 482 to 350 for PA-Ara, and the chromatograms were recorded in this condition (Fig. S1 to S7 in ESI†).
Results and discussions
Characterizations of PA-Ara prodrug molecule and PA-Ara assemblies
The final pure product was bluish violet with a yield of 37.1%. 1H-NMR, MS, and FTIR were used to analyze its molecule structure. The detailed 1H-NMR data is shown in Fig. 1A and is as follows: 10.79 (s, 1H, –NH), 8.05 (d, 1H, J = 7.60 Hz, 6-H), 7.20 (d, 1H, J = 7.60 Hz, 5-H), 6.05 (d, 1H, J = 3.84 Hz, 1′-H), 5.48 (d, 2H, 2′-OH, 3′-OH), 5.06 (t, 1H, 5′-OH), 4.04–4.07 (m, 1H, 2′-H), 3.91–3.94 (m, 1H, 3′-H), 3.80–3.85 (m, 1H, 4′-H),3.61 (t, 2H, 5′-H), 2.37 (t, 2H, a-H), 1.53 (t, 2H, b-H), 1.18–1.24 (m, 24H, c-H), 0.852 (s, 1H, d-H). All the 1H-NMR signals agreed well with the molecular structure of PA-Ara. The signal of hydrogen in the amide group directly confirmed the successful synthesis of a PA-Ara prodrug. In addition, the molecular weight information of the PA-Ara prodrug characterized by MS is shown in Fig. S8 in ESI.† Among all the peaks in Fig. S8,† the appearance of [M + H]+ (482.5) and [M + Na]+ (504.4) quasi-molecular ion peaks exactly proved the existence of C25H43O6N3, which was consistent with the theoretically calculated value. In addition, FTIR was also utilized to confirm the molecular structure of PA-Ara. The results are shown in Fig. 1B. The original peaks at 3475 cm−1 and 3440 cm−1 from the free amine of Ara-C were replaced by a brand new absorption band of N–H stretching appearing at 3353 cm−1 in the PA-Ara molecular spectrum. The peaks at 2919–2850 cm−1 were consistent with the stretching modes of methylene and methyl groups in the PA moiety of PA-Ara. Unlike the absorption of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1702 cm−1 in the PA molecule, the amine I absorption (CO stretching mode) seemed weak and apparently shifted to 1696 cm−1, and the amine II absorption (mainly NH bending and CH stretching modes) was centered at 1650 cm−1 and 1555 cm−1 for PA-Ara. The FTIR results further confirmed the successful synthesis of PA-Ara. Based on the various characterizations of PA-Ara, it can be concluded that the PA-Ara prodrug was synthesized successfully.
O at 1702 cm−1 in the PA molecule, the amine I absorption (CO stretching mode) seemed weak and apparently shifted to 1696 cm−1, and the amine II absorption (mainly NH bending and CH stretching modes) was centered at 1650 cm−1 and 1555 cm−1 for PA-Ara. The FTIR results further confirmed the successful synthesis of PA-Ara. Based on the various characterizations of PA-Ara, it can be concluded that the PA-Ara prodrug was synthesized successfully.
 | ||
| Fig. 1 (A) 1H-NMR spectra of PA-Ara. (B) FTIR spectra of Ara-C (a), PA (b) and PA-Ara (c). (C) TEM image of PA-Ara assembly. | ||
By a nanoprecipitation method, we obtained the assembly of PA-Ara. The as-prepared PA-Ara assembly appeared as a white suspension and the morphology of the PA-Ara assembly was observed with a transmission electron microscope. The TEM result is shown in Fig. 1C. This shows that our PA-Ara could self-assemble into spiral fibers in aqueous solution using a nanoprecipitation method. By surveying the literature involving PA-based assembly, we found that Zhang et al. had previously reported a cell membrane tracker named TR4 (containing palmitic acid, tetraphenylethylene and a hydrophilic peptide),28 which could assemble into nanofibers. However, their fibers did not have spiral morphology. This indicated that the Ara-C moiety in the PA-Ara was the indispensable factor for the spiral assembly in our case. Besides, the hydrogen bond formed by the CO and N–H in the amido bond also lets the amphiphilic prodrug molecules arrange in an orderly fashion to form a spiral assembly. The zeta potential of the PA-Ara assembly was measured to be −(19.8 ± 3.1) mV, demonstrating that the surface of the assembly had a negative charge which was helpful for its physical stability. The PA-Ara assembly suspension could remain stable for more than two weeks at room temperature.
Oil/water partition coefficient (P)
The oil/water partition coefficient (P) is a crucial parameter for oral agents, which plays an important role in the intracorporal process of drugs involving absorption, distribution, metabolism and excretion. The coefficient is generally presented as the logarithm to the base 10 (lgP).33 In principle, the stronger the lipid solubility of a compound, the greater the coefficient will be. In our case, the value of P of the PA-Ara prodrug was determined to be 1668 (corresponding to lgP = 3.22) which is significantly higher than that of Ara-C (0.16, corresponding to lgP = −0.80), indicating the remarkably improved lipophilicity of PA-Ara compared with Ara-C. The high lipophilicity of the PA-Ara prodrug could be attributed to the conjugation of palmitic acid. Thus, such high lipophilicity can sufficiently ensure the easy passive diffusion of the drugs, resulting in enhanced oral absorption.
In vitro stability
Good stability in the gastrointestinal tract is of particular importance for oral drugs. Therefore, we prepared artificial gastric juice and intestinal fluids to examine the gastrointestinal stability of our PA-Ara prodrug. The kinetic curves for the degradation of PA-Ara with time are shown in Fig. 2A. From Fig. 2A, it is clear that the PA-Ara prodrug exhibits good stability in both artificial gastric juice and intestinal fluids. Within 2 h of incubation in artificial gastric juice, the undegraded PA-Ara remained at (80.72 ± 2.52)%. In particular, the undegraded PA-Ara could remain at (77.26 ± 3.30)% even after 10 h in artificial intestinal fluids. These results suggested that a majority of the prodrug molecules could be stable in the process of being emptied from the stomach to the small intestine within 2 h where the prodrugs were absorbed mostly in the form of the original molecular structure. The slight degradation of PA-Ara in artificial gastric juice and intestinal fluids can be attributed to the pepsin and trypsin therein, respectively. In addition, we also investigated the chemical stability of PA-Ara in PBS of pH 2.0, 5.0, 7.4 and 8.0, which represent the pH of the stomach, duodenum, ileum and colon, respectively. As shown in Fig. 2B, our PA-Ara performed with excellent stability in various PBS except for a slight decline in pH 2.0. This indicates that our PA-Ara is stable with no pH-responsive degradation. Based on the stability results, our PA-Ara was highly suitable for oral administration.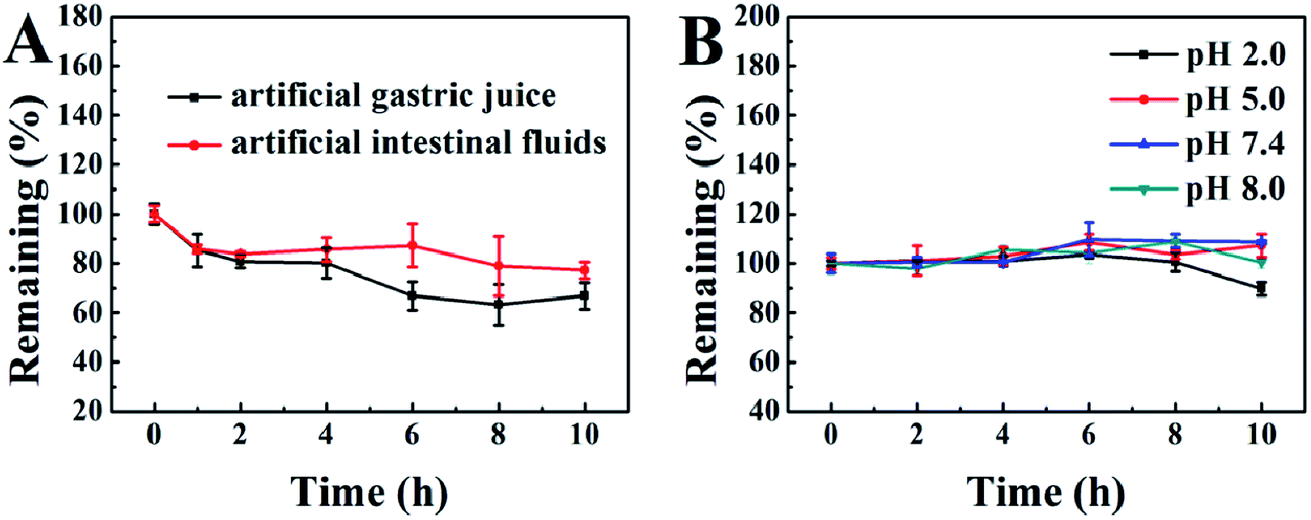 | ||
| Fig. 2 Gastrointestinal stability (A) and chemical stability (B) of the PA-Ara prodrug. | ||
We further investigated the stability of PA-Ara assemblies in artificial digestive juice by observing the changes in the morphology of the assemblies at different times. The as-prepared 15 mg mL−1 thick suspension was diluted 30 times with artificial gastric juice and intestinal fluids, respectively, and then the thin suspension was incubated at 37 °C. The samples were characterized by TEM after 1 h and 12 h of incubation, as shown in Fig. 3. When the assemblies were incubated in gastric juice for 1 h, the morphology of most assemblies remained unchanged in comparison with the original morphology in Fig. 1C. However, a few assemblies without spiral characteristic began to form (Fig. 3A). With the increase in incubation period to 12 h, all the original spiral fibers had changed into non-spiral fibers or rods (Fig. 3B). For the assemblies incubated in artificial intestinal fluids for 1 h (Fig. 3C), the spiral fibers had shrunk to be thinner in comparison with the original morphology in Fig. 1C. However, there was no obvious difference after further incubation to 12 h (Fig. 3D). In artificial gastric juice, the spiral structures of the PA-Ara assemblies were prone to uncoiling, in that some amide bonds or hydrogen bonds were hydrolyzed by acid environments and pepsin, which was similar to the destruction of the three dimensional structure of a protein. For the process of PA-Ara assemblies in vivo, the assemblies will first enter into the gastric juice and then arrive in the intestinal fluid. When the PA-Ara assemblies arrive in the small intestine (intestinal fluid), the assemblies dissolved in intestinal fluids could be slowly absorbed. Above all, no matter whether they were prodrug molecules or assemblies, they were both stable in artificial digestive juices within the required period, demonstrating that they were very suitable for oral application.
 | ||
| Fig. 3 TEM images of PA-Ara assembly after incubation in artificial gastric juice (A and B) or artificial intestinal fluids (C and D), respectively, for 1 h (A and C) and 12 h (B and D). | ||
Cytotoxicity in vitro
An MTT assay was further carried out to investigate the cytotoxicity in vitro of our PA-Ara prodrug. For comparison, free Ara-C was also studied. Fig. 4 shows the antiproliferative effects of Ara-C and PA-Ara prodrug on HL60 or K562 cells at 24 h and 48 h, respectively. For HL60 cells, the cell inhibition ratio of both Ara-C and PA-Ara performed in a dose-dependent fashion. In other words, the cell inhibition ratio increased with drug concentration. However, it was obvious that PA-Ara showed a significantly higher cell inhibition ratio than Ara-C for both 24 h and 48 h incubation. For example (24 h), the inhibition ratio of PA-Ara was 92.08% at 500 μM, which was significantly higher than that of Ara-C (59.95%). The increased inhibition ratio for PA-Ara was attributed to its increase in lipid solubility, endowed to a great extent by the fatty palmitic acid chain. The endowed lipid solubility changed the method of transmembrane transport, taking PA-Ara across the membrane in a passive transport way, where the greater the concentration difference between PA-Ara inside and outside the cell membrane, the faster the PA-Ara penetrated into the cell membrane. In contrast, Ara-C crossed the membrane assisted by nucleoside transporters, so its translocation efficiency was partially limited by the number of nucleoside transporters in addition to the concentration difference. Compared with 24 h incubation, when the incubation time was extended to 48 h, the inhibition ratio of Ara-C was higher than that at 24 h at the same concentration, while that for PA-Ara was slightly lower on the whole. This can be explained as follows. For Ara-C, on account of the restricting of nucleoside transporters, the rate of transportation was nearly constant, and therefore the inhibition ratio performed by rising steadily with time. However, for PA-Ara, the concentration difference between PA-Ara inside and outside the cell membrane became smaller and smaller as more and more PA-Ara molecules were passively transported into the cells, which thus caused a small decrease in inhibition ratio with time. Even so, the cytotoxicity of PA-Ara was overall superior to Ara-C with an antiproliferation inhibition ratio of 86.21% at 48 h. The performance of Ara-C in K562 cells was similar to that in HL60 cells for both 24 h and 48 h, indicating that the facilitated diffusion was not influenced by the differences between HL60 and K562 cells. On the contrary, PA-Ara performed antiproliferation activities against K562 cells both in an obviously concentration-dependent and time-dependent increasing way, which may be caused by the natural differences between K562 cells and HL60 cells. In a nutshell, no matter whether for HL60 cells or K562 cells, and no matter whether for 24 h or 48 h incubation, the endowed lipid solubility of PA-Ara promoted much better cytotoxic effects than the traditional agent Ara-C, providing a promising alternative therapy for leukemia.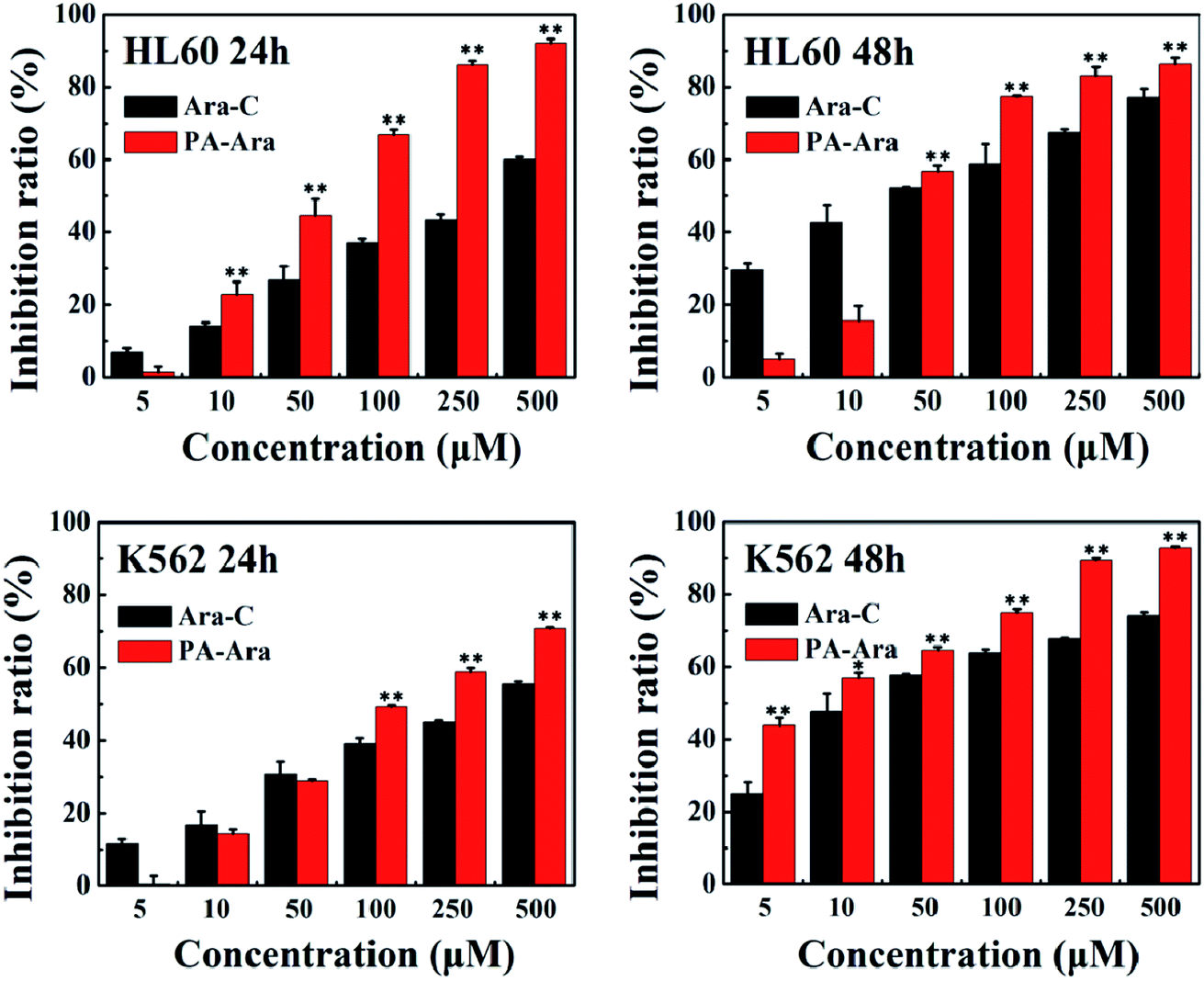 | ||
| Fig. 4 Cell inhibition ratios of different samples against HL60 and K562 cells after 24 h or 48 h incubation, *p < 0.05, **p < 0.01, n = 3. | ||
The IC50 value is the concentration of the drug when the cell inhibition ratio reaches 50%. It is a parameter often used to estimate the potency of each preparation. The smaller the IC50 value, the stronger the drug's ability to kill cells. Therefore, we further calculated the IC50 of Ara-C and PA-Ara. As shown in Table 1, the IC50 of PA-Ara was decreased vitally to HL60 and K562 cells in comparison with that of Ara-C. Especially for K562 cells, the IC50 of Ara-C was 2.18 times and 3.50 times high as PA-Ara at 24 h and 28 h, respectively, showing that PA-Ara behaved much better for anti-leukemia cell proliferation, agreeing well with what was discussed above.
| Cell line | Incubation time (h) | IC50 (Ara-C) | IC50 (PA-Ara) |
|---|---|---|---|
| HL60 | 24 | 289.30 | 52.45 |
| 48 | 34.02 | 46.74 | |
| K562 | 24 | 315.72 | 144.59 |
| 48 | 29.64 | 8.47 |
Pharmacokinetic studies in vivo
Improving oral bioavailability is crucial for oral preparation. Therefore, a pharmacokinetics assay was conducted to evaluate whether our PA-Ara assembly was suitable for oral administration. An HPLC-MS/MS approach was applied to determine the drug concentrations in plasma. We measured the concentration–time curve of Ara-C with oral administration.34 Therefore, only the curves of Ara-C and PA-Ara administrated intravenously and orally, respectively, were conducted in the present study. The results are shown in Fig. 5 and Table 2. Comparing the concentration–time curves of Ara-C and PA-Ara with different dosage forms and administrations, we could see that the concentrations of Ara-C administrated intravenously were much higher than for PA-Ara taken orally. However, the rats were all dead within 8 h for the Ara-C administrated intravenously, demonstrating the higher toxicity with the condition of the same dosage of Ara-C. In theory, AUC0–∞ (the mean area under the concentration–time curve from time 0 to the last time point) is a particularly important parameter for drugs, and a larger AUC0–∞ means better absorption and bioavailability. Due to AUC0–∞ being proportional to the dose, the AUC0–∞ of oral Ara-C solution was transformed from 1754.87 ng h mL−1 (15 mg kg−1) measured in our previous study to 1169.91 ng h mL−1 (10 mg kg−1),34 so the relative bioavailability (the ratio of the AUC0–∞ of oral cytarabine solution to that of the injection preparation) was only 3.23%. Surprisingly, the AUC0-∞ of our PA-Ara assembly suspension was calculated to be 22359.14 ng h mL−1, realizing a 19.11-fold improvement in comparison with that of oral Ara-C solution, and the relative bioavailability was as high as 61.77%. Hence, the absorption and oral bioavailability of Ara-C were unquestionably enhanced thanks to the rational design of the prodrug. In addition, after oral administration of a PA-Ara assembly, the concentrations of free Ara-C were much higher than of prodrug PA-Ara, which proved that PA-Ara prodrug could be metabolited into free Ara-C by amidase in vivo. In this way, this strategy would realize the sustained release of the drug and avoid huge changes in drug concentrations which may lead to side effects. According to the pharmacokinetic data, we calculated the pharmacokinetic parameters by DAS 2.0, and the results are displayed in Table 2. As presented in Table 2, the t1/2 (the elimination half-time of the drug) of free Ara-C from PA-Ara was calculated to be 12.89, which was significantly longer than that of Ara-C administrated orally (1.8).34 Thus, the strategy of protecting the active amino from deamination was proved to be wise for prolonging the circulation time of the drugs.
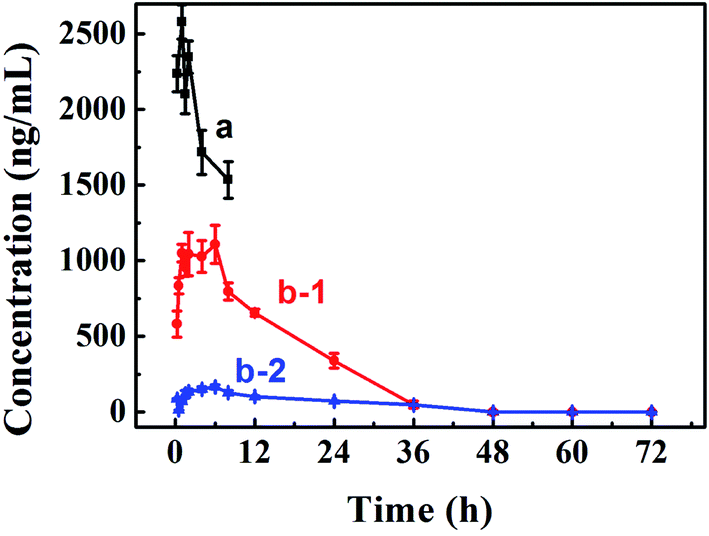 | ||
| Fig. 5 Pharmacokinetic concentration–time curves in rats after administration of different formulations. (a) The curve of Ara-C after intravenous administration of Ara-C saline solution. (b-1) The curve of Ara-C after oral administration of PA-Ara suspension. (b-2) The curve of PA-Ara after oral administration of PA-Ara suspension. | ||
| Pharmacokinetic parameters | Intravenous Ara-C | Oral suspension | ||
|---|---|---|---|---|
| Ara-C | PA-Ara | |||
| a AUC0–∞ is the mean area under the concentration–time curve from time 0 to the last time point.b Cmax is the peak plasma concentration of the drug.c Tmax is the time point when the plasma concentration reaches its peak.d t1/2 is the elimination half-time of the drug.e MRT0–∞ is the mean residence time which represents the average time that drug molecules reside in the body.f Clz is the volume of plasma containing the drug eliminated from the body per unit time, which stands for the clearance rate of the drug from blood. | ||||
| AUC0–∞/(ng h mL−1)a | 36199.75 |
22359.14 |
4630.77 | |
| Cmax/(ng mL−1)b | 2579.92 | 1108.59 | 167.13 | |
| Tmax/(h)c | 1 | 6 | 6 | |
| t1/2/(h)d | 11.46 | 12.89 | 19.68 | |
| MRT0–∞/(h)e | 14.09 | 11.87 | 29.09 | |
| Clz (L h−1 kg−1)f | 0.28 | 0.45 | 4.32 | |
Based on what has been discussed above, we can make it clear that the well-designed PA-Ara prodrug indeed made impressive progress in overcoming the drawbacks of short half-life and low bioavailability of oral cytarabine via molecular modification. By introducing the long fatty chain, inactivation caused by enzymes was efficiently avoided, and transmembrane transport increased remarkably, resulting in enhancements in oral bioavailability. Additionally, in view of the gastrointestinal stability we had investigated, the detection of major free Ara-C in the blood proved that the formed amide bond could be hydrolyzed in vivo to realize sustained release of free cytarabine rather than the robust entrance of Ara-C intravenously which may lead to serious toxicity. All of these results indicate the outstanding properties of our cytarabine prodrug preparation.
Conclusions
In summary, we rationally designed and synthesized a new amphiphilic low molecular weight cytarabine prodrug (PA-Ara) by conjugating palmitic acid to cytarabine, overcoming the drawbacks of cytarabine such as poor lipid solubility, deamination inactivation and low oral bioavailability for application by oral administration. The PA-Ara could be highly stable in PBS solution with different pH values for 10 h, and the remaining PA-Ara was more than 80% in artificial digestive juice within 2 h. This indicated that our PA-Ara is particularly suitable for oral administration. Compared with Ara-C, PA-Ara exhibited a significantly higher oil/water partition coefficient and an enhanced antiproliferative effect against HL60 and K562 cells. Results from a comparison of the pharmacokinetic behaviors of Ara-C injection and oral PA-Ara assembly suspension showed that the half-life of Ara-C for PA-Ara assembly was significantly prolonged. In particular, rats administrated intravenously were all dead within 8 h, whereas those taking the preparation orally were all alive after the assay, indicating the reduced toxicity of the as-prepared PA-Ara suspension compared to the Ara-C injection. Our PA-Ara suspension overcomes the drawbacks of oral Ara-C, exerts stronger anti-leukemia effects and significantly improves oral bioavailability, providing a promising/practical oral preparation as an alternative to injection and broadening the clinical application of cytarabine.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC, No. 21673128 and No. 21373126).References
- G. P. Bodey, E. J. Freireich, R. W. Monto and J. S. Hewlett, Cancer Chemother. Rep., Part 1, 1969, 53, 59–66 CAS.
- A. Ferrajoli, A. M. Liberati, P. Caricchi, E. Donti, E. Morra, M. Lazzarino, A. R. Betti, P. Bernasconi and G. Saglio, Eur. J. Haematol., 1995, 55, 184–188 CrossRef CAS PubMed.
- L. E. Robertson, R. Hall, M. J. Keating, E. Estey, H. M. Kantarjian, P. Mclaughlin, F. B. Hagemeister and W. Plunkett, Leuk. Lymphoma, 1993, 10, 43–48 CAS.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 7–30 CrossRef PubMed.
- D. W. Kufe, P. P. Major, E. M. Egan and G. P. Beardsley, J. Biol. Chem., 1980, 255, 8997–9000 CAS.
- J. J. Furth and S. S. Cohen, Cancer Res., 1968, 28, 2061–2067 CAS.
- R. L. Capizzi, J. C. White, B. L. Powell and F. Perrino, Semin. Hematol., 1991, 28, 54–69 CAS.
- A. Albert, Nature, 1958, 182, 421–423 CrossRef CAS PubMed.
- A. Satsangi, S. S. Roy, R. K. Satsangi, R. K. Vadlamudi and J. L. Ong, Mol. Pharmaceutics, 2014, 11, 1906–1918 CrossRef CAS PubMed.
- A. M. Mansour, J. Drevs, N. Esser, F. M. Hamada, O. A. Badary, C. Unger, I. Fichtner and F. Kratz, Cancer Res., 2003, 63, 4062–4066 CAS.
- H. D. Tang, C. J. Murphy, B. Zhang, Y. Q. Shen, M. H. Sui, E. A. Van Kirk, X. W. Feng and W. J. Murdoch, Nanomedicine, 2010, 5, 855–865 CrossRef CAS PubMed.
- Y. Q. Shen, E. L. Jin, B. Zhang, C. J. Murphy, M. H. Sui, J. Zhao, J. Q. Wang, J. B. Tang, M. H. Fan and E. Van Kirk, J. Am. Chem. Soc., 2010, 132, 4259–4265 CrossRef CAS PubMed.
- A. Maksimenko, F. Dosio, J. Mougin, A. Ferrero, S. Wack, L. H. Reddy, A. A. Weyn, E. Lepeltier, C. Bourgaux and B. Stella, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E217–E226 CrossRef CAS PubMed.
- M. G. Sarpietro, S. Ottimo, M. C. Giuffrida, F. Rocco, M. Ceruti and F. Castelli, Int. J. Pharm., 2011, 406, 69–77 CrossRef CAS PubMed.
- E. P. Cheon, J. H. Hong and H. K. Han, J. Pharm. Pharmacol., 2006, 58, 927–932 CrossRef CAS PubMed.
- E. P. Cheon and H. K. Han, Acta Pharmacol. Sin., 2007, 28, 268–272 CrossRef CAS PubMed.
- Y. B. Sun, J. Sun, S. L. Shi, Y. K. Jing, S. L. Yin, Y. Chen, G. Li, Y. J. Xu and Z. G. He, Mol. Pharmaceutics, 2009, 6, 315–325 CrossRef CAS PubMed.
- S. J. Sun, C. Luo, W. P. Cui, J. Sun and Z. G. He, J. Controlled Release, 2017, 264, 145–159 CrossRef PubMed.
- C. Luo, J. Sun, D. Liu, B. J. Sun, L. Miao, S. Musetti, J. Li, X. P. Han, Y. Q. Du, L. Li, L. Huang and Z. G. He, Nano Lett., 2016, 16, 5401–5408 CrossRef CAS PubMed.
- N. Keane, C. Freeman, R. Swords and F. J. Giles, Expert. Rev. Hematol., 2013, 6, 9–24 CrossRef CAS PubMed.
- P. Bias, R. Labrenz and P. Rose, Clin. Drug Invest., 2001, 21, 429–436 CrossRef CAS.
- M. I. Morasso, E. Hirmas, E. Firmani, R. Pezoa and E. Cid, Pharm. Acta Helv., 1980, 55, 270–273 CAS.
- X. Huang, C. Zhang, F. Lin, C. L. Zhou, W. W. Su and W. Peng, Acta Sci. Nat. Univ. Sunyatseni, 2015, 54, 115–118 Search PubMed.
- X. H. Cui, J. Xiang, W. Q. Zhu, A. J. Wei, Q. H. Le, J. J. Xu and X. D. Zhou, J. Glaucoma, 2016, 25, 487–492 CrossRef PubMed.
- J. Y. Zhang, Z. G. Shen, J. Zhong, T. T. Hu, J. F. Chen, Z. Q. Ma and J. Yun, Int. J. Pharm., 2006, 323, 153–160 CrossRef CAS PubMed.
- H. P. Sun, J. H. Su, Q. S. Meng, Q. Yin, L. L. Chen, W. W. Gu, P. C. Zhang, Z. W. Zhang, H. J. Yu, S. L. Wang and Y. P. Li, Adv. Mater., 2016, 28, 9581–9588 CrossRef CAS PubMed.
- H. Cabral1, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef PubMed.
- C. Q. Zhang, S. B. Jin, K. N. Yang, X. D. Xue, Z. P. Li, Y. G. Jiang, W. Q. Chen, L. R. Dai, G. Z. Zou and X. J. Liang, ACS Appl. Mater. Interfaces, 2014, 6, 8971–8975 CAS.
- T. M. Xie, B. Hulthe and S. Folestad, Chemosphere, 1984, 13, 445–459 CrossRef CAS.
- K. C. Kwan, Drug Metab. Dispos., 1997, 25, 1329–1336 CAS.
- M. Gibaldi, R. N. Boyes and S. Feldman, J. Pharm. Sci., 1971, 60, 1338–1340 CrossRef CAS PubMed.
- A. A. Vandeloosdrecht, R. H. J. Beelen, G. J. Ossenkoppele, M. G. Broekhoven and M. M. A. C. Langenhuijsen, J. Immunol. Methods, 1994, 174, 311–320 CrossRef CAS.
- A. Noubigh, A. Mgaidi and M. Abderrabba, J. Chem. Eng. Data, 2010, 55, 488–491 CrossRef CAS.
- J. Liu, J. Liu, D. J. Zhao, N. X. Ma and Y. X. Luan, RSC Adv., 2016, 6, 35991–35999 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01225c |
| This journal is © The Royal Society of Chemistry 2018 |