
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient and green synthesis of ferrocenyl-quinoline conjugates via a TsOH-catalyzed three-component reaction in water †
Rui-Qi Mou,
Mei Zhao,
Xue-Xin Lv,
Sheng-Yan Zhang and
Dian-Shun Guo *
*
College of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Shandong Normal University, Jinan 250014, P. R. China. E-mail: chdsguo@sdnu.edu.cn; Fax: +86 531 86180743; Tel: +86 531 86928773
First published on 6th March 2018
Abstract
An efficient and green synthesis of 4-ferrocenylquinoline derivatives through a TsOH-catalyzed three-component reaction of aromatic aldehydes, amines and ferrocenylacetylene in water has been successfully developed. This strategy is a powerful method for the construction of diverse ferrocenyl-quinoline conjugates from simple available starting materials as it minimized the use of metal catalyst and organic solvent in the reaction process. The conjugates feature unique structures and excellent electronic properties. Moreover, a plausible mechanism for this TsOH-catalyzed three-component reaction was proposed and assessed.
1 Introduction
Over the past two decades, organic synthesis in water has received considerable attention largely because of water as an economical and environmentally friendly solvent exhibiting unique reactivity and selectivity.1 On the other hand, the ever-increasing interest in multi-component reactions (MCRs) originates from their utilization in the fields of combinatorial chemistry, medicinal chemistry and materials science.2 Overall, to develop MCRs in water is very significant for efficiently constructing bioactive molecule libraries with structural diversity via simultaneously creating two or more chemical bonds in an eco-friendly process.Quinolines, as one of the most prevalent heterocyclic scaffolds, are widely existed in natural products,3 bioactive molecules,4 and functional materials.5 Especially, a majority of their derivatives show a broad range of biological properties involving antimalarial,6 antimicrobial,7 antituberculosis,8 and antitumor activities.9 While ferrocene possesses unique structural and electronic features, and its derivatives exhibit interesting biological properties.10 Most well-designed ferrocenyl-heterocycle conjugates are valuable candidates for antimalarial or anticancer therapies. For instance, ferroquine (FQ, SR97193), an alternative to chloroquine (CQ), has been found to display excellent antimalarial properties (Fig. 1);11 ferrocifen, a drug utilized on breast cancer treatment, has been assessed to be active in both hormone-dependent and hormone-independent breast cancers.12 It is more interesting that the incorporation of ferrocenyl function into some biologically active molecules could markedly enhance their bioactivities or generate novel medicinal properties.13
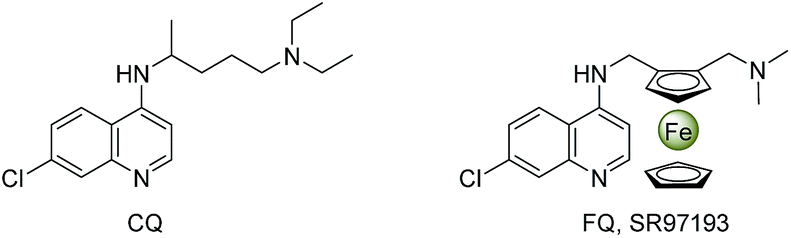 | ||
| Fig. 1 Chemical structures of chloroquine (CQ) and ferroquine (FQ, SR97193). | ||
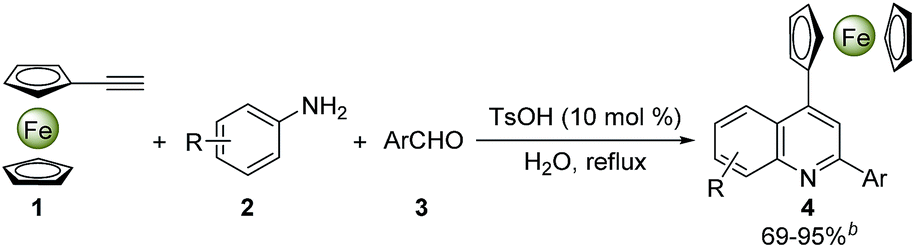
To date, great efforts have been focused on the synthesis of quinolines and their derivatives, but the preparation of ferrocenyl-quinoline conjugates has not been well-explored in the literature. The MCRs have nowadays become a preferred strategy to construct heterocyclic molecules in the light of their facile access to the structural diversity via a one-pot operation in high efficiency.2,14 A number of metal-mediated MCRs of aldehydes, amines and alkynes have been developed for the synthesis of quinoline derivatives. Kuninobu et al. reported an efficient synthesis of 2,4-disubstituted quinoline compounds from readily starting materials utilizing AgOTf and CuCl as catalysts.15 Tu and co-workers presented a method to yield quinoline scaffolds using FeCl3 as a catalyst.16 While some rare earth metal salts were also used as catalysts in this transformation under the harsh reaction conditions.17 Scheme 1 demonstrates the synthesis of 4-ferrocenylquinoline derivatives through the Ce(OTf)3-catalyzed MCRs of ferrocenylacetylene, aromatic aldehydes and amines under reflux or 110 °C in toluene or solvent-free conditions, which not only gave rise to the desired products in poor yields (22–76%) but also used the metal catalyst and organic solvent.18 Generally, most metal-based catalytic species are toxic and expensive, while organic solvents are flammable and harmful. Therefore, in view of the environmental safe and production cost, the usage of organic solvents and metal catalysts in organic synthesis should be minimized as far as possible. Herein, we would like to present a highly efficient and green synthesis of diverse 4-ferrocenylquinoline derivatives via a TsOH-catalyzed three-component reaction in water (Scheme 1). The broad substrate scope was assessed, and a possible reaction mechanism was also proposed. Furthermore, the typical crystal structures, UV-vis spectra and electrochemical properties of these ferrocenyl-quinoline conjugates were evaluated.
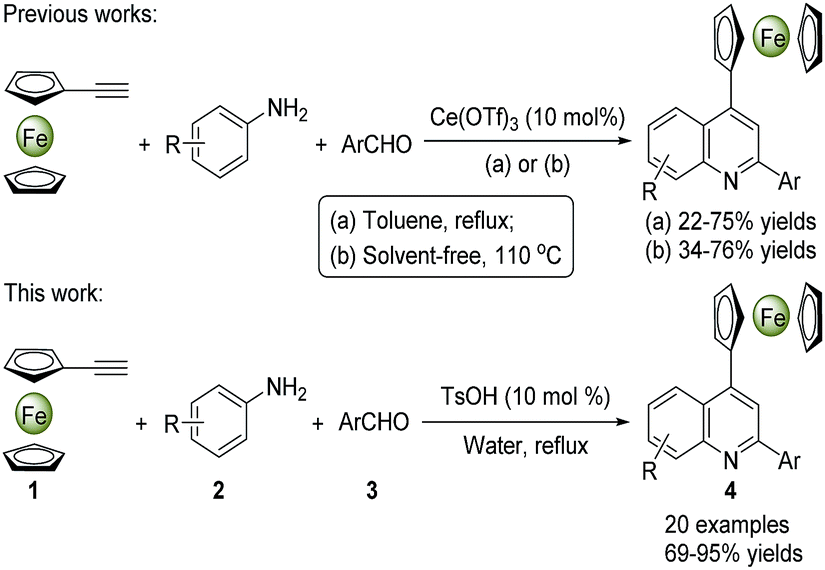 | ||
| Scheme 1 Synthesis of 4-ferrocenylquinoline derivatives via three-component reactions. | ||
2 Results and discussion
2.1 Optimization of reaction conditions
We initiated our study by the usage of ferrocenylacetylene 1, aniline 2a and benzaldehyde 3a as model substrates. First, a three-component reaction of 1, 2a and 3a was conducted in toluene using different organic acids as catalysts,19 we were pleased to find that the reaction can smoothly occur at 55 °C under air to furnish product 4a in 38–83% yields (Table 1, entries 1–4). Although both TfOH and TsOH are good catalysts, the latter became a better choice to assess the influence of various solvents in the light of the cost and easy operation as TsOH is a solid and inexpensive organic acid. To our surprise, this reaction can also perform in other diverse solvents involving DMF, THF, EtOH, and 95% EtOH (Table 1, entries 5–8). By extensive screening, we found that EtOH is the best solvent for the reaction with 86% yield (Table 1, entry 7) among the solvents assessed. Remarkably, this reaction could also give a good yield in 95% EtOH, indicating that the water might play an interesting role in this case. Thus, we further preferred to optimize the water as a solvent to carry out this transformation. When 50% EtOH in water and pure water were used, respectively, the reaction yields decreased greatly to 58% and 49% at the identical conditions (Table 1, entries 9 and 10). Meanwhile, to increase the loading of TsOH catalyst up to 10 mol%, the reaction performed in pure water with 79% yield (Table 1, entry 11) at 55 °C for 5 h. Moreover, we found that to raise the temperature not only can efficiently improve the yield up to 90% at 100 °C in pure water, but also greatly shorten the reaction time to 2 h (Table 1, entry 13). Similarly, an excellent yield of 93% was obtained in EtOH at the reflux condition for 2 h (Table 1, entry 14). It should be noted that the key side reaction is the hydration of ferrocenylacetylene catalyzed by TsOH to generate acetylferrocene byproduct, while higher temperature benefits the main reaction forming the desired product rather than the side reaction yielding the acetylferrocene.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Amount/mol% | Solvent | T/°C | t h−1 | Yieldb/% |
| a Reagents and condition: 1 (1.1 mmol), 2a (1.0 mmol), 3a (1.0 mmol), catalyst (0.05 mmol or 0.1 mmol), solvent (1.0 mL), under air.b Isolated yields. | ||||||
| 1 | HCO2H | 5 | Toluene | 55 | 6 | 38 |
| 2 | TFA | 5 | Toluene | 55 | 6 | 45 |
| 3 | TfOH | 5 | Toluene | 55 | 6 | 83 |
| 4 | TsOH | 5 | Toluene | 55 | 6 | 73 |
| 5 | TsOH | 5 | DMF | 55 | 6 | 82 |
| 6 | TsOH | 5 | THF | 55 | 6 | 70 |
| 7 | TsOH | 5 | EtOH | 55 | 6 | 86 |
| 8 | TsOH | 5 | 95% EtOH | 55 | 6 | 65 |
| 9 | TsOH | 5 | 50% EtOH | 55 | 6 | 58 |
| 10 | TsOH | 5 | H2O | 55 | 6 | 49 |
| 11 | TsOH | 10 | H2O | 55 | 5 | 79 |
| 12 | TsOH | 10 | H2O | 80 | 2.5 | 87 |
| 13 | TsOH | 10 | H2O | 100 | 2 | 90 |
| 14 | TsOH | 10 | EtOH | 78 | 2 | 93 |
2.2 Evaluation of substrate scopes
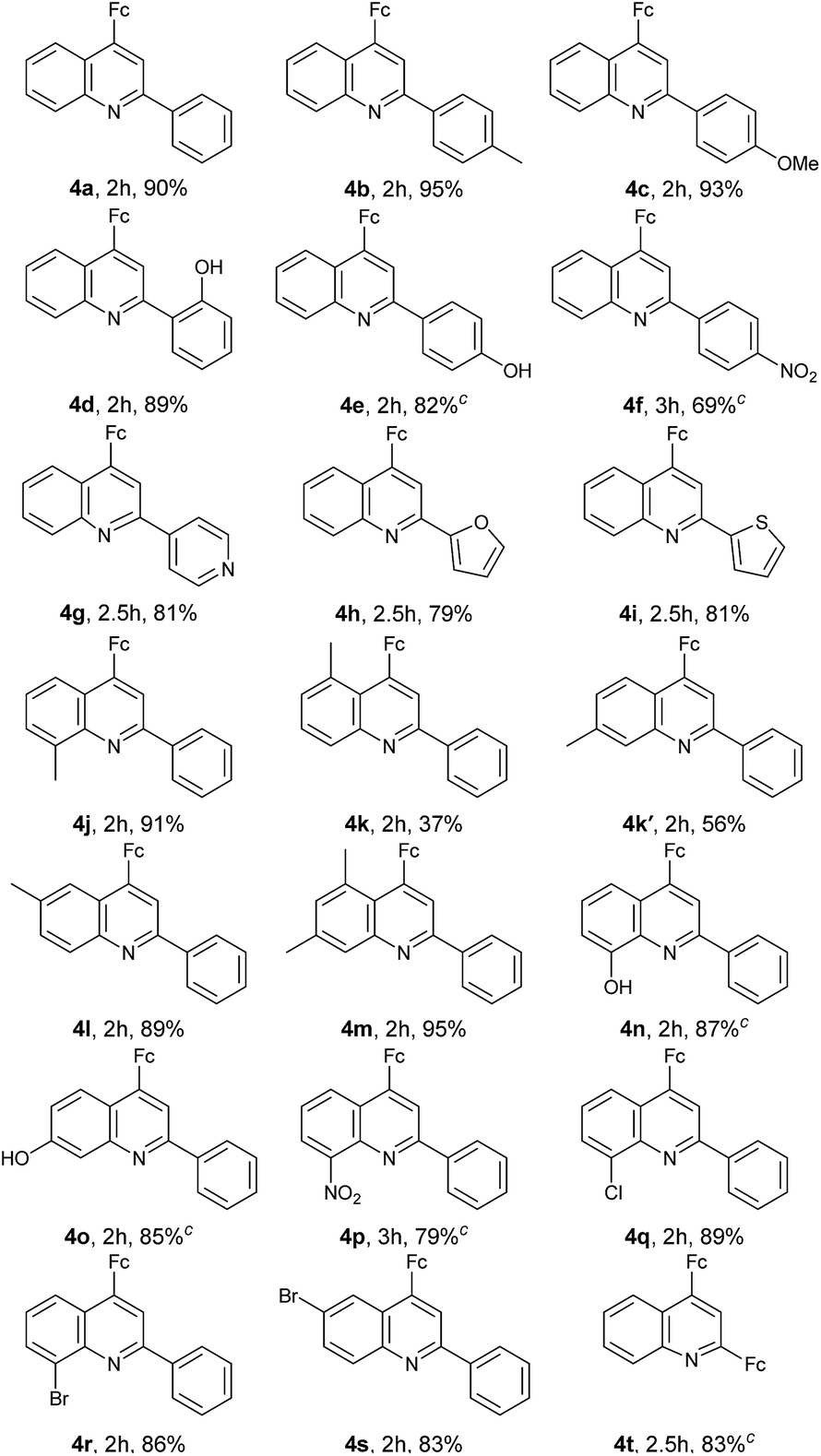
With the optimized reaction conditions in hand, the scope of ferrocenyl-quinoline conjugate generation was evaluated by a majority of aromatic amines and aldehydes in water. As shown in Table 2, in most cases, aromatic amines and aldehydes gave rise to the desired products 4a–4t in good to excellent yields. The aromatic aldehydes with an electron-donating function (4b–4e) generated higher yields than those attaching an electron-withdrawing group (4f and 4g). When heterocyclic aldehydes, isonicotinaldehyde, 2-furancarboxaldehyde, and 2-thiophenecarboxaldehyde were used, the reaction took place smoothly in water and furnished the desired products in 81% (4g), 79% (4h), and 81% (4i) yields. Similarly, the aromatic amines with an electron-rich moiety (4j–4o) afforded higher yields than that bearing an electron-deficient substituent (4p). However, it should be stressed that two regioisomers, 4k and 4k′, were isolated in a ratio of ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 when m-toluidine was used. This indicates that the ferrocenylacetylene can attack both the ortho position and the para position of the methyl group in the aza-Diels–Alder reaction (depicted in Fig. 4 hereinafter), but the former owns larger steric hindrance than the latter, resulting in the 2:3 ratio of 4k:4k′. The big steric hindrance between the ferrocenyl and the methyl in molecule 4k was also rationalized by the following crystal structural analysis. The amines with –Cl and –Br substituents also gave high yields of the products. Unfortunately, attempts on this reaction using phenylacetylene as well as internal alkynes are not successful. In addition, this transformation was also performed in EtOH and gave similar results (ESI†) as in water. All 4-ferrocenylquinoline derivatives prepared were fully confirmed by 1H NMR, 13C NMR and HR-MS analyses.
:3 when m-toluidine was used. This indicates that the ferrocenylacetylene can attack both the ortho position and the para position of the methyl group in the aza-Diels–Alder reaction (depicted in Fig. 4 hereinafter), but the former owns larger steric hindrance than the latter, resulting in the 2:3 ratio of 4k:4k′. The big steric hindrance between the ferrocenyl and the methyl in molecule 4k was also rationalized by the following crystal structural analysis. The amines with –Cl and –Br substituents also gave high yields of the products. Unfortunately, attempts on this reaction using phenylacetylene as well as internal alkynes are not successful. In addition, this transformation was also performed in EtOH and gave similar results (ESI†) as in water. All 4-ferrocenylquinoline derivatives prepared were fully confirmed by 1H NMR, 13C NMR and HR-MS analyses.
2.3 Crystal structures of 4a and 4k
To understand the precise morphology of 4-ferrocenyl quinoline derivatives 4, single crystal structures of two typical conjugates 4a and 4k were assessed by the X-ray crystallography (ESI†). In both molecules (Fig. 2), two cyclopentadienyl (Cp) rings of the ferrocenyl group adopt a nearly eclipsed fashion with average torsion angles of 3.3° and 18.3°, while they locate almost parallel to mutually with mean dihedral angles of 2.2° and 6.6°. Remarkably, the quinoline ring is not well coplanar with its connected Cp ring. For 4a, the ferrocenyl function leans toward the quinolyl plane characterized with a dihedral angle of 35.2° between the quinoline plane and the Cp ring. Surprisingly, the ferrocenyl group of 4k inclines toward the other side owing to the influence of the methyl group at 5 position of the quinoline ring and it is approximatively perpendicular with the quinolyl plane identified by a dihedral angle of 79.5°, while the methyl also tilts slightly with varied angles of 124.4° and 116.6° deviated from the standard value of 120°. This can rationalize the formation of 4k and 4k′ in ca. 2:3 ratio when m-toluidine was used. The quinolyl plane is not coplanar with the phenyl ring at its 2 position, creating dihedral angles of 12.2° and 24.9° for 4a and 4k, respectively. In the packing, the major interactions are intermolecular C–H⋯π contacts (ESI†). To fully understand the crystal packing driving forces, the Hirshfeld surfaces and fingerprint plots of 4a and 4k were further analyzed with CrystalExplorer.20 Fig. 3 depicts the surfaces mapped with close contacts between vicinal molecules for 4a and 4k. The key C–H⋯π interactions are showed as deep red spots, suggesting that they play an important role in their crystal packing, while there are various slight red spots observed in other orientations (ESI†), standing for weaker and longer C–H⋯π contacts. The main intermolecular contacts involve weak H⋯H and C⋯H/H⋯C interactions. They totally account for 93.4 and 89.5% of the surface–contact interaction coverage of 4a and 4k, respectively, in which C⋯H/H⋯C contacts comprise 37.2 and 26.5% of the total Hirshfeld surface area. For all C–H⋯C interactions in both molecules, C–H⋯π contacts appear in the fingerprint plot as a characteristic style and represent the closest contacts.
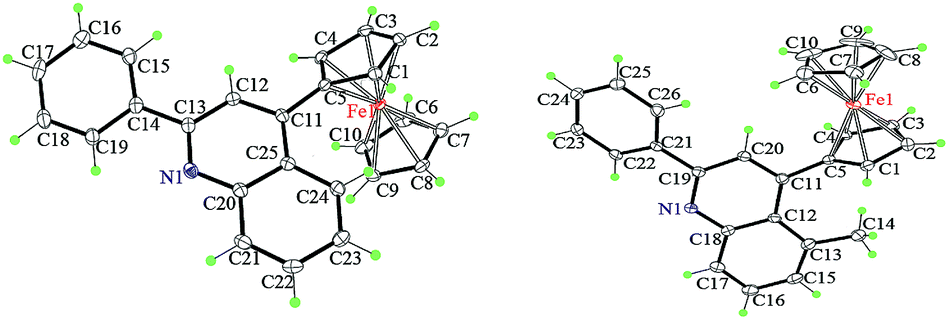 | ||
| Fig. 2 The crystal structures of 4a (left) and 4k (right), showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 30% probability level. | ||
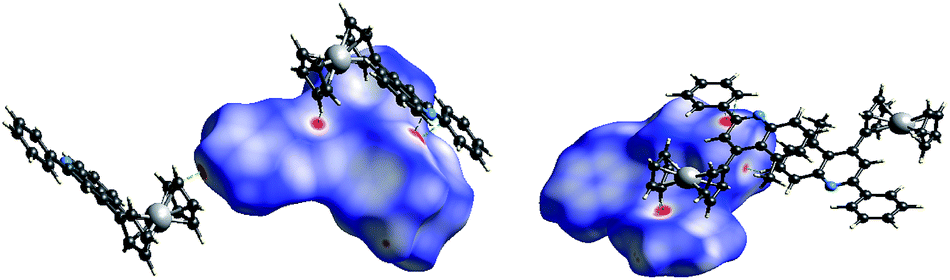 | ||
| Fig. 3 The Hirshfeld surface for 4a (left) and 4k (right). Vicinal molecules associated with close contacts are shown. | ||
2.4 Electrochemistry and UV-vis spectra of 4
Electrochemical assays of 4-ferrocenylquinoline derivatives 4 were carried out using cyclic voltammetry (CV). Their CV curves show the typical quasi-reversible wave of the ferrocene/ferrocenium (Fc/Fc+) redox couple (ESI†). Compared with the parent ferrocene, their E1/2 values are positively shifted for ca. 90 to 161 mV, varied markedly with different substituents at the quinoline ring (Table 3). This indicates that ferrocenyl-quinoline conjugates 4 are oxidized more difficultly than the parent ferrocene owing to the electron-deficient feature of the quinoline ring.21 In the case of 4t, although the ratio of Ipa/Ipc is 1.02, a bigger separation (129 mV) between anodic and cathodic peaks was obtained owing to the overlap of the CV curves resulted from two ferrocenyl groups at 2 and 4 position of quinoline ring.| No. | Epa/mV | Epc/mV | Epa−Epc/mV | E1/2/mV | Ipa/Ipc |
|---|---|---|---|---|---|
| a Conditions: 7.0 × 10−4 M of 4 and 0.1 M n-Bu4NPF6 in CH3CN, Pt disk working electrode, Pt auxiliary electrode, Hg/Hg2Cl2 reference electrode and scanning at 100 mV s−1. Errors: ± 10 mV.b The bigger value may be ascribed to the overlap of the CV peaks of two ferrocenyl functions at different positions. | |||||
| 4a | 559 | 485 | 76 | 522 | 1.02 |
| 4b | 554 | 484 | 70 | 519 | 1.01 |
| 4c | 567 | 481 | 86 | 524 | 0.99 |
| 4d | 590 | 504 | 86 | 547 | 0.99 |
| 4e | 560 | 480 | 80 | 520 | 0.96 |
| 4f | 573 | 502 | 71 | 538 | 0.97 |
| 4g | 562 | 492 | 70 | 527 | 0.98 |
| 4h | 563 | 485 | 78 | 524 | 0.95 |
| 4i | 560 | 488 | 72 | 524 | 0.99 |
| 4j | 554 | 480 | 74 | 517 | 1.00 |
| 4k | 550 | 476 | 74 | 513 | 1.00 |
| 4l | 558 | 478 | 80 | 518 | 0.98 |
| 4m | 531 | 450 | 80 | 490 | 0.95 |
| 4n | 561 | 490 | 71 | 526 | 0.94 |
| 4o | 562 | 494 | 70 | 528 | 0.99 |
| 4p | 604 | 518 | 86 | 561 | 0.96 |
| 4q | 580 | 507 | 73 | 544 | 0.95 |
| 4r | 579 | 502 | 77 | 541 | 0.96 |
| 4s | 576 | 502 | 74 | 539 | 0.98 |
| 4t | 625 | 496 | 129b | 561 | 1.02 |
UV-vis properties of 4a–4t were assessed using two concentrations (ESI†) as the UV absorbance of the ferrocenyl and quinolyl is very different. In the UV-vis spectra, most of compounds exhibit absorption peaks at ca. 202–209, 256–287, 310–321, 381–404, and 459–486 nm. The former three peaks can be ascribed to the E, K and B bands of the quinoline moiety, which produced a obviously bathochromic shift with 36–67 nm for the K band and 35–46 nm for the B band compared with the parent quinoline. The latter two peaks belong to the ferrocenyl function and also create a clear bathochromic shift compared with the ferrocene (325 and 440 nm). This behavior can be rationalized by the mutual contribution of the quinolyl and ferrocenyl functions to the expansion of the conjugate system in these compounds.
2.5 A plausible mechanism for generating 4
Synthetically considering the previous understanding of this type of three-component reactions as well as our experimental results, we have proposed a plausible mechanism for this TsOH-catalyzed reaction (Fig. 4). First, the aldehyde and amine react to generate aldimine I through a TsOH-catalyzed condensation in aqueous emulsions,22 where the aldimine I once formed, it was quickly expelled from the intermicellar hydrophilic environment. Next, the TsOH continues to activate the aldimine I to yield a protonated aldimine intermediate II, which then undergoes an aza-Diels–Alder reaction with ferrocenylacetylene, giving intermediate III. Finally, a deprotonation of III, followed an oxidation aromatization of dihydroquinoline IV to create the quinoline ring structure.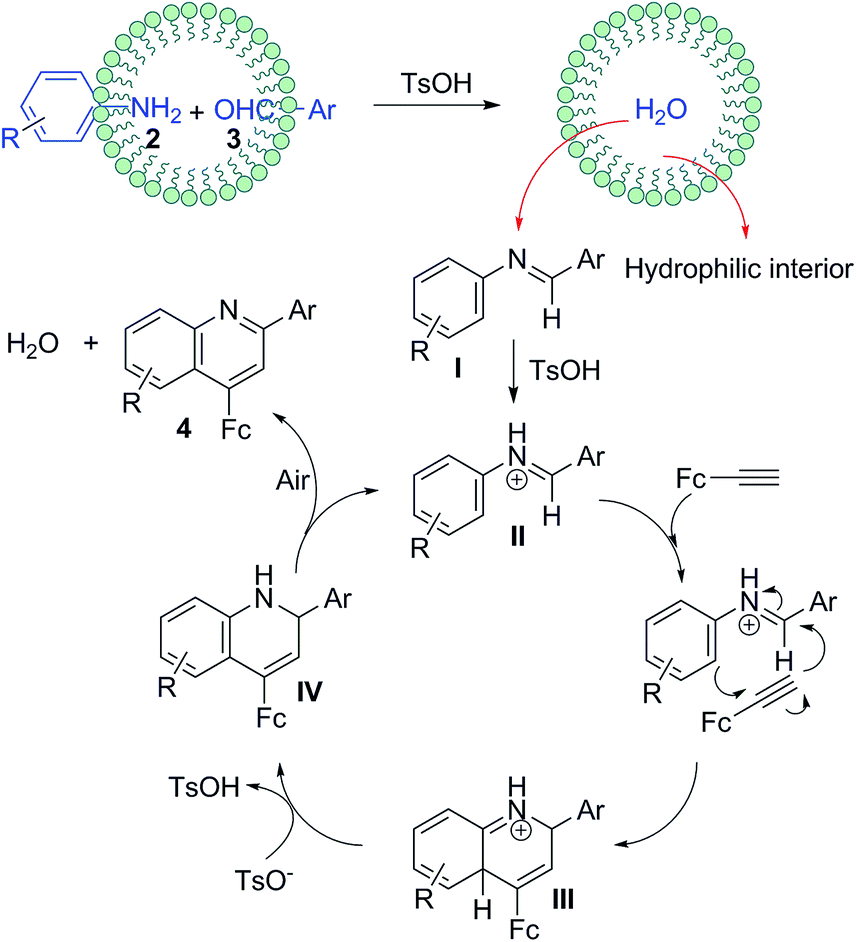 | ||
| Fig. 4 A plausible mechanism for producing 4. | ||
To rationalize the mechanism proposed above, we utilized aldimine Ia instead of aniline 2a and benzaldehyde 3a to perform the model reaction in the presence or absence of TsOH in water at 100 °C under air (ESI†). In the former case, the similar yield (91%) was obtained in a shorter time (1.3 h), while the reaction cannot occur in the latter case, confirming that the TsOH catalyst not only accelerates the formation of aldimine Ia, but also promotes the following transformation into the desired products 4. It should be noted that such an aza-Diels–Alder reaction only suits for the electron-rich ferrocenylacetylene, however phenylacetylene does not work in the identical conditions. In addition, when an aldimine substrate 5, obtained from the reaction of benzaldehyde and 2,6-dimethylaniline, was treated with the ferrocenylacetylene in the same conditions (Scheme 2) to furnish a new intermediate product 6 confirmed by 1H NMR (ESI†), while the desired final product could not be found. This indicates that the aza-Diels–Alder reaction has not occurred as two o-positions of the imino moiety were blocked by two methyl groups, in which the corresponding dihydroquinoline intermediate IV is unable to generate. Alternatively, a usual electrophilic addition reaction of aldimine 5 to ferrocenylacetylene might take place with a deprotonation rearrangement process to yield an alkenylated imine compound 6. This proves the possibility of the ferrocenylacetylene attacking the protonated aldimine. Moreover, the formation of the aldimine I in water is easier and faster than in non-polar solvent, which might be ascribed to the promotion of the self-assembly micelle.
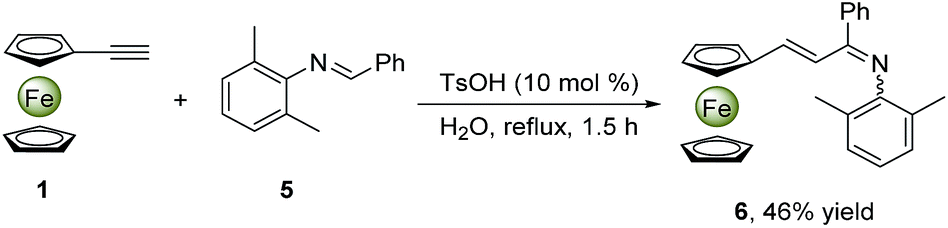 | ||
| Scheme 2 Synthesis of alkenylated imine 6. | ||
3 Conclusions
In summary, we have successfully developed a TsOH-catalyzed three-component reaction in water media for efficient and green synthesis of ferrocenyl-quinoline conjugates. The usage of an inexpensive acid catalyst and an eco-friendly solvent to promote such a transformation is very significant for the development of smart atom-economic strategies. This strategy can be used for easily creating complex organometallic heterocyclic molecules from simple starting materials.4 Experimental
4.1 Reagents and instruments
All starting materials and solvents were commercially available and used without further purification. 1H NMR and 13C NMR spectra were measured with BRUKER ADVANCE 300/400 spectrometers (CDCl3 or DMSO-d6, TMS as internal standard). UV-vis spectra were measured on UV-2600 UV-vis spectrometer. Melting points were recorded on Yanaco MP-500 micro melting point instrument and uncorrected. Electrospray ionization mass spectra (ESI-MS) were obtained on ma Xis UHR-TOF system. Electrochemical tests were performed with CHI 660 electrochemical analyzer. Crystal data were obtained by Agilent SuperNova X-Ray single crystal diffractometer.4.2 General procedure for the synthesis of 4-ferrocenyl-quinoline conjugates 4
A suspension of ferrocenylacetylene (1.1 mmol), aldehyde (1.0 mmol), amine (1.0 mmol) and TsOH (0.1 mmol) in water (1.0 mL) was stirred at 100 °C under air. When the reaction completed (monitored by TLC), the resulting mixture was cooled to room temperature and extracted with ethyl acetate. The organic phase was washed with saturated sodium bicarbonate solution and brine, and dried over anhydrous MgSO4. The volatile was evaporated under reduced pressure, and the residue was purified via flash column chromatography on silica-gel, to give products 4.Note: for 4e, 4f, 4n, 4o, 4p and 4t, the reaction was carried out in 1.0 mL of H2O/EtOH (9:1, v/v).
4.3 4-Ferrocenyl-2-phenylquinoline (4a)
This compound was prepared according to the general procedure from aniline (0.093 g, 1.0 mmol) and benzaldehyde (0.106 g, 1.0 mmol) and purified by flash column chromatography (2 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.3), to give 4a in 90% yield (75%)18a as a red solid, mp 145–146 °C. 1H NMR (CDCl3, 300 MHz): δ 8.61 (d, J = 9.0 Hz, 1H), 8.23–8.20 (m, 3H), 8.16 (s, 1H), 7.72 (t, J = 6.6 Hz, 1H), 7.58–7.51 (m, 4H), 4.84 (s, 2H), 4.54 (s, 2H), 4.24 (s, 5H); 13C NMR (CDCl3, 75 MHz): δ 156.5, 148.9, 146.9, 139.9, 130.4, 129.2, 128.9, 127.5, 125.7, 125.6, 119.7, 112.9, 83.8, 70.5, 70.0, 69.4. HR-MS (ESI): calcd for C25H19FeN [M]+: 390.0945; found: 390.0975.
4.4 4-Ferrocenyl-2-(4-methylphenyl)quinoline (4b)
Following the procedure for 4a, 4-methylbenzaldehyde (0.120 g, 1.0 mmol) was used instead of benzaldehyde (2 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.4), to give 4b in 95% yield (63%)18a as a red solid, mp 127–129 °C. 1H NMR (CDCl3, 300 MHz): δ 8.58 (d, J = 8.4 Hz, 1H), 8.22 (d, J = 8.4 Hz, 1H), 8.14–8.11 (m, 3H), 7.71 (t, J = 7.2 Hz, 1H), 7.53 (t, J = 7.6 Hz, 1H), 7.38 (d, J = 7.6 Hz, 2H), 4.82 (s, 2H), 4.53 (s, 2H), 4.23 (s, 5H), 2.47 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 156.5, 149.0, 146.6, 139.3, 137.2, 130.3, 129.6, 129.2, 127.4, 126.0, 125.7, 125.4, 119.6, 83.9, 70.5, 70.0, 69.3, 21.4. HR-MS (ESI): calcd for C26H21FeN [M + H]+: 404.1151; found: 404.1102.
4.5 4-Ferrocenyl-2-(4-methoxyphenyl)quinoline (4c)
Following the procedure for 4a, 4-methoxybenzaldehyde (0.136 g, 1.0 mmol) was used instead of benzaldehyde (2 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.4), to give 4c in 93% yield (64%)18a as a red solid, mp 163–164 °C. 1H NMR (CDCl3, 400 MHz): δ 8.55 (d, J = 8.2 Hz, 1H), 8.18–8.16 (m, 3H), 8.09 (s, 1H), 7.68 (t, J = 6.9 Hz, 1H), 7.50 (t, J = 7.2 Hz, 1H), 7.07 (d, J = 6.9 Hz, 2H), 4.79 (s, 2H), 4.50 (s, 2H), 4.20 (s, 5H), 3.89 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 160.8, 156.0, 149.0, 146.9, 132.3, 130.1, 129.3, 128.9, 125.8, 125.7, 125.4, 119.3, 114.3, 83.9, 70.5, 70.0, 69.4, 55.5. HR-MS (ESI): calcd for C26H21FeNO [M + H]+: 420.1051; found: 420.1060.
4.6 4-Ferrocenyl-2-(2-hydroxyphenyl)quinoline (4d)
Following the procedure for 4a, 2-hydroxybenzaldehyde (0.122 g, 1.0 mmol) was used instead of benzaldehyde (2 h, ethyl acetate/hexane = 1:10, v/v, Rf = 0.3), to give 4d in 89% yield (44%)18b as an orange solid, mp 189–190 °C. 1H NMR (CDCl3, 400 MHz): δ 8.57 (d, J = 8.4 Hz, 1H), 8.30 (s, 1H), 8.06–8.00 (m, 2H), 7.71 (t, J = 7.6 Hz, 1H), 7.54 (t, J = 7.6 Hz, 1H), 7.39 (t, J = 7.0 Hz, 1H), 7.09 (d, J = 7.7 Hz, 1H), 6.97 (t, J = 7.2 Hz, 1H), 4.82 (s, 2H), 4.53 (s, 2H), 4.22 (s, 5H); 13C NMR (CDCl3, 100 MHz): δ 156.8, 148.3, 145.3, 131.8, 130.0, 128.2, 126.5, 126.0, 125.9, 125.6, 119.0, 117.8, 83.3, 70.7, 70.1, 69.7. HR-MS (ESI): calcd for C25H19FeNO [M + H]+: 406.1060; found: 406.1050.
4.7 4-Ferrocenyl-2-(4-hydroxyphenyl)quinoline (4e)
Following the procedure for 4a, 4-hydroxybenzaldehyde (0.122 g, 1.0 mmol) was used instead of benzaldehyde (2 h, ethyl acetate/hexane = 1:5, v/v, Rf = 0.3), to give 4e in 82% yield (60%)18b as a brown solid, mp 186–188 °C. 1H NMR (CDCl3, 300 MHz): δ 8.60 (d, J = 8.4 Hz, 1H), 8.24 (d, J = 8.4 Hz, 1H), 8.07–8.00 (m, 3H), 7.71 (t, J = 7.6 Hz, 1H), 7.53 (t, J = 7.6 Hz, 1H), 6.95 (d, J = 8.4 Hz, 2H), 4.82 (s, 2H), 4.53 (s, 2H), 4.23 (s, 5H); 13C NMR (DMSO-d6, 100 MHz): δ 159.5, 155.6, 148.8, 146.6, 130.0, 129.8, 129.2, 125.9, 125.3, 118.6, 116.3, 116.1, 83.3, 70.7, 70.3, 70.0. HR-MS (ESI): calcd for C25H19FeNO [M + H]+: 406.1060; found: 406.0994.
4.8 4-Ferrocenyl-2-(4-nitrophenyl)quinoline (4f)
Following the procedure for 4a, 4-nitrobenzaldehyde (0.151 g, 1.0 mmol) was used instead of benzaldehyde (3 h, ethyl acetate/hexane = 1:10, v/v, Rf = 0.4), to give 4f in 69% yield (38%)18a as a red solid, mp 178–179 °C. 1H NMR (CDCl3, 300 MHz): δ 8.69 (d, J = 8.4 Hz, 1H), 8.41 (s, 4H), 8.24 (d, J = 7.0 Hz, 1H), 8.16 (s, 1H), 7.77 (t, J = 7.1 Hz, 1H), 7.63 (t, J = 7.5 Hz, 1H), 4.85 (s, 2H), 4.58 (s, 2H), 4.24 (s, 5H); 13C NMR (CDCl3, 100 MHz): δ 153.7, 148.9, 148.3, 148.0, 145.7, 130.5, 129.8, 128.3, 126.6, 125.9, 124.1, 119.3, 83.2, 70.5, 70.1, 69.8. HR-MS (ESI): calcd for C25H18FeN2O2 [M + H]+: 435.0803; found: 435.0796.
4.9 4-Ferrocenyl-2-(4-pyridinyl)quinoline (4g)
Following the procedure for 4a, isonicotinaldehyde (0.107 g, 1.0 mmol) was used instead of benzaldehyde (2.5 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.4), to give 4g in 81% yield as a red solid, mp 155–157 °C. 1H NMR (CDCl3, 300 MHz): δ 8.90 (s, 2H), 8.66 (d, J = 8.4 Hz, 1H), 8.23 (d, J = 8.4 Hz, 1H), 8.16 (s, 3H), 7.77 (t, J = 7.5 Hz, 1H), 7.62 (t, J = 7.5 Hz, 1H), 4.84 (s, 2H), 4.57 (s, 2H), 4.24 (s, 5H); 13C NMR (CDCl3, 100 MHz): δ 153.6, 150.5, 149.0, 147.8, 146.9, 130.6, 129.7, 126.6, 126.5, 125.8, 121.7, 119.0, 83.3, 70.5, 70.0, 69.7. HR-MS (ESI): calcd for C24H18FeN2 [M + H]+: 391.0884; found: 391.0898.
4.10 4-Ferrocenyl-2-(2-furanyl)quinoline (4h)
Following the procedure for 4a, 2-furancarboxaldehyde (0.096 g, 1.0 mmol) was used instead of benzaldehyde (2.5 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.3), to give 4h in 79% yield (48%)18a as a red solid, mp 182–183 °C. 1H NMR (CDCl3, 400 MHz): δ 8.51 (d, J = 8.3 Hz, 1H), 8.19–8.04 (m, 2H), 7.66–7.64 (m, 2H), 7.48 (t, J = 7.4 Hz, 1H), 7.25 (s, 1H), 6.60 (s, 1H), 4.79 (s, 2H), 4.49 (s, 2H), 4.20 (s, 5H); 13C NMR (CDCl3, 100 MHz): δ 153.9, 148.8, 148.3, 146.9, 144.0, 130.0, 129.4, 126.1, 125.8, 125.5, 118.0, 112.2, 109.8, 83.5, 70.6, 70.0, 69.4. HR-MS (ESI): calcd for C23H17FeNO [M + Na]+: 402.0558; found: 402.0561.
4.11 4-Ferrocenyl-2-(2-thiophenyl)quinoline (4i)
Following the procedure for 4a, 2-thio-phenecarbaldehyde (0.112 g, 1.0 mmol) was used instead of benzaldehyde (2.5 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.3), to give 4i in 81% yield (53%)18a as a red solid, mp 156–157 °C. 1H NMR (CDCl3, 300 MHz): δ 8.53 (d, J = 8.4 Hz, 1H), 8.12 (t, J = 8.4 Hz, 1H), 8.10 (s, 1H), 7.81 (s, 1H), 7.69 (t, J = 7.5 Hz, 1H), 7.53–7.49 (m, 2H), 7.22 (d, J = 4.5 Hz, 1H), 4.81 (s, 2H), 4.53 (s, 2H), 4.23 (s, 5H); 13C NMR (CDCl3, 75 MHz): δ 151.5, 148.8, 146.8, 145.6, 129.9, 129.4, 128.2, 128.1, 126.2, 125.7, 125.5, 121.0, 118.4, 83.6, 70.5, 70.0, 69.4. HR-MS (ESI): calcd for C23H17FeNS [M + H]+: 396.0509; found: 396.0521.
4.12 4-Ferrocenyl-8-methyl-2-phenylquinoline (4j)
Following the procedure for 4a, o-toluidine (0.107 g, 1.0 mmol) was used instead of aniline (2 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.4), to give 4j in 91% yield as a red solid, mp 131–133 °C. 1H NMR (CDCl3, 400 MHz): δ 8.40 (d, J = 8.4 Hz, 1H), 8.31 (d, J = 6.6 Hz, 2H), 8.20 (s, 1H), 7.56 (s, 3H), 7.48 (t, J = 6.6 Hz, 1H), 7.41 (t, J = 7.0 Hz, 1H), 4.80 (s, 2H), 4.49 (s, 2H), 4.21 (s, 5H), 2.94 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 154.6, 147.9, 146.8, 140.2, 138.1, 129.4, 129.2, 128.9, 127.5, 126.0, 125.3, 123.7, 119.1, 84.4, 70.7, 70.0, 69.2, 18.7. HR-MS (ESI): calcd for C26H21FeN [M + H]+: 404.1151; found: 404.1104.
4.13 4-Ferrocenyl-5-methyl-2-phenylquinoline (4k) and 4-ferrocenyl-7-methyl-2-phenylquinoline (4k′)
Following the procedure for 4a, m-toluidine (0.107 g, 1.0 mmol) was used instead of aniline (2 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.4 for 4k and 0.35 for 4k′), to give 4k (37%, mp 178–180 °C) and 4k′ (56%, mp 144–146 °C) as a red solid. For 4k, 1H NMR (CDCl3, 400 MHz): δ 8.66 (s, 1H), 8.26 (d, J = 7.4 Hz, 2H), 8.09 (d, J = 7.4 Hz, 1H), 7.58–7.51 (m, 4H), 7.19 (d, J = 7.0 Hz, 1H), 4.54 (s, 2H), 4.41 (s, 2H), 4.29 (s, 5H), 2.10 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 155.0, 135.5, 129.5, 129.3, 129.0, 128.8, 128.6, 127.6, 127.4, 124.6, 118.5, 113.7, 110.0, 92.8, 72.4, 69.7, 67.7, 24.4. HR-MS (ESI): calcd for C26H21FeN [M + H]+: 404.1151; found: 404.1121. For 4k′, 1H NMR (CDCl3, 300 MHz): δ 8.50 (d, J = 8.6 Hz, 1H), 8.20 (d, J = 7.2 Hz, 2H), 8.08 (s, 1H), 8.02 (s, 1H), 7.60–7.52 (m, 3H), 7.39 (d, J = 8.6 Hz, 1H), 4.82 (s, 2H), 4.52 (s, 2H), 4.23 (s, 5H), 2.60 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 156.5, 149.1, 140.0, 139.5, 129.3, 129.1, 128.8, 127.9, 127.5, 127.3, 125.4, 124.0, 118.9, 83.8, 70.5, 69.9, 69.4, 21.7. HR-MS (ESI): calcd for C26H21FeN [M + H]+: 404.1151; found: 404.1115.
4.14 4-Ferrocenyl-6-methyl-2-phenylquinoline (4l)
Following the procedure for 4a, p-toluidine (0.107 g, 1.0 mmol) was used instead of aniline (2 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.4), to give 4l in 89% yield (52%)18a as a red solid, mp 169–171 °C. 1H NMR (CDCl3, 300 MHz): δ 8.40 (s, 1H), 8.20 (d, J = 7.2 Hz, 3H), 8.11 (s, 1H), 7.62–7.53 (m, 3H), 7.50 (d, J = 7.2 Hz, 1H), 4.83 (s, 2H), 4.54 (s, 2H), 4.24 (s, 5H), 2.59 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 155.7, 147.5, 145.8, 139.9, 135.4, 131.5, 130.0, 129.0, 128.8, 127.4, 125.9, 124.7, 119.8, 84.0, 70.5, 70.0, 69.3, 22.1. HR-MS (ESI): calcd for C26H21FeN [M + H]+: 404.1151; found: 404.1104.
4.15 4-Ferrocenyl-5,7-dimethyl-2-phenylquinoline (4m)
Following the procedure for 4a, 3,5-dimethylaniline (0.121 g, 1.0 mmol) was used instead of aniline (2 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.4), to give 4m in 95% yield as an orange solid, mp 153–154 °C. 1H NMR (CDCl3, 400 MHz): δ 8.59 (s, 1H), 8.24 (d, J = 7.2 Hz, 2H), 7.94 (s, 1H), 7.57 (t, J = 8.0 Hz, 2H), 7.51–7.47 (m, 1H), 7.05 (s, 1H), 4.54 (s, 2H), 4.40 (s, 2H), 4.28 (s, 5H), 2.48 (s, 3H), 2.08 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 155.1, 150.0, 145.6, 139.7, 138.7, 135.1, 131.8, 129.2, 129.0, 127.8, 127.3, 123.9, 119.6, 93.0, 72.4, 69.7, 67.6, 24.2, 21.3. HR-MS (ESI): calcd for C27H23FeN [M + H]+: 418.1258; found: 418.1275.
4.16 4-Ferrocenyl-8-hydroxy-2-phenylquinoline (4n)
Following the procedure for 4a, 2-aminophenol (0.109 g, 1.0 mmol) was used instead of aniline (2 h, ethyl acetate/hexane = 1:10, v/v, Rf = 0.5), to give 4n in 87% yield (60%)18b as a red solid, mp 159–160 °C. 1H NMR (CDCl3, 300 MHz): δ 8.19–8.10 (m, 3H), 8.01 (d, J = 8.6 Hz, 1H), 7.56–7.47 (m, 3H), 7.41 (t, J = 8.1 Hz, 1H), 7.15 (d, J = 7.4 Hz, 1H), 4.80 (s, 2H), 4.49 (s, 2H), 4.17 (s, 5H); 13C NMR (CDCl3, 100 MHz): δ 153.9, 152.6, 147.7, 139.0, 138.8, 129.5, 128.9, 127.3, 126.7, 126.1, 120.0, 116.2, 109.6, 83.4, 70.4, 70.0, 69.6. HR-MS (ESI): calcd for C25H19FeNO [M + H]+: 406.1060; found: 406.1009.
4.17 4-Ferrocenyl-7-hydroxy-2-phenylquinoline (4o)
Following the procedure for 4a, 3-aminophenol (0.109 g, 1.0 mmol) was used instead of aniline (2 h, ethyl acetate/hexane = 1:5, v/v, Rf = 0.3), to give 4o in 85% yield (63%)18b as an orange solid, mp 170–171 °C. 1H NMR (CDCl3, 300 MHz): δ 9.71 (s, 1H), 8.43 (d, J = 9.1 Hz, 1H), 8.16 (d, J = 7.2 Hz, 2H), 7.92 (s, 1H), 7.55–7.50 (m, 3H), 7.45 (s, 1H), 7.17 (d, J = 9.1 Hz, 1H), 4.78 (s, 2H), 4.49 (s, 2H), 4.19 (s, 5H); 13C NMR (DMSO-d6, 100 MHz): δ 158.8, 155.7, 150.7, 146.7, 139.5, 129.8, 129.2, 127.6, 127.5, 120.0, 119.0, 116.5, 111.3, 83.5, 70.7, 70.3, 69.9. HR-MS (ESI): calcd for C25H19FeNO [M + H]+: 406.1060; found: 406.0999.
4.18 4-Ferrocenyl-8-nitro-2-phenylquinoline (4p)
Following the procedure for 4a, 2-nitroaniline (0.138 g, 1.0 mmol) was used instead of aniline (3 h, ethyl acetate/hexane = 1:10, v/v, Rf = 0.3), to give 4p in 79% yield (34%)18b as a deep red solid, mp 150–152 °C. 1H NMR (CDCl3, 400 MHz): δ 8.73 (d, J = 8.2 Hz, 1H), 8.28–8.16 (m, 3H), 7.92 (d, J = 7.0 Hz, 1H), 7.57–7.45 (m, 4H), 4.77 (s, 2H), 4.53 (s, 2H), 4.18 (s, 5H); 13C NMR (CDCl3, 100 MHz): δ 157.7, 149.2, 147.8, 140.3, 138.3, 130.3, 129.6, 129.0, 127.7, 127.1, 123.8, 123.0, 120.4, 82.7, 70.7, 70.2, 70.0. HR-MS (ESI): calcd for C25H18FeN2O2 [M + H]+: 435.0803; found: 435.0796.
4.19 8-Chloro-4-ferrocenyl-2-phenylquinoline (4q)
Following the procedure for 4a, 2-chloroaniline (0.127 g, 1.0 mmol) was used instead of aniline (2 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.3), to give 4q in 89% yield (52%)18a as an orange solid, mp 168–170 °C. 1H NMR (CDCl3, 400 MHz): δ 8.49 (d, J = 7.1 Hz, 1H), 8.32 (s, 2H), 8.24 (s, 1H), 7.83 (d, J = 5.5 Hz, 1H), 7.56 (s, 2H), 7.55–7.50 (m, 1H), 7.49–7.41 (m, 1H), 4.79 (s, 2H), 4.52 (s, 2H), 4.21 (s, 5H); 13C NMR (CDCl3, 100 MHz): δ 156.4, 147.8, 145.1, 139.3, 134.5, 129.7, 129.5, 129.0, 127.8, 127.5, 125.3, 124.9, 120.2, 83.6, 70.8, 70.1, 69.6. HR-MS (ESI): calcd for C25H18FeNCl [M + H]+: 424.0555; found: 424.0561.
4.20 8-Bromo-4-ferrocenyl-2-phenylquinoline (4r)
Following the procedure for 4a, 2-bromoaniline (0.171 g, 1.0 mmol) was used instead of aniline (2 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.3), to give 4r in 86% yield as an orange solid, mp 155–156 °C. 1H NMR (CDCl3, 400 MHz): δ 8.53 (d, J = 8.4 Hz, 1H), 8.34 (d, J = 7.5 Hz, 1H), 8.24 (s, 1H), 8.05 (d, J = 7.2 Hz, 1H), 7.57 (d, J = 7.2 Hz, 1H), 7.54–7.45 (m, 1H), 7.35 (d, J = 7.9 Hz, 1H), 4.80 (s, 2H), 4.53 (s, 2H), 4.21 (s, 5H); 13C NMR (CDCl3, 100 MHz): δ 156.5, 147.7, 145.7, 139.2, 133.0, 129.7, 129.0, 127.7, 127.4, 126.2, 125.7, 120.0, 83.9, 71.1, 70.5, 69.9. HR-MS (ESI): calcd for C25H18FeNBr [M + H]+: 468.0050; found: 468.0100.
4.21 6-Bromo-4-ferrocenyl-2-phenylquinoline (4s)
Following the procedure for 4a, 4-bromoaniline (0.171 g, 1.0 mmol) was used instead of aniline (2 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.4), to give 4s in 83% yield (64%)18a as an orange solid, mp 221–222 °C. 1H NMR (CDCl3, 300 MHz): δ 8.98 (s, 1H), 8.19 (d, J = 6.9 Hz, 2H), 8.08 (s, 2H), 7.80 (d, J = 7.4 Hz, 1H), 7.63–7.48 (m, 3H), 4.81 (s, 2H), 4.57 (s, 2H), 4.25 (s, 5H); 13C NMR (CDCl3, 75 MHz): δ 156.8, 147.5, 146.2, 139.4, 132.6, 132.0, 129.5, 128.9, 128.4, 127.5, 127.0, 120.1, 119.5, 83.1, 70.3, 70.1, 69.7. HR-MS (ESI): calcd for C25H18FeNBr [M]+: 466.9972; found: 466.9993.
4.22 2,4-Diferrocenylquinoline (4t)
Following the procedure for 4a, ferrocenealdehyde (0.214 g, 1.0 mmol) was used instead of benzaldehyde (2.5 h, ethyl acetate/hexane = 1:20, v/v, Rf = 0.3), to give 4t in 83% yield (54%)18b as a red solid, mp 189–190 °C. 1H NMR (CDCl3, 300 MHz): δ 8.47 (d, J = 8.4 Hz, 1H), 8.09 (d, J = 8.4 Hz, 1H), 7.89 (s, 1H), 7.66 (t, J = 7.6 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 5.13 (s, 2H), 4.80 (s, 2H), 4.53–4.51 (m, 4H), 4.26 (s, 5H), 4.13 (s, 5H); 13C NMR (CDCl3, 100 MHz): δ 158.5, 148.9, 145.2, 129.6, 129.0, 125.7, 124.9, 120.1, 84.1, 83.9, 70.6, 70.4, 70.0, 69.7, 69.2, 67.9. HR-MS (ESI): calcd for C29H23Fe2N [M + H]+: 498.0608; found: 498.0556.
4.23 Crystal structure determination
Orange single crystals were obtained by slow evaporation of solutions of 4a and 4k in dichloromethane/hexane = 1:1 (v/v) at room temperature. Selected single crystals of 4a and 4k were mounted on glass fibers, respectively. The intensity data were measured at 100 K on an Agilent SuperNova CCD-based diffractometer (CuKα radiation, λ = 1.54184 Å).23 Empirical absorption corrections were applied using SCALE3 ABSPACK. The structures were solved by direct methods and difference Fourier syntheses, and refined by full-matrix least-squares technique on F2 using SHELXS-97,24 and SHELXL-97.25 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms attached to refined atoms were placed in geometrically idealized positions and refined using a riding model with C–H = 0.93, and 0.96 Å for aromatic, and methyl H, respectively, Uiso(H) = 1.5Ueq(C) for methyl H, and Uiso(H) = 1.2Ueq(C) for all other H atoms. Crystallographic data for 4a and 4k have been deposited with the Cambridge Crystallography Data Centre (CCDC No. 1584004 and 1584006, respectively).
4.24 Hirshfeld surface analysis
The Hirshfeld surfaces and 2D fingerprint plots for 4a and 4k were generated using the program CrystalExplorer 3.1.20 All bond lengths to hydrogen atoms were normalized to the standard neutron value (C–H = 1.083 Å).264.25 Electrochemistry and UV-vis measurements
Cyclic voltammetry (CV) measurements were carried out using a solution of 4 (7.0 × 10−4 M) in CH3CN with 0.1 M n-Bu4NPF6 as supporting electrolyte, Hg/Hg2Cl2 as the reference electrode, platinum working and auxiliary electrodes, and a scan rate at 100 mV s−1.UV-vis measurements were carried out using a solution of 4 (7.0 × 10−4 M or 3.0 × 10−5 M) in anhydrous MeOH.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (grant No. 21372147) and the Undergraduate Innovative Research Training Program of China (grant No. 201710445059).Notes and references
-
(a) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed
; (b) M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 RSC
-
(a) J. Zhu and H. Bienayme, Multicomponent Reactions, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed
-
(a) G. Jones, Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon, Oxford, UK, 1996, vol. 5, pp. 167–245 Search PubMed
-
(a) J. B. Chaires, J. Ren, M. Henary, O. Zegrocka, G. R. Bishop and L. Strekowski, J. Am. Chem. Soc., 2003, 125, 7272–7283 CrossRef CAS PubMed
-
(a) F. O'Donnell, T. J. P. Smyth, V. N. Ramachandran and W. F. Smyth, Int. J. Antimicrob. Agents, 2010, 35, 30–38 CrossRef PubMed
-
(a) M. Foley and L. Tilley, Pharmacol. Ther., 1998, 79, 55–87 CrossRef CAS PubMed
-
(a) V. V. Kouznetsov, C. M. M. Gómez, M. G. Derita, L. Svetaz, E. del Olmo and S. A. Zacchino, Bioorg. Med. Chem., 2012, 20, 6506–6512 CrossRef CAS PubMed
-
(a) A. Mahajan, L. Kremer, S. Louw, Y. Guéradel, K. Chibale and C. Biot, Bioorg. Med. Chem. Lett., 2011, 21, 2866–2868 CrossRef CAS PubMed
-
(a) P. F. Salas, C. Herrmann, J. F. Cawthray, C. Nimphius, A. Kenkel, J. Chen, C. de Kock, P. J. Smith, B. O. Patrick, M. J. Adam and C. Orvig, J. Med. Chem., 2013, 56, 1596–1613 CrossRef CAS PubMed
-
(a) B. Albada and N. Metzler-Nolte, Chem. Rev., 2016, 116, 11797–11839 CrossRef CAS PubMed
-
(a) D. De, F. M. Krogstad, L. D. Byers and D. J. Krogstad, J. Med. Chem., 1998, 41, 4918–4926 CrossRef CAS PubMed
- G. Jaouen, A. Vessières and S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817 RSC
-
(a) E. Alessio, Bioinorganic Medicinal Chemistry, Wiley-VCH, Weinheim, Germany, 1st edn, 2011 Search PubMed
-
(a) R. Suresh, S. Muthusubramanian, R. Senthilkumaran and G. Manickam, J. Org. Chem., 2012, 77, 1468–1476 CrossRef CAS PubMed
- Y. Kuninobu, Y. Inoue and K. Takai, Chem. Lett., 2007, 36, 1422–1423 CrossRef CAS
- K. Cao, F.-M. Zhang, Y.-Q. Tu, X.-T. Zhuo and C.-A. Fan, Chem.–Eur. J., 2009, 15, 6332–6334 CrossRef CAS PubMed
-
(a) X. Zhang, B. Liu, X. Shu, Y. Gao, H. Lv and J. Zhu, J. Org. Chem., 2012, 77, 501–510 CrossRef CAS PubMed
-
(a) S. Chen, L. Li, H. Zhao and B. Li, Tetrahedron, 2013, 69, 6223–6229 CrossRef CAS
-
(a) Y.-B. Xie, S.-P. Ye, W.-F. Chen, Y.-L. Hu, D.-J. Li and L. Wang, Asian J. Org. Chem., 2017, 6, 746–750 CrossRef CAS
-
(a) S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, CrystalExplorer, 2007 Search PubMed
-
(a) M. Zhao, G.-K. Shao, D.-D. Huang, X.-X. Lv and D.-S. Guo, J. Organomet. Chem., 2017, 851, 79–88 CrossRef CAS
-
(a) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed
- Agilent, CrysAlis PRO, Aglient Technologies, Yarnton, Oxfordshire, England, 2012.
- G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, University of Gottingen, Lower Saxony, Germany, 1997 Search PubMed
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University of Gottingen, Lower Saxony, Germany, 1997 Search PubMed
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, 1–19 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1584004 and 1584006. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra01004h |
| This journal is © The Royal Society of Chemistry 2018 |