
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHelix loop-mediated isothermal amplification of nucleic acids
Rui Mao ab,
Lifei Qiab,
Zhuo Wang*a,
Hongtao Liu*ac and
Yuguang Du*a
ab,
Lifei Qiab,
Zhuo Wang*a,
Hongtao Liu*ac and
Yuguang Du*a
aState Key Laboratory of Biochemical Engineering, Key Laboratory of Biopharmaceutical Production & Formulation Engineering, PLA, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: wangzhuo@ipe.ac.cn; liuhongtao@ipe.ac.cn; ygdu@ipe.ac.cn; Fax: +86-10-82545070; Tel: +86-10-82545070
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cZhengzhou Institute of Emerging Industrial Technology, Zhengzhou 450000, P. R. China
First published on 23rd May 2018
Abstract
Isothermal nucleic acid amplification has played a key role in the point of care test (POCT). In this study, a helix loop-mediated isothermal amplification (HAMP) method with high specificity, efficiency and rapidity was developed. The MERS-Cov orf1b gene was chosen for the validation and optimization of HAMP. The HAMP analysis was performed at a constant temperature of 61–65 °C and yielded a self-primed spiral structure with no introduction of exogenous gene sequence by two pairs of specially designed primers. The primers for helix loop formation were composed of two complementary primers including the helix forward primer and the helix reverse primer, the 3′ ends of which were complementary to their respective target nucleic acids. HAMP assay can be monitored by fluorescence signals with the addition of Eva Green in the reaction mixture. In addition, an accelerated HAMP was developed after the addition of acceleration probe, which could be finished within 75 min with a sensitivity of 10 copies per reaction. Further, a reverse transcription-HAMP (RT-HAMP) was proven to be feasible for RNA detection by combining the reverse transcriptase with DNA polymerase. Finally, both the HAMP and RT-HAMP assay were visually conducted by using Hydroxynaphthol blue (HNB) as a chromogenic indicator. All in all, it is suggested that the HAMP assay would have great potential in POCT applications.
1. Introduction
Nucleic acid amplification is one of the most valuable tools in life science research because of its applications in molecular diagnosis, forensic investigations, and biomedical developments. Polymerase chain reaction (PCR) was the first and is still the most popular amplification technology for nucleic acid analysis. However, PCR is usually complex and time-consuming, requiring sophisticated thermal cycling instruments and well-trained operators.1 These disadvantages have limited its application to some special fields such as point-of-care use in resource-limited areas.2In an effort to reduce the resource requirements of PCR, a variety of isothermal amplification techniques have been developed and applied widely, including self-sustained sequence replication reaction (3SR),3 rolling circle amplification (RCA),4 strand displacement amplification (SDA),5 helicase-dependent amplification (HDA),6 loop-mediated isothermal amplification (LAMP)7 and recombinase polymerase amplification (RPA),8 Cross-priming amplification (CPA),9 and Polymerase Spiral Reaction (PSR),10 etc. 3SR, NASBA, SDA, HDA and RPA, require multiple enzymes and rigorous optimization, which may increase the assay cost. As to the single-enzyme based methods, LAMP and CPA require four or more primers, which may pose challenges for primer design,11 although specific primer-design software is available.
Among the above-mentioned methods, PSR, based on auto nucleic acid synthesis with spiral structure (the scheme is presented in Fig. 1a), has been successfully applied to the rapid detection of toxA gene.12 However, an exogenous botanic gene was introduced to the primer design, which may influence the selection of appropriate primers. Furthermore, the overlap part in the starting procedure of helix self-priming elongation is relatively short, and this may affect the self-annealing and elongation efficiency, which may easily lead to the amplification bias and false positive/negative results.
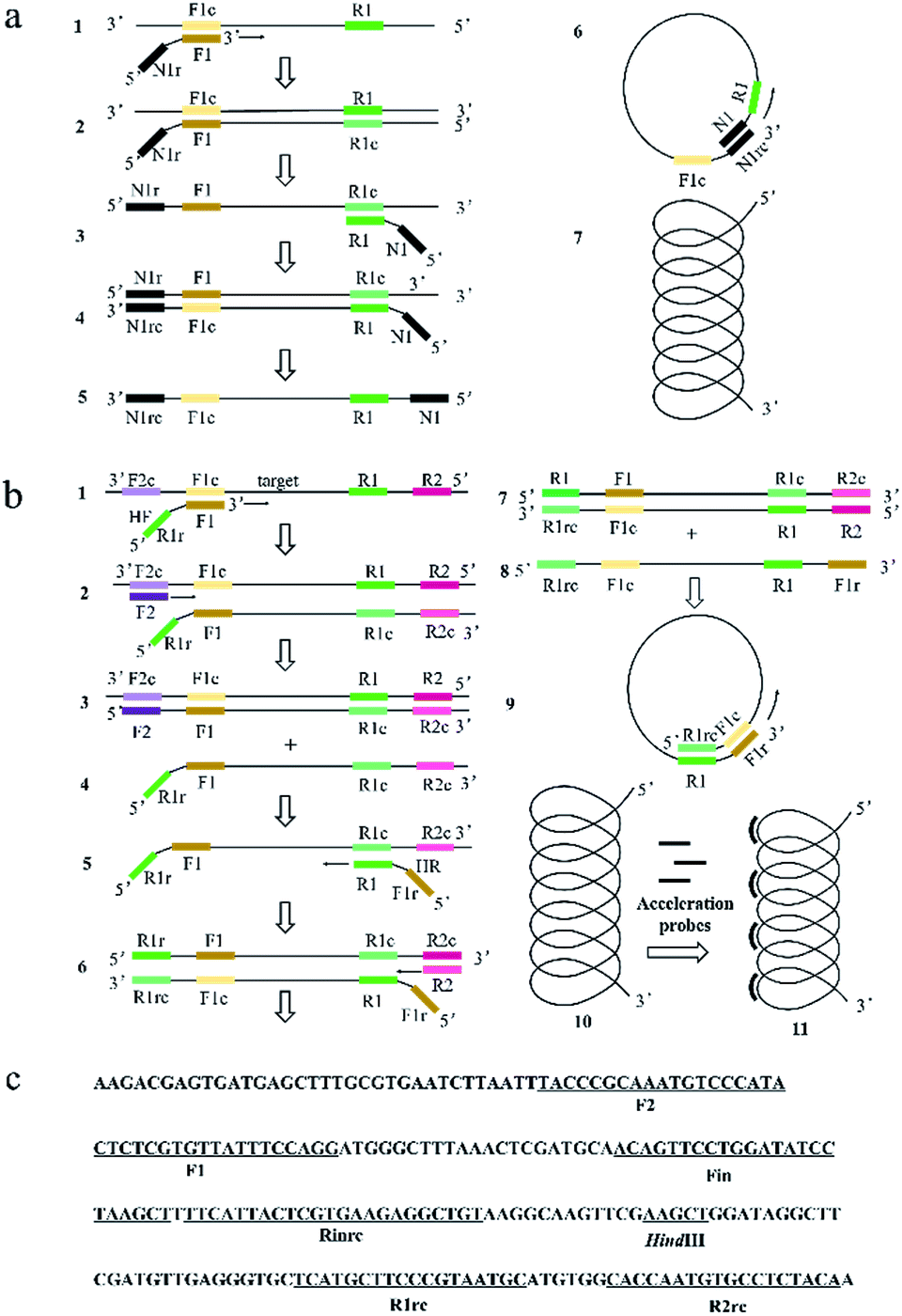 | ||
| Fig. 1 The schematic presentation of PSR and HAMP system and primer design of HAMP system. (a) Representative scheme about the PSR system; (b) representative scheme about the HAMP system; (c) localization of primers and restriction enzyme cutting sites in the nucleotide sequence of MERS-orf1b (part) for HAMP method. | ||
In this study, a helix loop-mediated isothermal amplification (HAMP) method inspired by spiral structure in PSR was developed. Similar to LAMP, HAMP was performed with two pairs of primers at a constant temperature. Unlike PSR, there is no exogenous sequences introduced in primer design to start a self-priming helix structure formation. The structure can be formed by slightly modified PCR primer and the overlap part of helix loop in HAMP is larger compared with that in PSR which would enhance the stability of the self-priming structure. In addition, the acceleration probe for HAMP reaction will dramatically improve the amplification efficiency and sensitivity. Moreover, an Mg2+ indicator, Hydroxynaphthol blue (HNB) was introduced into the reaction system to offer a visual judgement method for end-point result based on the colour change. Overall, the HAMP method may offer an alternative for the point of care test.
2. Materials and methods
2.1. Reagents and materials
A 550 bp DNA fragment of the MERS-orf1b genome (GenBank: KU308549.1) with a T7 promoter sequence cloned into a pUC57-simple vector was provided by BGI Biological Engineering Technology and Services Co. Ltd (Shenzhen, China). Other DNA fragments of MERS-CoV, such as orf1a (GenBank: KX108946.1), N2 (GenBank: KU242424.1), N3 (GenBank: KJ156873.1), upE (GenBank: KX108944.1) were also provided by BGI. The copy number of the plasmid was determined using DNA copy number calculator available at http://cels.uri.edu/gsc/cndna.html. The artificial RNA of MERS-orf1b was prepared by RiboMax™ Large Scale RNA Production System following the recommended instruction.Primers and probes (listed in Table 1) were provided by BGI Biological Engineering Technology and Services Co. Ltd (Shenzhen, China). The Bst 2.0 WarmStart DNA polymerase, AMV Reverse Transcriptase and 10 × ThermoPol reaction buffer (including 200 mM Tris–HCl, 100 mM KCl, 100 mM (NH4)2SO4, 20 mM MgSO4, and 1% Triton X-100) were purchased from New England BioLabs (Ipswich, MA, USA). Deoxynucleotide triphosphates (dNTPs) and RiboMax™ Large Scale RNA Production Systems were from Promega (Madison, WI, USA). Eva Green and GelRed were obtained from Biotium (Hayward, CA, USA). Restricted enzymes were purchased from Thermo Fisher Scientific (Shanghai, China). DNA purification kit and Nuclease-free water were purchased from Tiangen Biotech Co., Ltd. (Beijing, China). Other reagents, unless specified, were obtained from Sigma-Aldrich (St. Louis, MO, USA).
| Primer | 5′ → 3′ |
|---|---|
| Mo1b-HF | GTACGAAGGGCATTACGCTCTCGTGTTATTTCCAGG |
| Mo1b-HR | GGACCTTTATTGTGCTCTCGCATTACGGGAAGCATG |
| Mo1b-F2 | TACCCGCAAATGTCCCATA |
| Mo1b-R2 | TGTAGAGGCACATTGGTG |
| Mo1b-Fin | ACAGTTCCTGGATATCCTAAGCT |
| Mo1b-Rin | ACAGCCTCTTCACGAGTAATG |
2.2. Instruments
The real-time fluorescence detection of nucleic acid was performed using StepOne™ Real-Time PCR detection system (The Applied Biosystems) at 1 min intervals under a constant temperature and followed by the melting curve analysis procedure. Gels were imaged by Tanon 4100 Gel Image Analysis System (Tanon Science & Technology Co., Ltd.). Nucleic acid concentration was determined by using the Quawell UV-Vis Spectrophotometer Q5000 (Quawell Technology, Inc.).2.3. Reaction mixture for HAMP and accelerated HAMP
HAMP reactions were carried out in a 25 μl reaction mixture system as followed: 8 U Bst 2.0 WarmStart DNA polymerase, 2.5 μl 10 × ThermoPol reaction buffer, 1 M betaine, 6 mM MgSO4, 1.4 mM each dNTP and appropriate nucleic acid template. For one reaction, 1.6 μM HF, 1.6 μM HR, 0.2 μM F2 and 0.2 μM R2 primers were used. In addition, 0.8 μM Fin and Rin primers were added to accelerate the HAMP reaction. At last, nuclease-free water was added to obtain a final volume of 25 μl. For application of HAMP on detecting of RNA targets, 5 U AMV Reverse Transcriptase instead of water was further added into the reaction mixture. The reactions were performed at 63 °C on StepOne™ real-time PCR system or water bath.HAMP amplification products were determined by gel electrophoresis, fluorescence-based detection using the real time PCR or direct visual detection using Mg2+ indicator.
For gel electrophoresis analysis, the amplification products were firstly digested using the restriction enzyme HindIII found in MERS-orf1b gene, and then analysed by the electrophoresis in 1% agarose gel in 1 × TAE buffer (40 mM Tris–acetate, 1 mM EDTA, pH 8.0). DNA was stained with GelRed (Biotium, USA) and further visualised by Gel Image Analysis System.
For fluorescence-based detection, Eva Green, a sequence-unspecific nucleic acid labelling dye used for real time of monitoring PCR and LAMP reactions,13 was used for quantitatively analysing amplification results of HAMP based on the fluorescence intensity detected by the StepOne™ real-time PCR detection system.
For visual detection, 1 μl Hydroxynaphthol blue (HNB) solution (3 mM) was added to the reaction mixture. A positive reaction was indicated by a colour change from violet to sky blue, while a negative reaction remained on violet colour.
2.4. Sensitivity and specificity evaluation of HAMP
The Plasmid DNA of pUC57-orf1b was used as the template with 10-fold serial dilution and resulted in 108, 107, 106, 105, 104, and 103 copies of the template per reaction respectively. And the artificial RNA of MERS-orf1b gene was synthetized by RiboMax™ Large Scale RNA Production System. For the amplification of RNA template, the same serial dilution was performed using the RNA of MERS-orf1b. And 108, 107, 106, 105, 104, 103 and 102 copies of RNA per reaction were used to evaluate the sensitivity of reverse transcription-HAMP (RT-HAMP). The Plasmid DNA of orf1a, N2, N3 and UpE were used for specificity evaluation of HAMP.All experiments described above were replicated to ensure the reproducibility.
3. Results and discussion
3.1. The HAMP method
Similar to LAMP and PSR, HAMP is dependent on auto-cycling strand displacement by a DNA polymerase with high strand displacement activity and a set of two specially designed primers and two outer primers. The reaction mechanisms of PSR and HAMP were shown in Fig. 1a and b, respectively. As to the primer design of HAMP, the sequences (typically 20–24 nt) at both ends of the target region for amplification in a DNA were designated as F1c and R1. Two sequences (typically 20–24 nt) inside both ends of F1c and R1 were designated as Finc and Rin (not shown in Fig. 1b) and two sequences (typically 17–21 nt) outside the ends of F1c and R1 were designated as F2c and R2. Given this structure above, the sequences of Helix forward primer (HF) and Helix reverse primer (HR) were designed as the following: the primer HF contained two parts, i.e., the reverse sequence of R1 (R1r) and the complementary sequence of F1c (F1). The primer HR contained two parts: the reverse complementary sequence of F1c (F1r) and the sequence of R1. Evidently, the sequences of HF and HR were reverse to each other. The two inner primers (accelerate probe) consisted of Rin and the sequence (Fin) complementary to Finc. The two outer primers consisted of the sequence of R2 and the complementary sequence of F2c (F2).The mechanism of HAMP is shown in Fig. 1b. Firstly, the two strands of DNA double helix are separated in the presence of betaine at 61–65 °C. Then, the F1 segment of HF primer hybridizes to the F1c for the target sequence amplification (structure 1 to structure 2). The outer primer F2 hybridizes with F2c to initiate the DNA synthesis and then displace the newly-produced DNA strand containing HF primer sequence at 5′ end (structure 4). Next, the R1 segment of HR primer hybridizes to the R1c sequence in the newly-produced DNA (structure 5). The outer primer R2 hybridizes with R2c sequence in the newly-produced DNA to initiate the DNA synthesis (structure 6), and then displace the DNA strand containing HR primer sequence at 5′ end (structure 8). The R1rc and F1c of 5′ regions are reverse complementary to R1 and F1r of 3′ regions on the DNA strand of structure 8. Thus, the strand could curl to form a helix-loop to start self-primed DNA synthesis (structure 9). Finally, a single helix structure is formed (structure 10) and the non-complementary pairing region can be annealed by accelerated probes and a partially double helix structure may be formed (structure 11).
Basically, the primer design for HAMP reaction is relatively simple. The two target sites (F1 and R1) and two outer primer sites (F2 and R2) could all be selected by the common PCR primer design software (e.g., DNASTAR, DNAMAN). Similar to PSR, the reactions of polymerization and displacement in HAMP system are activated by a single target to produce multiple target copies and dependent on auto-cycling strand displacement by a self-primed single helix structure. However, unlike PSR, no exogenous sequences are introduced in the HAMP reaction. Furthermore, a longer overlap part in the helix loop is formed in the HAMP reaction, which may promote the efficiency of self-annealing and elongation.
The amplification can be further accelerated by the addition of acceleration probes, which may improve the reaction efficiency and sensitivity by accelerating isothermal amplification and reducing detection time.
3.2. Characterization of HAMP assay
MERS-CoV orf1b gene (Fig. 1c) was chosen for systemic verification of HAMP using the selected primers (Table 1). As shown in Fig. 2b–c, the crossing threshold (Ct) values of 3 independent reactions for detecting 106 copies of MERS-CoV orf1b gene were all about 51 min (Fig. 2b) and the melting temperature were 84.32 °C (Fig. 2c). Both the real-time amplification curve and melting curve analysis indicated a good repeatability of HAMP. Furthermore, the result of negative control indicated that non-specific amplification failed to occur.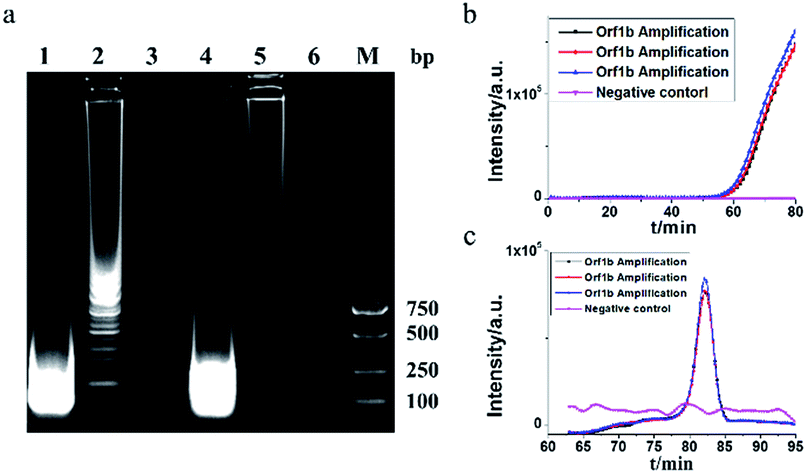 | ||
| Fig. 2 Characterization of HAMP results. (a) Gel electrophoresis for HAMP products with or without digestion by the restriction enzyme HindIII. Lane 1, digested HAMP products; lane 2, HAMP products prior to digestion; lane 3, negative control of HAMP; lane 4, digested HAMP products applying acceleration probes; lane 5, HAMP products applying acceleration probes prior to digestion; lane 6, negative control of HAMP reaction applying acceleration probes; lane M, molecular weight marker. (b) HAMP amplification plot by monitoring the fluorescence intensity. (c) Melting curve analysis on HAMP products. | ||
The HAMP amplification product was further determined by enzyme digestion of HindIII, which cuts the correct amplification product into 146 bp fragments. As shown in Fig. 2a, the digested product showed a strong band approximately 100–200 bp. Two bands from undigested products were excised from the gel and sequenced. The sequencing result confirmed that the DNA bands were corresponded to the predicted HAMP amplification product of MERS-CoV orf1b sequence.
3.3. Sensitivity of HAMP for dsDNA detection
The MERS-CoV orf1b plasmid DNA was used as target nucleic acids to determine the sensitivity of HAMP. A serial dilution of MERS-CoV orf1b plasmid was used as templates. As shown in Fig. 3a, the amplification by HAMP was completed within 80 min. As low as 103 copies of template was detected by HAMP, indicating a good sensitivity, although the amplification efficiency is relatively low at this concentration of template. Meanwhile, the standard amplification curve of HAMP showed a fine regression coefficient (R2 = 0.9969) (Fig. 3b), suggesting the potential application of HAMP for quantitative analysis. In addition, primary evaluation of HAMP specificity was conducted by applying the fragments of MERS-CoV (orf1a, N2, N3 and UpE) and no amplification was observed (data not shown).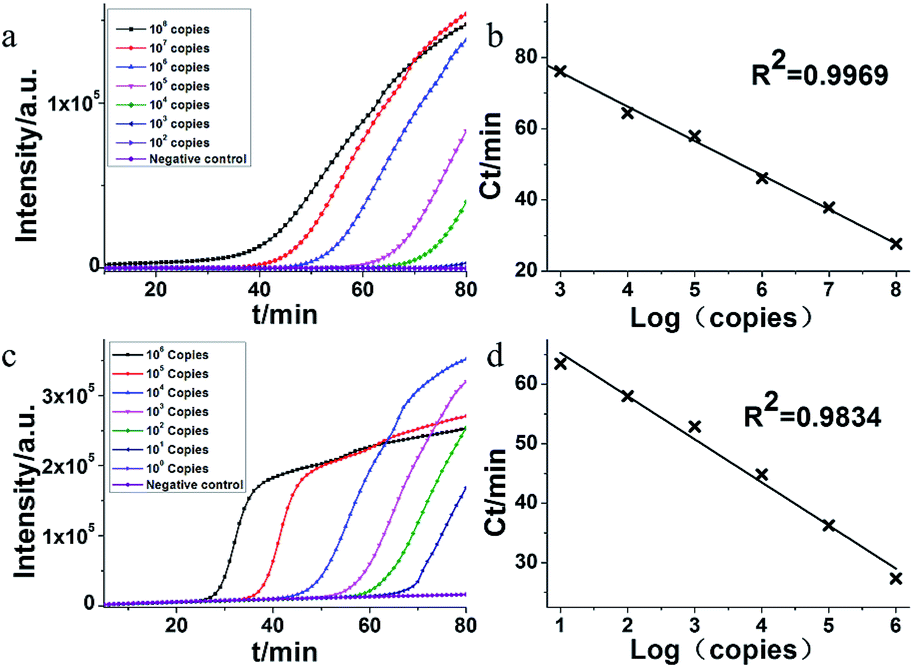 | ||
| Fig. 3 Sensitivity of serially diluted MERS-CoV dsDNA templates by HAMP and acceleration probe added HAMP. (a) Representative isothermal amplification plot by real time PCR analysis on serially diluted dsDNA. Amplification was performed at 63 °C for 90 min. (b) Standard amplification curve obtained by plotting the Ct value versus the plasmid copy number. The DNA concentration was as followed: 108 copies, 107 copies, 106 copies, 105 copies, 104 copies, 103 copies, 102 copies and negative control (nuclease-free water). (c) Representative isothermal amplification plot by real time PCR analysis on serially diluted dsDNA with addition of acceleration probe. Amplification was performed at 63 °C for 65 min. (d) Standard amplification curve obtained by plotting the Ct value versus the plasmid copy number. The DNA concentration was as followed: 106 copies, 105 copies, 104 copies, 103 copies, 102 copies, 101 copies, 100 copies and negative control (nuclease-free water). | ||
3.4. Effect of acceleration probes on HAMP
The acceleration probes were designed to further improve amplification efficiency of HAMP. The probe targeted the MERS-CoV orf1b sequence between F1 and R1, and annealed to the single strand part of helix loop during HAMP amplification (Fig. 1a, structure 11). The amplification product of HAMP using acceleration probes was also analysed by enzyme digestion and gel electrophoresis. The digested product is similar to that of HAMP, and the negative control showed no amplification product (Fig. 2a). The amplification products were also excised form the gel and further confirmed by sequencing. These results indicated that addition of acceleration probe did not influence the specificity of HAMP reaction. But different bands were formed by comparing their products (lane 1 and lane 4), implying that the annealing of acceleration probe may produce different structures of nucleic acids with similar nucleic acid sequence.The amplification efficiency of HAMP was further analysed. Results showed that the addition of acceleration probes shortened the threshold time in the amplification curve by approximately 20 min compared to that in HAMP with the same template concentration, indicating a dramatic improvement in the HAMP efficiency (Fig. 3a and c). However, the regression coefficient on the standard amplification curve (R2 = 0.9834) was worse than that in HAMP (Fig. 3d).
Theoretically, the developed HAMP produced no non-specific products when the appropriate accelerated primers were introduced. Noticeably, accelerated primers can recognize the specific sequences included in the amplification products, hence the DNA synthesis will exhibit higher efficiency in HAMP analysis. The accelerated primers were also applied to PSR.12 Since the isothermal nucleic acid amplification was induced by the same helix loop structure, the HAMP method showed similar amplification efficiency and sensitivity in comparison with PSR.
3.5. RT-HAMP for RNA detection
The feasibility of HAMP for RNA detection was demonstrated by combining the reverse transcriptase with DNA polymerase. In this assay, a serial dilution of the artificial RNA of MERS-orf1b gene was uses as templates. As shown in Fig. 4, the detection limit of RT-HAMP was 104 copies within 70 min with a regression coefficient (R2 = 0.9723). The result suggested that the RT-HAMP was suitable for RNA target amplification. The amplification efficiency of RT-HAMP is likely determined by the activity of AMV reverse transcriptase and DNA polymerase, more suitable enzymes may be screened to optimize our amplification system in the future work.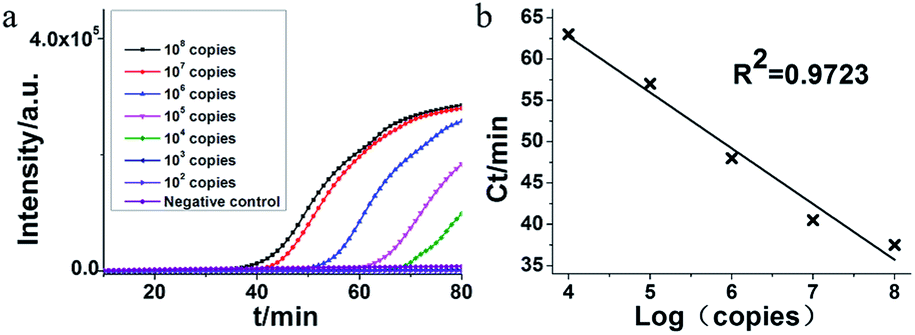 | ||
| Fig. 4 Sensitivity verification of RT-HAMP for RNA amplification. (a) Representative isothermal amplification plot by RT-PCR analysis on serially diluted artificial RNA. Amplification was performed at 63 °C for 90 min. (b) Standard amplification curve obtained by plotting the Ct value versus the plasmid copy number. The RNA concentration was as followed: 108 copies, 107 copies, 106 copies, 105 copies, 104 copies, 103 copies, 102 copies and negative control (nuclease-free water). | ||
3.6. Visual detection of HAMP and RT-HAMP
HNB was applied to the visual detection of HAMP and RT-HAMP. As shown in Fig. 5, visual detection of HAMP and RT-HAMP for nucleic acids worked well in this study after HNB was chosen as colorimetric agent, which would make it applicable in resource limit areas. | ||
| Fig. 5 Visual detection of HAMP and RT-HAMP for nucleic acids. The concentration of both plasmid DNA and mimic RNA was 106 copies in the amplification system. Amplification was performed at 63 °C for 60 min. Reaction 1 and 2, MERS-orf1b plasmid DNA; reaction 3 and 4, negative control of 1 and 2; reaction 5 and 6, MERS-orf1b mimic RNA; reaction 7 and 8, negative control of 5 and 6. | ||
Nucleic acid amplification usually produced a large amount of pyrophosphate ion by-products, which may react with Mg2+ to form insoluble magnesium pyrophosphate, accompanied by the decrease in Mg2+ concentration in reaction mixture.14 On the other hand, some chromogenic agents such as HNB can chelate Mg2+ to form a stable chemical complex with the purple colour produced. With the consumption of Mg2+, the solution colour will change into sky blue. Based on the above, the colorimetric monitoring method was developed for LAMP reaction,15,16 and proved to be also suitable in HAMP.
4. Conclusions
In recent years, the point of care test (POCT) has been paid more attention for its huge potential in global healthcare.17 At the same time, the POCT should meet the guidelines proposed by the World Health Organization (WHO) for developing diagnostic techniques, namely, ASSURED (Affordable, Sensitive, Specific, User friendly, Robust and Rapid, Equipment free and Deliverable).18 In accordance with the guidelines by WHO, the HAMP system developed in this study allowed a good specificity and a sensitivity of 10 DNA copies per reaction within 75 min, and the method is suitable for HNB-based visual detection by naked eye. It is suggested that HAMP assay would be of great potential in the application to POCT.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Chinese Academy of Sciences for the funding of this research (Grant No. CXJJ-15Z012).Notes and references
- S. Yang and R. E. Rothman, Lancet Infect. Dis., 2004, 4, 337–348 CrossRef PubMed.
- Y. Zhao, F. Chen, Q. Li, L. Wang and C. Fan, Chem. Rev., 2015, 115, 12491–12545 CrossRef PubMed.
- M. Gebinoga and F. Oehlenschlager, Eur. J. Biochem., 1996, 235, 256–261 Search PubMed.
- P. M. Lizardi, X. H. Huang, Z. R. Zhu, P. Bray-Ward, D. C. Thomas and D. C. Ward, Nat. Genet., 1998, 19, 225–232 CrossRef PubMed.
- M. C. Little, J. Andrews, R. Moore, S. Bustos, L. Jones, C. Embres, G. Durmowicz, J. Harris, D. Berger, K. Yanson, C. Rostkowski, D. Yursis, J. Price, T. Fort, A. Walters, M. Collis, O. Llorin, J. Wood, F. Failing, C. O'Keefe, B. Scrivens, B. Pope, T. Hansen, K. Marino, K. Williams and M. Boenisch, Clin. Chem., 1999, 45, 777–784 Search PubMed.
- M. Vincent, Y. Xu and H. M. Kong, EMBO Rep., 2004, 5, 795–800 CrossRef PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, 7 CrossRef.
- O. Piepenburg, C. H. Williams, D. L. Stemple and N. A. Armes, PLoS Biol., 2006, 4, 1115–1121 Search PubMed.
- R. Fang, X. Li, L. Hu, Q. You, J. Li, J. Wu, P. Xu, H. Zhong, Y. Luo, J. Mei and Q. Gao, J. Clin. Microbiol., 2009, 47, 845–847 CrossRef PubMed.
- W. Liu, D. R. Dong, Z. Yang, D. Y. Zou, Z. L. Chen, J. Yuan and L. Y. Huang, Sci. Rep., 2015, 5, 8 Search PubMed.
- W. Su, X. Gao, L. Jiang and J. Qin, J. Chromatogr. A, 2015, 1377, 13–26 CrossRef PubMed.
- D. R. Dong, D. Y. Zou, H. Liu, Z. Yang, S. M. Huang, N. W. Liu, X. M. He, W. Liu and L. Y. Huang, Front. Microbiol., 2015, 6, 1100 Search PubMed.
- G. Seyrig, R. D. Stedtfeld, D. M. Tourlousse, F. Ahmad, K. Towery, A. M. Cupples, J. M. Tiedje and S. A. Hashsham, J. Microbiol. Methods, 2015, 119, 223–227 CrossRef PubMed.
- Y. Mori, M. Kitao, N. Tomita and T. Notomi, J. Biochem. Biophys. Methods, 2004, 59, 145–157 CrossRef PubMed.
- S. J. Oh, B. H. Park, J. H. Jung, G. Choi, D. C. Lee, H. Kim do and T. S. Seo, Biosens. Bioelectron., 2016, 75, 293–300 CrossRef PubMed.
- M. Goto, E. Honda, A. Ogura, A. Nomoto and K. Hanaki, BioTechniques, 2009, 46, 167–172 CrossRef PubMed.
- D. A. Giljohann and C. A. Mirkin, Nature, 2009, 462, 461–464 CrossRef PubMed.
- M. Urdea, L. A. Penny, S. S. Olmsted, M. Y. Giovanni, P. Kaspar, A. Shepherd, P. Wilson, C. A. Dahl, S. Buchsbaum, G. Moeller and D. C. Hay Burgess, Nature, 2006, 444(suppl 1), 73–79 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |