
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical design and prediction of properties for dinitromethyl, fluorodinitromethyl, and (difluoroamino)dinitromethyl derivatives of triazole and tetrazole†
Teng Fei ,
Yao Du and
Siping Pang*
,
Yao Du and
Siping Pang*
School of Materials Science & Engineering, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: pangsp@bit.edu.cn
First published on 13th March 2018
Abstract
Fluorine- and oxygen-rich compounds are promising as energetic materials for composite propellants, explosives, and pyrotechnics. As an effective and timesaving tool for screening the structures of potential energetic compounds, computer simulation has been widely used to predict the detonation or physicochemical properties of energetic molecules with relatively high precision. In this study, twelve series of dinitromethyl, fluorodinitromethyl, and (difluoroamino)dinitromethyl derivatives of triazole and tetrazole were designed by C- or N-functionalization. Their properties, including density, heat of formation, and detonation properties, were evaluated extensively using volume-based thermodynamic calculations and density functional theory. Among the investigated compounds, 1-(fluorodinitromethyl)-3-nitro-1,2,4-triazole (B3), 1-(fluorodinitromethyl)-4-nitro-1,2,3-triazole (F3), 4,5-bis(fluorodinitromethyl)-1,2,3-triazole (H3), and 5-(fluorodinitromethyl)-tetrazole (I3) displayed excellent integrated performance, that is, high density (≥1.95 g cm−3), oxygen balance (≥2.97%), detonation velocity (>8900 m s−1), and detonation pressure (>40.0 GPa). These results are expected to facilitate the synthesis of a new generation of fluorine- and oxygen-rich energetic compounds. More importantly, our design strategy of constructing nitrogen-rich molecular skeletons with highly dense substituents and highly positive heats of formation by C- or N-functionalization is a valuable approach for developing novel high-energy-density materials with excellent performance.
Introduction
High-energy-density materials (HEDMs) react very rapidly upon initiation to release large amounts of energy, which makes them suitable for a number of civil and industrial applications.1,2 Obvious challenges in the development of HEDMs include the search for materials with higher performance, better safety, and improved environmental compatibility. In general, a useful HEDM exhibits the following properties: (a) high detonation performance; (b) acceptable sensitivity toward external stimuli; (c) thermal stability to ≥150 °C; (d) stability upon storage for long periods of time; and (e) ease and safety of synthesis.3,4 Therefore, novel HEDMs that are nitrogen-rich and have high density, favorable stability, and good environmental compatibility are highly sought after. From the viewpoint of molecular structure, energetic compounds are composed of organic skeletons and energetic groups. As the basis of energetic compounds, organic skeletons are the source of heat and gases, and provide sites for the introduction of energetic groups. The explosion process of energetic compounds can be regard as a complicated oxidation–reduction reaction. In detonation, the energy release manifests itself in the form of high pressures and shock waves, the organic skeletons produce heat and gases by oxidation reaction. Therefore, organic skeleton is the fuel (the reducing agents) in the molecule of energetic compound while the energetic groups are the oxidizers and a good skeleton is of paramount importance for the synthesis of new energetic compounds. The modification of organic skeletons with energetic groups, such as NO2, can further increase the overall nitrogen content, density. The incorporation of NH2 almost always desensitizes but also dramatically increases the heats of formation of high-nitrogen compounds, while N3 groups almost always lowers the thermal stability. According to numerous previous studies, the enhancement or reduction of detonation properties are the result of synergistic effect between organic skeletons and energetic groups.5,6 For the combination of different organic skeletons and energetic groups, a certain degree is beneficial to enhance detonation properties (energy level). However, in some cases, it may get the opposite result. Therefore, it depends on the specific situation.Recently, high density nitrogen-rich compounds have received widespread attention as a new type of energetic material.3,7–9 The molecular skeletons of nitrogen-rich compounds are mainly composed of nitrogen-based heterocyclic rings, such as triazine,10 tetrazine,11 triazole,12,13 tetrazole,14,15 and other heterocycles.16,17 High nitrogen content compounds have a large number of N–N and C–N bonds and therefore exhibit large positive heats of formation (HOF). These compounds often show remarkable insensitivity toward electrostatic discharge, friction, and impact. Nitrogen-rich compounds are quite different from classical explosives molecules (such as TNT, RDX, and HMX). They derive most of their energy from their high positive HOF while the latter gain their energy from oxidation of the carbon backbone. Such transformations are accompanied with an enormous energy release due to the wide difference in the average bond energies of N–N (160 kJ mol−1) and N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (418 kJ mol−1) compared to that of N
N (418 kJ mol−1) compared to that of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N (954 kJ mol−1). Based on the structural characteristics of nitrogen-rich compounds, there are two aspects to consider during modification. First, an organic molecule with a high positive HOF and nitrogen content should be chosen as a skeleton, for example, 1,2,4-triazole, 1,2,3-triazole, and tetrazole (109, 272, and 237 kJ mol−1, respectively). Second, dense energetic substituents, such as multi-nitro functional groups, should be introduced at a limited number of modification positions. In the process of seeking more powerful and energetic functional groups, dinitromethyl [–CH(NO2)2] is of particular interest because this group has relatively high density, satisfactory oxygen content, and promising detonation properties.18 Introducing the –CH(NO2)2 group into heterocyclic compounds has been demonstrated as an efficient way of constructing HEDMs. In addition to the nitro groups supplying additional oxygen atoms and energy, the –CH(NO2)2 group has a greater stabilizing influence than the –C(NO2)3 group owing to hydrogen-bond interactions.19 Furthermore, it has been found that partially fluorinated nitro groups exhibit potential for applications in the field of energetic materials.19–23 Molecules in which the fluorodinitromethyl moiety [–CF(NO2)2] is attached via a carbon atom to an aliphatic, aromatic, or heterocyclic skeleton have been revealed to have useful energetic properties and stabilities.22,24–26 Notably, the incorporation of the (difluoroamino)dinitromethyl moiety [–C(NF2)(NO2)2] is another effective tool for constructing energetic compounds, as it provides a high energy content while maintaining a high density and acceptable thermal and processing properties.27 In view of these findings, it is observed that the multi-nitro functional groups have a great development from simple to complex (Scheme 1). In recent years, although the synthesis of some CH(NO2)2-, CF(NO2)2-, and C(NF2)(NO2)2-based compounds has been reported, the number of compounds is not sufficient to determine general rules and characteristics. Moreover, as less attention has been paid to compounds bearing a –CF(NO2)2 or –C(NF2)(NO2)2 moiety at a mono-heterocyclic nitrogen atom, the design or synthesis of new energetic compounds based on triazole or tetrazole skeletons with –CF(NO2)2 or –C(NF2)(NO2)2 groups is rare. In addition, there is a lack of comprehensive understanding of the structure–property relationships for –CH(NO2)2, –CF(NO2)2, and –C(NF2)(NO2)2 derivatives.
N (954 kJ mol−1). Based on the structural characteristics of nitrogen-rich compounds, there are two aspects to consider during modification. First, an organic molecule with a high positive HOF and nitrogen content should be chosen as a skeleton, for example, 1,2,4-triazole, 1,2,3-triazole, and tetrazole (109, 272, and 237 kJ mol−1, respectively). Second, dense energetic substituents, such as multi-nitro functional groups, should be introduced at a limited number of modification positions. In the process of seeking more powerful and energetic functional groups, dinitromethyl [–CH(NO2)2] is of particular interest because this group has relatively high density, satisfactory oxygen content, and promising detonation properties.18 Introducing the –CH(NO2)2 group into heterocyclic compounds has been demonstrated as an efficient way of constructing HEDMs. In addition to the nitro groups supplying additional oxygen atoms and energy, the –CH(NO2)2 group has a greater stabilizing influence than the –C(NO2)3 group owing to hydrogen-bond interactions.19 Furthermore, it has been found that partially fluorinated nitro groups exhibit potential for applications in the field of energetic materials.19–23 Molecules in which the fluorodinitromethyl moiety [–CF(NO2)2] is attached via a carbon atom to an aliphatic, aromatic, or heterocyclic skeleton have been revealed to have useful energetic properties and stabilities.22,24–26 Notably, the incorporation of the (difluoroamino)dinitromethyl moiety [–C(NF2)(NO2)2] is another effective tool for constructing energetic compounds, as it provides a high energy content while maintaining a high density and acceptable thermal and processing properties.27 In view of these findings, it is observed that the multi-nitro functional groups have a great development from simple to complex (Scheme 1). In recent years, although the synthesis of some CH(NO2)2-, CF(NO2)2-, and C(NF2)(NO2)2-based compounds has been reported, the number of compounds is not sufficient to determine general rules and characteristics. Moreover, as less attention has been paid to compounds bearing a –CF(NO2)2 or –C(NF2)(NO2)2 moiety at a mono-heterocyclic nitrogen atom, the design or synthesis of new energetic compounds based on triazole or tetrazole skeletons with –CF(NO2)2 or –C(NF2)(NO2)2 groups is rare. In addition, there is a lack of comprehensive understanding of the structure–property relationships for –CH(NO2)2, –CF(NO2)2, and –C(NF2)(NO2)2 derivatives.
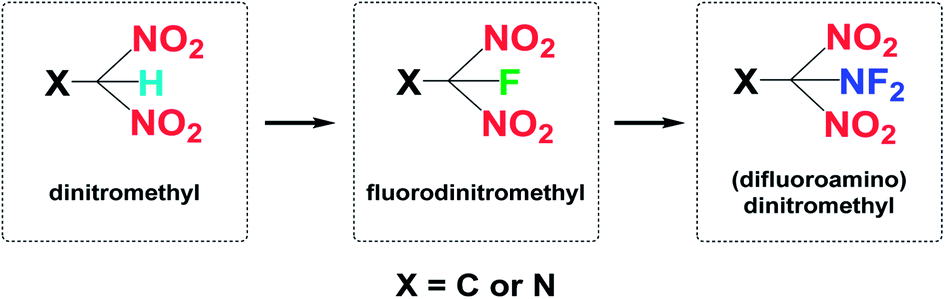 | ||
| Scheme 1 The development history of multi-nitro functional groups. | ||
From a synthetic perspective, compounds bearing –CF(NO2)2 moieties are typically prepared from –C(NO2)2 salts by electrophilic fluorination. Generally, powerful gaseous reagents, such as FClO3 (ref. 27) and N2/F2,28 or solid reagents, such as XeF2,24 (NH4)SO4F,29 RIF2,30 2-fluoro-2,2-dinitroethanol,31 and Selectfluor®,32 have been used. Notably, the discovery of F2NOSO2 (ref. 27 and 33) as an effective reagent for carrying out difluoroamination34 of nitroalkane anions was a key factor in advancing the emerging field of synthetic fluorine chemistry. Most fluorinating agents are highly toxic with high vapor pressures, which can cause irreparable harm to experimenters. Therefore, it is necessary to predict their performance in advance. Fortunately, modern theoretical studies based on quantum chemical treatments have gained acceptance as a useful research tool for screening HEDM candidates, thereby avoiding expensive and dangerous experimental tests. Such studies can provide an understanding of the relationships between molecular structures and properties, which in turn can be used to design better and more efficient laboratory tests.35–38 It is worth reminding that the expected high toxicity of fluoro nitromethyl derivatives the results are not really relevant to the energetic properties. Therefore, it is of great significance to investigate the energetic properties of fluoro nitromethyl derivatives.
Based on the above considerations, in this work, twelve series of triazole or tetrazole derivatives were designed by replacing hydrogen atoms on triazole or tetrazole with one or two –CH(NO2)2, –CF(NO2)2, or –C(NF2)(NO2)2 group via C- or N-functionalization (Scheme 2). Density functional theory and volume-based thermodynamics calculations were employed to predict and evaluate the properties of these compounds, including density (ρ), HOF, stability, and detonation properties. The structure–activity relationships between the organic skeletons and the energetic functional groups were also studied systematically. It is expected that our results will provide useful information for the molecular design of novel HEDMs.
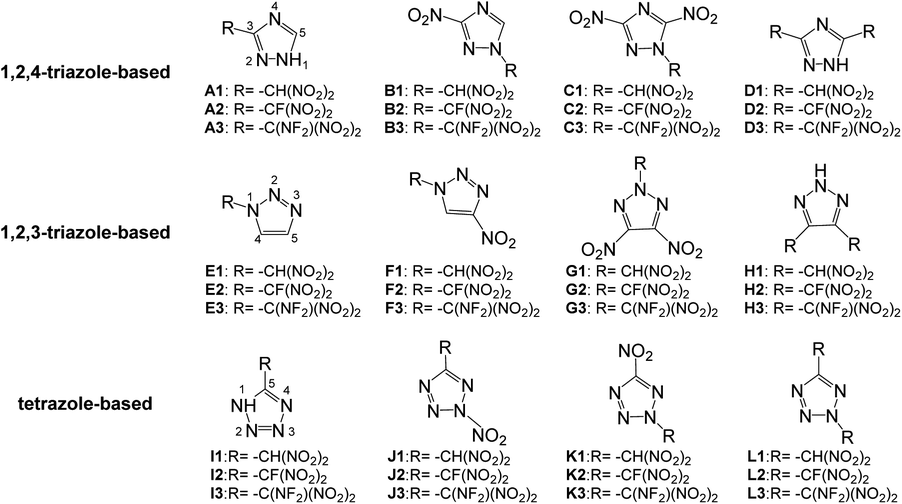 | ||
| Scheme 2 Molecules of CH(NO2)2-, CF(NO2)2- and C(NF2)(NO2)2-modified derivatives of triazole and tetrazole. | ||
Computational methods
The calculations were performed with the Gaussian 09 package.39 DFT-B3LYP methods with the 6-311G++(d,p) basis set were adopted to optimize the molecular structures and predict the HOFs. Previous studies have shown that the 6-311G++(d,p) basis set is able to precisely predict the molecular structures and energies of energetic organic compounds.40 Normal-mode analysis for each structure gave true local energy minima on the potential energy surfaces with no imaginary frequencies. Numerous studies have shown that the HOFs theoretically predicted via isodesmic reactions are in good agreement with the experimental values for many organic systems.41–43 Isodesmic reaction processes, in which the number of each kind of formal bond is invariable for the reactants and products, are designed to decrease the calculation errors for HOFs.44,45For an isodesmic reaction, the heat of reaction ΔH298 K at 298 K can be calculated using the following equation:
| ΔH298 K = ∑ΔHf,p − ∑ΔHf,R |
| ΔH298 K = ΔH298 K + Δ(PV) = ΔE0 + ΔZPE + ΔHT + ΔnRT |
| ΔHf,solid = ΔHf,gas − ΔHsub |
According to Politzer et al.,47,49 the heat of sublimation can be computed from the corresponding molecular surface area and electrostatic interaction index (vσtot2) for energetic compounds. The empirical expression for this approach is:
| ΔHsub = aA2 + b(vσtot2)0.5 + c |
For each –CH(NO2)2, –CF(NO2)2, and –C(NF2)(NO2)2 derivative of triazole and tetrazole, the theoretical density was initially determined from the molecular weight (M) divided by V0.001, where V0.001 is defined as the volume inside the 0.001 electrons bohr−3 density contour, which was obtained by Monte Carlo integration using the Multiwfn program. By introducing the interaction index vσtot2, the density of an energetic compound can be corrected according to the following equation:54
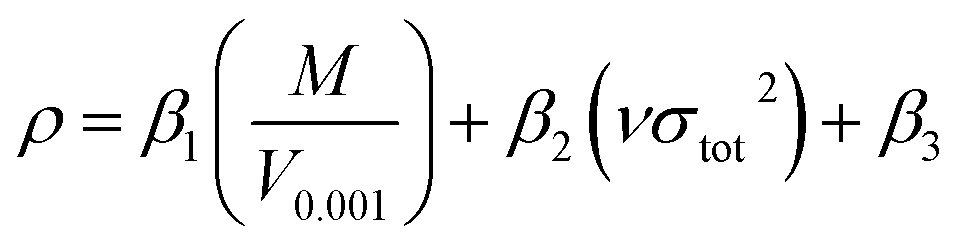
The bond dissociation energy (BDE) can provide useful information for understanding the stability of a molecule. Generally, the smaller the energy for breaking a bond is, the weaker the bond is, making it easier for the bond to act as a trigger bond. For many organic molecules, the terms “bond dissociation energy” (BDE) and “bond dissociation enthalpy” often appear interchangeably in the literature.56
At 0 K, the homolytic BDE is given as:
| BDE0(A − B) → E0(A˙) + E0(B˙) − E0(A − B) |
The BDE with a ZPE correction can be calculated using the following equation:
| BDE(A − B)ZPE = BDE0(A − B) + ΔEZPE |
Impact sensitivity (h50) is often used to judge whether high energy density compounds are sensitive or insensitive to external impact. A series of empirical methods have been proposed to predict the sensitivity of these materials approximately. Specifically, Politzer and coworkers developed a method using crystal volume factors to predict impact sensitivities of high energy density compounds which give acceptable accuracy.57 The impact sensitivity (h50) can be predicted by the equation:
| h50 = ασ+2 + βv + γ |
Energetic properties, such as the detonation pressure (P) and detonation velocity (D), were calculated from the HOF and the calculated ρ using the EXPLO5 v6.01 program.58
The optimizations were performed without any symmetry restrictions using the default convergence criteria in the program. All of the optimized structures were characterized as true local energy minima on the potential energy surface without imaginary frequencies.
Results and discussion
Structural design and molecular geometry
This study was aimed at the molecular design and prediction of properties for novel HEDMs. Based on previous experiments and calculations, the ρ and oxygen balance (OB) of 1,2,4-triazole, 1,2,3-triazole, and tetrazole are not satisfactory (1.15, 1.19, and 1.48 g cm−3; and −1.27%, −1.27%, and −0.68%, respectively). Introducing –CH(NO2)2, –CF(NO2)2, or –C(NF2)(NO2)2 groups into the molecular skeletons has been demonstrated as an efficient way to construct HEDMs without incurring an excessive performance penalty, as nitro groups not only supply additional oxygen atoms, but also have a stabilizing influence. Therefore, starting with 1,2,4-triazole, 1,2,3-triazole, and tetrazole as initial skeletons, the structural design of a series of new fluorine- and oxygen-rich molecules with monocyclic structures was investigated. In the first design strategy, a hydrogen atom in 1,2,4-triazole, 1,2,3-triazole, or tetrazole was replaced with one –CH(NO2)2, –CF(NO2)2, or –C(NF2)(NO2)2 group to form new energetic molecules (N-functionalization: A1–A3 and E1–E3, C-functionalization: I1–I3). In the second phase, a nitro group was introduced with the goal of ensuring that the resultant new molecules had high energy densities (C-functionalization: B1–B3 and F1–F3, N-functionalization: J1–J3). In the third phase, to further enhance the energy level, two nitro groups were introduced by C-functionalization, while a –CH(NO2)2, –CF(NO2)2, or –C(NF2)(NO2)2 group was introduced by N-functionalization (C1–C3 and G1–G3). Considering that the tetrazole molecule has only two sites that can be modified, we reversed the positions of the nitro group and the –CH(NO2)2, –CF(NO2)2 or –C(NF2)(NO2)2 group to examine the influence of different positions (K1–K3). In the final stage, two –CH(NO2)2, –CF(NO2)2, or –C(NF2)(NO2)2 groups were introduced by C-functionalization to form new disubstituted molecules without nitro groups (D1–D3 and H1–H3). In series L, we investigated the influence of simultaneously introducing the same substituents by C- and N-functionalization.The geometric structures of all compounds were optimized based on the true local energy minima on the potential energy surface without imaginary frequencies. Analyzing the molecular orbitals of a molecule provides valuable information on its electronic structure. Therefore, the molecular electrostatic potential (ESP), highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies, and the energy gap (ΔELUMO–HOMO) were obtained for each compound. To help understand the stereo structures of the –CH(NO2)2, –CF(NO2)2, and –C(NF2)(NO2)2 derivatives of triazole and tetrazole, the optimized structures of A1–L3 are shown in Fig. 1. The surface analyses of A1–L3 are summarized in the ESI.†
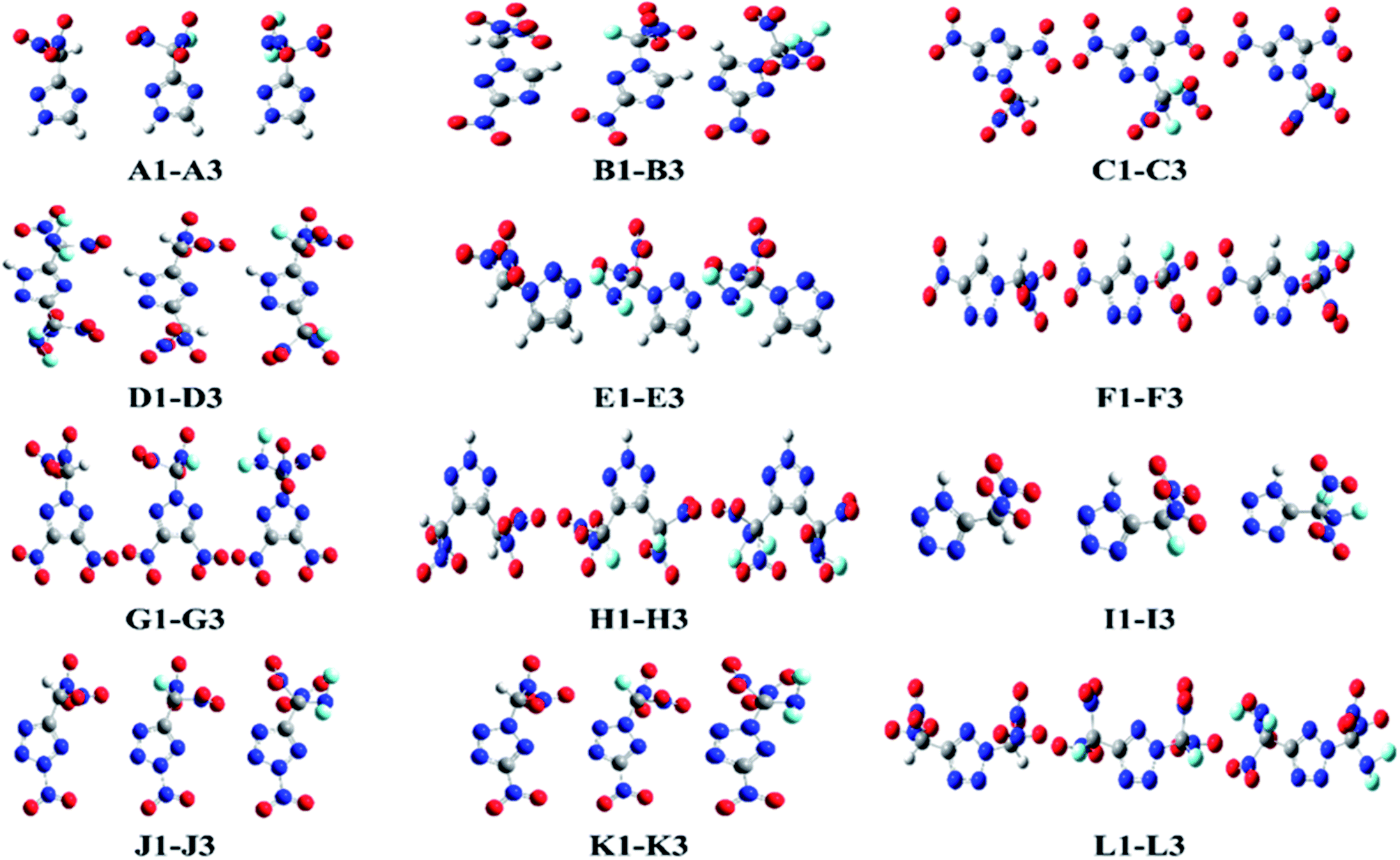 | ||
| Fig. 1 The optimized structures of the title compounds. | ||
The ESP on the molecular surface not only provides meaningful insight into charge distribution and intermolecular interactions, but also has an important relationship with impact sensitivity. Positive regions and higher intensities of electropositive potential surfaces are related to higher impact sensitivities.59 A quantitative molecular surface analysis was performed using the Multiwfn program,60 and the extreme value points of the ESP on the molecular surface are plotted in Fig. 2. The surface local minima and maxima are shown as blue and red points, respectively, and only the global minima and maxima are labeled. Overall, the surface maxima are present near hydrogen atoms on the triazole or tetrazole ring, whereas the surface minima are present near –NO2, –CH(NO2)2, –CF(NO2)2, or –C(NF2)(NO2)2 groups, which may be the primary electrophilic sites. Clearly, all these compounds have relatively average negative potentials and positive potentials, which might result in better stability. Table 1 lists the HOMO, LUMO, and ΔELUMO–HOMO for the –CH(NO2)2, –CF(NO2)2, and –C(NF2)(NO2)2 derivatives of triazole and tetrazole. ΔELUMO–HOMO is an important parameter that can be used to evaluate the reactivity in chemical or photochemical processes that involve electron transfer or leap. It can be seen clearly that the replacement of a hydrogen atom with a –F or –NF2 group leads to a slight decrease in the HOMO relative to that observed for the corresponding CH(NO2)2-substituted compound. A similar trend is observed for the LUMO energy levels of these derivatives. For series B, F, and I, the CF(NO2)2-substituted derivatives have a lower ΔELUMO–HOMO values than the CH(NO2)2-substituted derivatives. In contrast, in the other series, the CF(NO2)2-substituted derivatives have higher ΔELUMO–HOMO values than the CH(NO2)2-substituted derivatives. It is interesting to note that the C(NF2)(NO2)2-substituted derivatives of series C, H, and I have higher ΔELUMO–HOMO values than the CH(NO2)2-substituted derivatives, whereas the other series show the opposite trend. A2, C2, C3, E2, B2, H3, I3, K2, and L2 have higher energy gaps than CH(NO2)2-substituted 1,2,4-triazole, 1,2,3-triazole, or tetrazole, indicating a shift toward higher frequencies in their electronic absorption spectra. However, the other CF(NO2)2- or C(NF2)(NO2)2-substituted derivatives have lower energy gaps than the corresponding CH(NO2)2-substituted derivatives, reflecting a shift toward lower frequencies in their electronic absorption spectra. Among these derivatives, A2 has the highest ΔELUMO–HOMO value, whereas B3 has the smallest. Overall, different substituted molecules present a comparison of the energetics.61
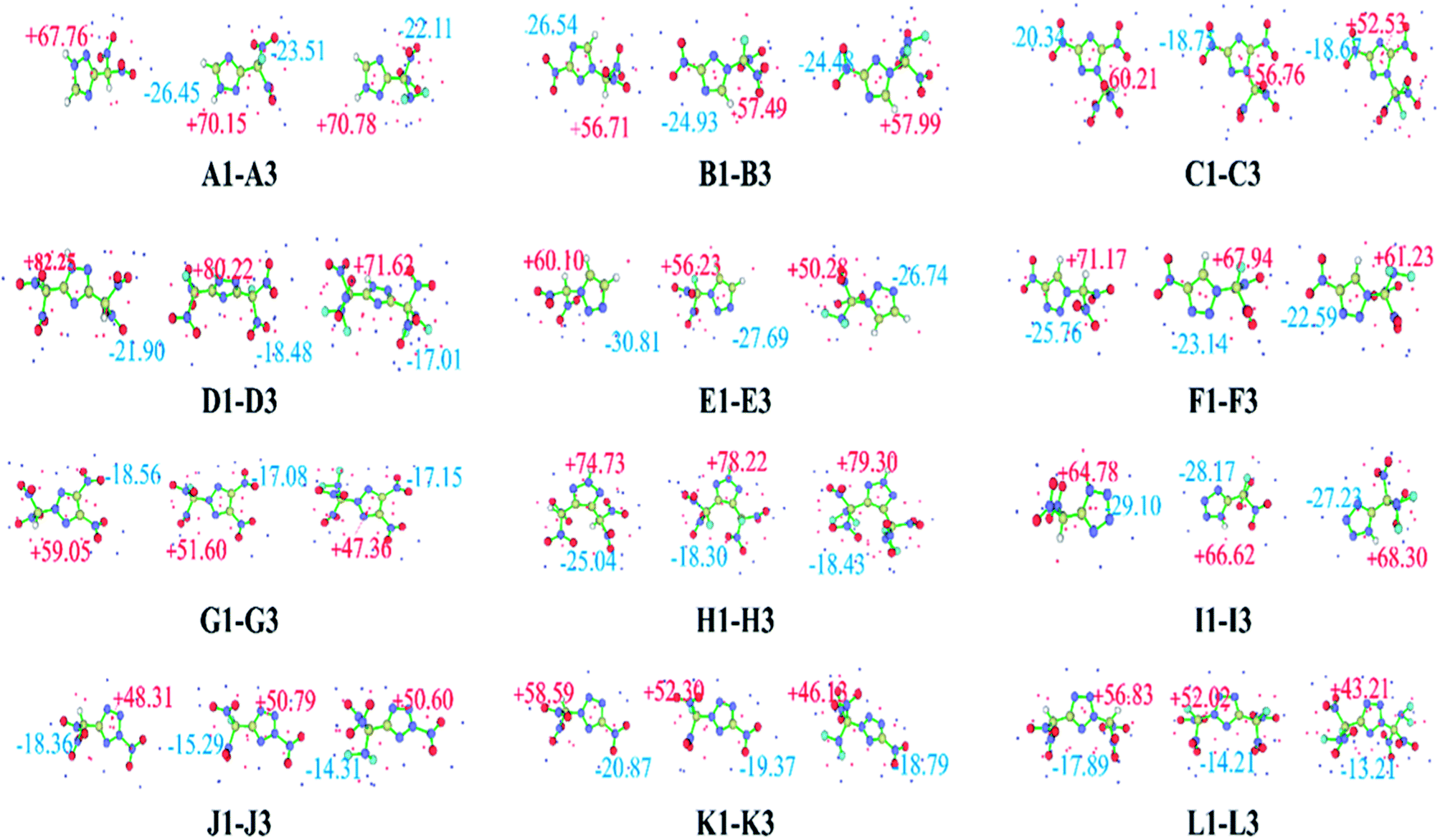 | ||
| Fig. 2 Electrostatic potentials mapped of the title compounds. | ||
| Comp. | EHOMO | ELUMO | ΔELUMO–HOMO | Comp. | EHOMO | ELUMO | ΔELUMO–HOMO |
|---|---|---|---|---|---|---|---|
| A1 | −0.31631 | −0.11205 | 0.20426 | G1 | −0.34676 | −0.15240 | 0.19436 |
| A2 | −0.32377 | −0.11716 | 0.20661 | G2 | −0.35104 | −0.15806 | 0.19298 |
| A3 | −0.32373 | −0.12066 | 0.20307 | G3 | −0.35035 | −0.16228 | 0.18807 |
| B1 | −0.33251 | −0.15590 | 0.17661 | H1 | −0.33365 | −0.14557 | 0.18808 |
| B2 | −0.33467 | −0.16281 | 0.17186 | H2 | −0.34661 | −0.14360 | 0.20301 |
| B3 | −0.33646 | −0.16686 | 0.16960 | H3 | −0.34125 | −0.14937 | 0.19188 |
| C1 | −0.34916 | −0.17998 | 0.16918 | I1 | −0.33677 | −0.15648 | 0.18029 |
| C2 | −0.35313 | −0.17914 | 0.17399 | I2 | −0.33991 | −0.16013 | 0.17978 |
| C3 | −0.35288 | −0.17530 | 0.17758 | I3 | −0.34370 | −0.15985 | 0.18385 |
| D1 | −0.33529 | −0.14032 | 0.19497 | J1 | −0.35144 | −0.15880 | 0.19264 |
| D2 | −0.34538 | −0.14679 | 0.19859 | J2 | −0.35855 | −0.16214 | 0.19641 |
| D3 | −0.34575 | −0.16406 | 0.18169 | J3 | −0.35318 | −0.16358 | 0.18960 |
| E1 | −0.31282 | −0.12939 | 0.18343 | K1 | −0.34888 | −0.15245 | 0.19643 |
| E2 | −0.31957 | −0.13471 | 0.18486 | K2 | −0.35294 | −0.15591 | 0.19703 |
| E3 | −0.32029 | −0.13702 | 0.18327 | K3 | −0.35432 | −0.15953 | 0.19479 |
| F1 | −0.33928 | −0.14619 | 0.19309 | L1 | −0.35167 | −0.14766 | 0.20401 |
| F2 | −0.34330 | −0.15161 | 0.19169 | L2 | −0.36179 | −0.15595 | 0.20584 |
| F3 | −0.34371 | −0.15386 | 0.18985 | L3 | −0.35750 | −0.16099 | 0.19651 |
Stability
BDEs can provide useful information for understanding the stability of a molecule.56 Generally, a smaller BDE corresponds to a weaker bond, which makes it easier for the bond to function as a trigger bond. To investigate the thermal stabilities of the compounds of interest, the C–NO2 bond in the –CH(NO2)2, –CF(NO2)2, or –C(NF2)(NO2)2 functional groups were considered to be the weakest bond. The BDEs of the C–NO2 bonds are listed in Table 2.| Comp. | BDE of C–NO2 | Comp. | BDE of C–NO2 | Comp. | BDE of C–NO2 |
|---|---|---|---|---|---|
| a C–NO2 bond in the –CF(NO2)2 linked in position 5.b C–NO2 bond in the CF(NO2)2 linked in position 5.c C–NO2 bond in the C(NF2)(NO2)2 linked in position 5.d C–NO2 bond in the dinitromethyl by C,N-linked.e C–NO2 bond in the CF(NO2)2 by C,N-linked.f C–NO2 bond in the C(NF2)(NO2)2 by C,N-linked. | |||||
| A1 | 256.84 | E1 | 194.21 | J1 | 312.18 |
| A2 | 262.98 | E2 | 232.95 | J2 | 317.57 |
| A3 | 251.92 | E3 | 207.42 | J3 | 313.55 |
| B1 | 229.76 | F1 | 194.44 | K1 | 207.27 |
| B2 | 252.61 | F2 | 227.17 | K2 | 224.41 |
| B3 | 216.87 | F3 | 210.04 | K3 | 222.36 |
| C1 | 204.24 | G1 | 219.90 | L1Nd | 267.84 |
| C2 | 215.50 | G2 | 228.61 | L2Ne | 278.33 |
| C3 | 234.49 | G3 | 221.25 | L3Nf | 250.21 |
| D1 | 269.32 | H1 | 290.10 | L1C | 312.17 |
| D2 | 286.54 | H2 | 295.95 | L2C | 323.73 |
| D3 | 256.46 | H3 | 288.33 | L3C | 304.91 |
| D1Ha | 231.53 | I1 | 281.72 | ||
| D2Hb | 248.76 | I2 | 286.51 | ||
| D3Hc | 224.65 | I3 | 295.42 | ||
As is evident in Table 2, in the same compound series, the BDE of the C–NO2 bonds in –CF(NO2)2 is higher than that in –CH(NO2)2. Therefore, it can be deduced that substitution of the –CF(NO2)2 group is very useful for increasing the thermal stabilities of 1,2,4-triazole, 1,2,3-triazole, and tetrazole. The –C(NF2)(NO2)2 group is also observed to increase the thermal stability in some series, namely, E, F, and I. Notably, the C–NO2 bonds in functional groups with C-linkages have higher BDEs than those in the corresponding groups with N-linkages (e.g., J1 vs. K1, J2 vs. K2, and J3 vs. K3). Moreover, in series L, which contain both C- and N-linked functional groups, the C–NO2 bonds in functional groups with C-linkages have higher BDEs than those in groups with N-linkages (BDEL1C > BDEL1N, BDEL2C > BDEL2N, and BDEL2C > BDEL2N). Overall, all of the examined compounds exhibit good thermal stabilities, as they have relatively high BDE values for the weakest bond and meet the requirement that HEDM candidates should have a dissociation barrier larger than 80–120 kJ mol−1. Based on these results, it is necessary to study other physical and chemical properties of these compounds.
Density
Density (ρ) is a very important physical property of any energetic material. In general, a high density causes better performance.22 For example, the D of an explosive is proportional to its ρ, and the P is proportional to the square of its ρ. For an energetic material, a high ρ means that more energy per unit volume can be packed into a volume-limited space, thereby yielding the maximum violent explosion. Here, the densities of the molecules were defined as the molecular weight divided by the average molecular volume and then corrected using the interaction index vσtot2. The molecular volumes, uncorrected densities, and corrected densities are listed in Table 3.| Comp. | Mw (g mol−1) | Volume (cm3 mol−1) | ρuncorrected (g cm−3) | ρcorrected (g cm−3) | Comp. | Mw (g mol−1) | Volume (cm3 mol−1) | ρuncorrected (g cm−3) | ρcorrected (g cm−3) |
|---|---|---|---|---|---|---|---|---|---|
| A1 | 173.09 | 103.61 | 1.67 | 1.66 | G1 | 263.08 | 141.38 | 1.86 | 1.83 |
| A2 | 191.08 | 106.36 | 1.80 | 1.78 | G2 | 281.07 | 144.27 | 1.95 | 1.92 |
| A3 | 224.08 | 119.16 | 1.88 | 1.87 | G3 | 314.08 | 157.22 | 2.00 | 1.97 |
| B1 | 218.09 | 122.16 | 1.79 | 1.81 | H1 | 277.11 | 153.51 | 1.81 | 1.80 |
| B2 | 236.08 | 124.91 | 1.89 | 1.91 | H2 | 313.09 | 159.41 | 1.96 | 1.93 |
| B3 | 269.08 | 137.88 | 1.95 | 1.97 | H3 | 379.10 | 183.45 | 2.07 | 2.04 |
| C1 | 263.08 | 140.21 | 1.88 | 1.86 | I1 | 174.08 | 100.03 | 1.74 | 1.75 |
| C2 | 281.07 | 142.49 | 1.97 | 1.95 | I2 | 192.07 | 102.62 | 1.87 | 1.89 |
| C3 | 314.08 | 155.07 | 2.03 | 2.02 | I3 | 225.07 | 115.79 | 1.94 | 1.95 |
| D1 | 277.11 | 155.23 | 1.79 | 1.76 | J1 | 219.07 | 119.74 | 1.83 | 1.80 |
| D2 | 313.09 | 160.85 | 1.95 | 1.92 | J2 | 237.06 | 122.61 | 1.93 | 1.89 |
| D3 | 379.10 | 186.11 | 2.04 | 2.01 | J3 | 270.07 | 135.42 | 1.99 | 1.96 |
| E1 | 173.09 | 104.18 | 1.66 | 1.66 | K1 | 219.07 | 119.46 | 1.83 | 1.82 |
| E2 | 191.08 | 106.93 | 1.79 | 1.77 | K2 | 237.06 | 122.37 | 1.94 | 1.92 |
| E3 | 224.08 | 119.87 | 1.87 | 1.85 | K3 | 270.07 | 135.26 | 2.00 | 1.98 |
| F1 | 218.09 | 122.69 | 1.78 | 1.78 | L1 | 278.10 | 152.34 | 1.83 | 1.83 |
| F2 | 236.08 | 125.55 | 1.88 | 1.88 | L2 | 314.08 | 158.03 | 1.99 | 1.94 |
| F3 | 269.08 | 138.50 | 1.94 | 1.95 | L3 | 380.09 | 183.76 | 2.07 | 2.02 |
The ρ for the designed compounds were found to be in the range of 1.66 (A1 and E1) to 2.04 g cm−3 (H3). Clearly, replacement of a hydrogen atom with a –F or –NF2 group leads to a greater increase in density than that observed for the corresponding CH(NO2)2-substituted compounds. For example, compound C3, which contains a –C(NF2)(NO2)2 group, has a much higher density (2.01 g cm−3) than compounds C2 (1.95 g cm−3) and C1 (1.86 g cm−3). Within each 1,2,4-triazole, 1,2,3-triazole, and tetrazole series, the densities of the compounds decrease as a function of the substituent, according to C(NF2)(NO2)2 > CF(NO2)2 > CH(NO2)2. This observation indicates that the introduction of a highly dense –F or –NF2 group is a very effective method for increasing the density of energetic molecules. For the same skeleton (e.g., A1 vs. B1 vs. C1, A2 vs. B2 vs. C2, and A3 vs. B3 vs. C3), more nitro groups results in higher densities. For example, compound C1, which contains four nitro groups, exhibits a much higher density (2.01 g cm−3) than compounds B1 (1.81 g cm−3) and A1 (1.66 g cm−3), which contain three and two nitro groups, respectively. However, for similar skeletons that contain the same number of nitro groups (e.g., C1 vs. D1, C2 vs. D2, and C3 vs. D3), the introduction of two –CH(NO2)2, –CF(NO2)2, or –C(NF2)(NO2)2 groups via C-functionalization leads to a slight decrease in the density of the latter compounds. For example, C1 has a density of 1.86 g cm−3, whereas the density of D1 is only 1.76 g cm−3. In addition, for tetrazole-based compounds, the simultaneous introduction of two –CH(NO2)2, –CF(NO2)2, or –C(NF2)(NO2)2 via C- and N-functionalization leads to a slight increase in density relative to that of compounds with only C- or N-functionalization (L1 vs. J1 or K1, L2 vs. J2 or K2, and L3 vs. J3 or K3). For example, compound L3, which contains two –C(NF2)(NO2)2 groups via simultaneous C- and N-functionalization, has a higher density (2.02 g cm−3) than compounds J3 (1.96 g cm−3; C-functionalization) and K3 (1.98 g cm−3; N-functionalization).
This finding illustrates that the incorporation of highly dense groups into high-density skeletons via C- or N-functionalization is an effective strategy for obtaining higher density compounds. Overall, compounds C3 (2.01 g cm−3), D3 (2.01 g cm−3), H3 (2.04 g cm−3), and L3 (2.02 g cm−3) exhibited the highest calculated densities of >2.0 g cm−3, indicating that these compounds have high potential as new HEDMs.
Heat of formation
The solid-phase HOF (ΔHf,solid) is an important property for predicting the detonation properties of energetic materials. Therefore, to estimate the detonation velocity and pressure of an explosive molecule through theoretical calculations, the ΔHf,solid value must be obtained. Here, we investigated the effects of different substituents on the gas-phase HOF (ΔHf,gas) and ΔHf,solid of 1,2,4-triazole, 1,2,3-triazole, and tetrazole derivatives. To obtain more accurate calculation results, the basic ring skeleton of the parent compound was kept invariable during the designed isodesmic reactions. The isodesmic reactions used to obtain ΔHf,gas for the investigated compounds are as follows (Scheme 3): | ||
| Scheme 3 Designed isodesmic reactions for the title compounds. | ||
Table 4 lists the total energies (E0), ZPEs, thermal corrections (HT), and ΔHf,gas for the reference compounds in the isodesmic reactions. The ΔHf,gas value of each compound was calculated from the atomization reaction at the G2 level. Table 5 summarizes the calculated ΔHf,solid values and the parameters related to the ESPs of all the designed compounds, including E0, ZPE, HT, ΔHf,gas, molecular surface area (A), degree of balance between negative and positive potentials (ν), square of the variability in the ESP (σtot2), heat of sublimation (ΔHsub), and ΔHf,gas.
| Comp. | E0 | ZPE | HT | ΔHf,gas | Comp. | E0 | ZPE | HT | ΔHf,gas |
|---|---|---|---|---|---|---|---|---|---|
| a DA: 1,2,4-triazole.b DB: 3-nitro-1,2,4-triazole.c DC: 3,5-dinitro-1,2,4-triazole.d DD = DA.e DE: 1H-1,2,3-triazole.f DF: 4-nitro-1,2,3-triazole.g DG: 4,5-dinitro-1,2,3-triazole.h DH: 2H-1,2,3-triazole.i DI: 1H-tetrazole.j DJ: 2-nitro-tetrazole.k DK: 5-nitro-2H-tetrazole.l DL: 2H-tetrazole.m GN1: 1,1-dinitroethane.n GN2: 1-fluoro-1,1-dinitroethane.o GN3: N,N-difluoro-1,1-dinitroethan-1-amine.p GN4: dinitromethanamine.q GN5: fluorodinitromethanamine.r GN6: N,N-difluoro-1,1-dinitromethanediamine.s Experimental values taken from ref. 13 and 62. | |||||||||
| CH4 | −40.3796 | 0.0448 | 10.03 | −74.60s | DIi | −257.6536 | 0.0467 | 11.65 | 335.14 |
| NH3 | −56.4155 | 0.0344 | 10.00 | −45.94s | DJj | −461.7117 | 0.0480 | 18.28 | 436.03 |
| DAa | −241.6631 | 0.0596 | 11.84 | 194.03 | DKk | −461.7459 | 0.0496 | 17.86 | 349.68 |
| DBb | −445.7561 | 0.0620 | 18.17 | 200.18 | DLl | −257.6604 | 0.0474 | 11.47 | 327.24 |
| DCc | −649.8453 | 0.0640 | 24.87 | 217.48 | GN1m | −487.7724 | 0.0804 | 20.70 | −115.69 |
| DDd | −241.6631 | 0.0596 | 11.84 | 194.03 | GN2n | −586.8482 | 0.0718 | 24.94 | −313.80 |
| DEe | −241.6365 | 0.0587 | 11.96 | 267.61 | GN3o | −740.9992 | 0.0786 | 30.07 | −118.55 |
| DFf | −445.7307 | 0.0613 | 18.14 | 263.40 | GN4p | −503.8041 | 0.0694 | 22.91 | −73.50 |
| DGg | −649.8217 | 0.0640 | 22.91 | 281.94 | GN5q | −602.8809 | 0.0607 | 24.67 | −278.57 |
| DHh | −241.6448 | 0.0596 | 11.82 | 251.79 | GN6r | −757.0300 | 0.0673 | 29.92 | −83.80 |
| Comp. | E0 | ZPE | HT | A | ν | σtot2 | ΔHf,gas | ΔHsub | ΔHf,solid |
|---|---|---|---|---|---|---|---|---|---|
| A1 | −689.06 | 243.34 | 28.58 | 177.07 | 0.11 | 317.26 | 141.20 | 88.89 | 52.31 |
| A2 | −788.13 | 220.11 | 30.79 | 179.67 | 0.08 | 372.88 | −42.91 | 86.48 | −129.39 |
| A3 | −942.28 | 237.13 | 36.16 | 193.45 | 0.08 | 396.33 | 145.87 | 92.13 | 53.74 |
| B1 | −893.13 | 246.41 | 35.53 | 205.75 | 0.18 | 262.03 | 203.85 | 107.37 | 96.48 |
| B2 | −992.2 | 223.14 | 37.72 | 208.41 | 0.20 | 221.32 | 27.42 | 107.37 | −79.95 |
| B3 | −1146.35 | 239.91 | 43.18 | 222.73 | 0.20 | 199.53 | 215.81 | 111.85 | 103.96 |
| C1 | −1097.22 | 251.37 | 42.4 | 231.44 | 0.09 | 268.52 | 233.29 | 106.39 | 126.91 |
| C2 | −1196.28 | 227.9 | 44.42 | 231.21 | 0.09 | 216.6 | 233.29 | 103.32 | −51.27 |
| C3 | −1350.42 | 243.77 | 50.26 | 243.59 | 0.11 | 189.65 | 52.05 | 110.7 | 158.57 |
| D1 | −1136.45 | 328.11 | 46.37 | 254.12 | 0.05 | 483.02 | 115.08 | 118.99 | −3.91 |
| D2 | −1334.59 | 281.64 | 50.97 | 260.08 | 0.05 | 355.62 | −253.38 | 117.15 | −370.53 |
| D3 | −1642.9 | 316.55 | 61.34 | 286.26 | 0.08 | 218.84 | 115.31 | 133.39 | −18.08 |
| E1 | −689.01 | 238.7 | 28.64 | 177.48 | 0.12 | 309.62 | 273.99 | 90.03 | 183.96 |
| E2 | −788.08 | 215.28 | 30.95 | 179.9 | 0.14 | 202.68 | 81.29 | 85.18 | −3.89 |
| E3 | −942.23 | 232.06 | 36.48 | 193.38 | 0.13 | 193.81 | 229.75 | 89.37 | 140.38 |
| F1 | −893.1 | 244.41 | 35.47 | 206.65 | 0.09 | 421.15 | 278.87 | 103.22 | 175.65 |
| F2 | −992.17 | 220.88 | 37.85 | 209.37 | 0.14 | 259.25 | 89.45 | 102.38 | −12.93 |
| F3 | −1146.32 | 237.5 | 43.5 | 222.92 | 0.16 | 213.8 | 284.19 | 108.53 | 175.66 |
| G1 | −1097.2 | 251.11 | 42.72 | 235.15 | 0.08 | 256.55 | 291.28 | 105.62 | 185.66 |
| G2 | −1196.26 | 227.71 | 45.03 | 237.85 | 0.11 | 163.51 | 110.93 | 105.21 | 5.72 |
| G3 | −1350.41 | 244.26 | 50.59 | 251.83 | 0.14 | 130.06 | 304.47 | 112.65 | 191.82 |
| H1 | −1136.44 | 329.82 | 45.68 | 241.05 | 0.10 | 313.8 | 148.82 | 116.2 | 32.62 |
| H2 | −1334.57 | 282.41 | 50.42 | 247.54 | 0.05 | 318.34 | −209.73 | 108.58 | −318.31 |
| H3 | −1642.88 | 317.16 | 60.47 | 267.61 | 0.05 | 330.14 | 178.68 | 120.9 | 57.78 |
| I1 | −705.05 | 209.13 | 28.5 | 171.55 | 0.15 | 272.86 | 289.59 | 89.19 | 200.40 |
| I2 | −804.11 | 185.91 | 30.51 | 174.02 | 0.15 | 285.68 | 113.59 | 91.34 | 22.25 |
| I3 | −958.27 | 203.41 | 35.66 | 188.29 | 0.14 | 238.96 | 301.19 | 92.71 | 208.48 |
| J1 | −909.1 | 211.53 | 35.76 | 204.06 | 0.09 | 208.03 | 399.06 | 89.52 | 309.54 |
| J2 | −1008.17 | 188.04 | 38.17 | 206.98 | 0.08 | 175.4 | 221.30 | 86.61 | 134.69 |
| J3 | −1162.32 | 205.11 | 43.49 | 220.71 | 0.08 | 164.38 | 408.55 | 92.38 | 316.17 |
| K1 | −909.12 | 213.41 | 35.15 | 203.36 | 0.11 | 272.75 | 362.10 | 95.86 | 266.24 |
| K2 | −1008.18 | 189.8 | 37.51 | 206.11 | 0.15 | 165.98 | 184.51 | 94.33 | 90.18 |
| K3 | −1162.33 | 206.42 | 43.02 | 219.97 | 0.17 | 133.26 | 376.91 | 99.37 | 277.55 |
| L1 | −1152.42 | 293.81 | 45.78 | 250.82 | 0.07 | 252.15 | 298.32 | 111.89 | 186.43 |
| L2 | −1350.56 | 246.75 | 50.51 | 256.52 | 0.09 | 124.28 | −339.73 | 108.62 | −448.35 |
| L3 | −1658.86 | 280.26 | 61.46 | 284.08 | 0.12 | 75.96 | 323.39 | 123.53 | 199.86 |
It has been found that the presence of –CF(NO2)2 group is not beneficial for obtaining energetic molecules with highly positive HOFs.22,63 Clearly, the replacement of a hydrogen atom with a –F group leads to a greater decrease in ΔHf,solid than that observed for the corresponding CH(NO2)2-substituted compounds. Overall, although the new CF(NO2)2-substituted compounds exhibit relatively high densities (>1.77 g cm−3), their HOFs were not highly positive, indicating that the –CF(NO2)2 group has both positive and negative effects on the properties, i.e., high densities are obtained, but the HOFs are not highly positive.
By contrast, the –C(NF2)(NO2)2 group has a positive effect on ΔHf,solid. For example, compound L3 has a much higher ΔHf,solid (199.86 kJ mol−1) than corresponding CF(NO2)2-substituted compound L2 (−448.35 kJ mol−1). Moreover, all the C(NF2)(NO2)2-substituted tetrazole-based compounds exhibit high ΔHf,solid values (186.43–316.17 kJ mol−1). These large positive HOFs mainly result from synergy between the strain inherent to the tetrazole skeleton and the substituent group effect.
Predicted detonation performance
Generally, higher the OBs correspond to larger detonation velocities and pressures. A positive OB is significant for explosive molecules, which can be used as oxidizers. However, too much oxygen is not favorable for improving the explosive performance of energetic compounds, as additional oxygen will produce O2, which consumes a considerable amount of energy during the explosion of energetic materials. Thus, the ideal OB is equal to zero. In this work, the OB values of CHFNO compounds (molecular formula CaHbFcNdOe) were calculated as follows: OB (%) = 1600 × [e − 2a − (b − c)/2]/Mw.64 Most compounds exhibited positive OB values (2.56–17.77%), whereas some energetic molecules had negative OB values (from −32.35 to −7.34%), mainly due to relatively higher carbon and hydrogen contents. As shown in Table 6, substitution with –CF(NO2)2 and –C(NF2)(NO2)2 is helpful for improving the OB values of the designed compounds. This finding suggests that the incorporation of more –NO2 groups into a molecule substantially improves the HOF and the OB, eventually resulting in higher exothermicities for combustion and detonation processes. In particular, the OB values of B2, I2, and F2 are equal to zero, which indicates that they combust completely without releasing toxic gases, such as carbon monoxide, during decomposition.| Comp. | OBa (%) | Db (m s−1) | Pc (GPa) | Comp. | OBa (%) | Db (m s−1) | Pc (GPa) |
|---|---|---|---|---|---|---|---|
| a Oxygen balance (%) for CaHbFcNdOe:1600 × [e − 2a − (b−c)/2]/Mw. Mw: molecular weight of the title compounds.b Calculated detonation velocities.c Calculated detonation pressures. | |||||||
| A1 | −32.35 | 8022.1 | 24.72 | G1 | 9.12 | 8548.1 | 31.55 |
| A2 | −20.93 | 8080.1 | 28.03 | G2 | 14.23 | 8237.0 | 29.26 |
| A3 | −14.28 | 8747.7 | 35.77 | G3 | 15.28 | 8430.4 | 31.44 |
| B1 | −7.34 | 8726.7 | 33.40 | H1 | −8.66 | 8632.1 | 32.51 |
| B2 | 0 | 8811.9 | 37.77 | H2 | 2.56 | 8665.1 | 35.95 |
| B3 | 2.97 | 8978.8 | 40.00 | H3 | 6.33 | 8900.1 | 40.03 |
| C1 | 9.12 | 8607.8 | 32.20 | I1 | −9.19 | 8645.2 | 31.66 |
| C2 | 14.23 | 8511.6 | 31.97 | I2 | 0 | 8820.1 | 38.09 |
| C3 | 15.28 | 8267.2 | 29.65 | I3 | 3.55 | 8968.1 | 40.53 |
| D1 | −8.66 | 8449.7 | 30.60 | J1 | 10.96 | 8555.9 | 30.99 |
| D2 | 2.56 | 8492.8 | 35.27 | J2 | 16.87 | 8070.1 | 27.39 |
| D3 | 6.33 | 8751.9 | 38.21 | J3 | 17.77 | 8341.4 | 30.34 |
| E1 | −32.35 | 8106.9 | 27.08 | K1 | 10.96 | 8577.8 | 31.35 |
| E2 | −20.93 | 8286.3 | 29.65 | K2 | 16.87 | 8106.5 | 27.75 |
| E3 | −14.28 | 8789.8 | 36.36 | K3 | 17.77 | 8360.2 | 30.67 |
| F1 | −7.34 | 8714.5 | 33.15 | L1 | 5.75 | 8552.8 | 31.12 |
| F2 | 0 | 8777.8 | 37.03 | L2 | 15.28 | 8388.7 | 22.15 |
| F3 | 2.97 | 8976.6 | 40.02 | L3 | 16.84 | 8245.7 | 29.92 |
| RDX | −21.66 | 8748 | 34.9 | HMX | −21.66 | 9320 | 39.6 |
D and P are two important performance parameters for an energetic material. Based on the calculated HOFs and the measured ρ, the detonation properties of A1–L3 were determined using the EXPLO5 (v 6.01) program, as summarized in Table 6 alongside a comparison with common explosives. As shown in Table 6, the calculated detonation velocities are in the range of 8022.1–8978.8 m s−1, which are remarkably higher than that of TNT (6881 m s−1). Among these compounds, B3 has the highest detonation velocity (8978.8 m s−1), which exceeds that of RDX (8748 m s−1). Despite having a higher calculated density (2.04 g cm−3), the D of H3 (8900.1 m s−1) is not superior to that of B3 (8798.8 m s−1), which may be explained by the lower HOF value of the former (i.e., 57.78 kJ mol−1 for H3 and 103.96 kJ mol−1 for B3). In terms of detonation pressures, the P values of the designed compounds are in the range of 22.15 (L2) to 40.53 GPa (I3). The highest P value (I3, 40.53 GPa) is much larger than those of RDX (34.9 GPa) and HMX (39.6 GPa). As the number of substituents increases in the A and D series, the D and P values of the 1,2,4-triazole derivatives increase. This observation indicates that substitution with –CF(NO2)2 and –C(NF2)(NO2)2 group is useful for increasing the densities and detonation properties of 1,2,4- triazole derivatives. For the J and K series, the detonation properties are different when the position of the substituent in the tetrazole ring is changed. Moreover, in the L series, some of the derivatives with two –CH(NO2)2, –CF(NO2)2, or –C(NF2)(NO2)2 groups via simultaneous C- and N-linkages have lower energy levels than the corresponding derivatives with one –CH(NO2)2, –CF(NO2)2, or –C(NF2)(NO2)2 group via a C- or N-linkage. This finding reveals that simultaneous C- and N-functionalization does not necessarily enhance detonation performance.
Impact sensitivity
In addition to the energy properties (e.g., ρ, D, and P), the sensitivity of high explosives is also an important subject of keen interest to researchers in the field of energetic materials. Impact sensitivity (h50) is often used to judge whether energetic compounds are sensitive or insensitive to external impact. Therefore, the impact sensitivities of the designed energetic compounds were also studied by using such a calculation equation for h50 values and the structure sensitivity relationships were analyzed.From Table 7, it can be seen that with the exception of C1 and C2, the other compounds exhibit relatively acceptable impact sensitivities with h50 values ranging from 12.06 to 44.65 cm, which are comparable with that of RDX (26 cm) and HMX (32 cm). It is obvious that the compounds that contain more nitro groups show the higher impact sensitivities. Among them, C1 and C2 display very high impact sensitivity (the h50 value is 6.00 and 6.44 cm, respectively), mainly due to its completely nitrated structure and the absence of hydrogen bonding interactions in the molecule. In contrast, CF(NO2)2-substituted B2 show the lowest sensitivity to impact, that is, 44.65 cm for h50. Clearly, replacement of a hydrogen atom with a –F or –NF2 group leads to a slightly decrease in impact sensitivity than that observed for the corresponding CH(NO2)2-substituted compounds. Therefore, the impact sensitivities of the compounds decrease as a function of the substituent, according to C(NF2)(NO2)2 > CF(NO2)2 > CH(NO2)2.
| Comp. | σ+2 | ν | h50 (cm) | Comp. | σ+2 | ν | h50 (cm) |
|---|---|---|---|---|---|---|---|
| A1 | 276.07 | 0.11 | 22.08 | G1 | 233.99 | 0.08 | 14.43 |
| A2 | 339.84 | 0.08 | 23.89 | G2 | 142.62 | 0.11 | 22.56 |
| A3 | 363.67 | 0.08 | 32.50 | G3 | 108.51 | 0.14 | 29.25 |
| B1 | 199.87 | 0.18 | 38.98 | H1 | 278.27 | 0.10 | 19.03 |
| B2 | 158.47 | 0.20 | 44.65 | H2 | 301.47 | 0.05 | 26.76 |
| B3 | 143.21 | 0.20 | 44.57 | H3 | 312.30 | 0.05 | 36.92 |
| C1 | 241.53 | 0.09 | 6.00 | I1 | 223.83 | 0.15 | 30.72 |
| C2 | 193.99 | 0.09 | 6.44 | I2 | 233.83 | 0.15 | 30.94 |
| C3 | 165.16 | 0.11 | 22.66 | I3 | 197.11 | 0.14 | 30.19 |
| D1 | 456.90 | 0.05 | 16.85 | J1 | 186.28 | 0.09 | 17.98 |
| D2 | 336.92 | 0.05 | 17.90 | J2 | 159.55 | 0.08 | 25.40 |
| D3 | 199.02 | 0.08 | 15.18 | J3 | 149.46 | 0.08 | 25.53 |
| E1 | 265.87 | 0.12 | 24.16 | K1 | 239.86 | 0.11 | 20.64 |
| E2 | 169.22 | 0.14 | 28.76 | K2 | 135.72 | 0.15 | 31.70 |
| E3 | 163.19 | 0.13 | 27.64 | K3 | 104.52 | 0.17 | 36.74 |
| F1 | 377.97 | 0.09 | 16.37 | L1 | 232.95 | 0.07 | 12.06 |
| F2 | 217.45 | 0.14 | 27.82 | L2 | 112.46 | 0.09 | 16.63 |
| F3 | 170.75 | 0.16 | 34.31 | L3 | 65.36 | 0.12 | 25.13 |
Potential HEDM candidates
As a function of the substituent, the OB and ρ of each series of compounds decreases according to C(NF2)(NO2)2 > CF(NO2)2 > CH(NO2)2. However, in terms of the detonation properties, the trends observed as a function of the substituent for some series of compounds were not satisfactory. As shown in Fig. 3, the values of D and P increase according to C(NF2)(NO2)2 < CF(NO2)2 < CH(NO2)2 for series C, and CF(NO2)2 < C(NF2)(NO2)2 < CH(NO2)2 for series G. As shown in Scheme 1, the compounds in series C and G have similar molecular structures and the same molecular weights. In these compounds, two nitro groups were introduced by C-functionalization, whereas one –CH(NO2)2, –CF(NO2)2, or –C(NF2)(NO2)2 group was introduced by N-functionalization. Using this way of combination, the energy level cannot be enhanced. These results are worth exploring in future studies. In contrast, for series J and K, the values of D and P first decrease sharply and then increase slightly, indicating that a synergistic effect between a nitro group and –CF(NO2)2 or –C(NF2)(NO2)2 cannot enhance the energy levels of molecules based on tetrazole. Therefore, this result is contrary to previous assumptions. Furthermore, the trend observed for series L indicates that the simultaneous introduction of two of the same substituent by C- and N-functionalization also cannot enhance detonation properties. This behavior may result from unbalanced molecular symmetry. According to the above results, these series of compounds are not recommended for synthesis in subsequent studies. It is well known that a good nitrogen-rich HEDM candidate should have not only excellent detonation properties but also good stability. On this basis, only B3, F3, H3, and I3 are expected to have good detonation performance (D and P), higher stability (BDE) and lower sensitivity (h50) similar to those of RDX and HMX. Therefore, B3, F3, H3, and I3 may be considered as potential HEDM candidates with improved stability and performance. In addition, compounds A2,63 B1,18 and K118 have been successfully synthesized. The calculated value (density, detonation velocity and detonation pressure) is slightly lower than the experimental value, but within acceptable limits (Table 8). However, the syntheses of other energetic compounds examined in this study have not yet been reported. Thus, further investigations are still needed.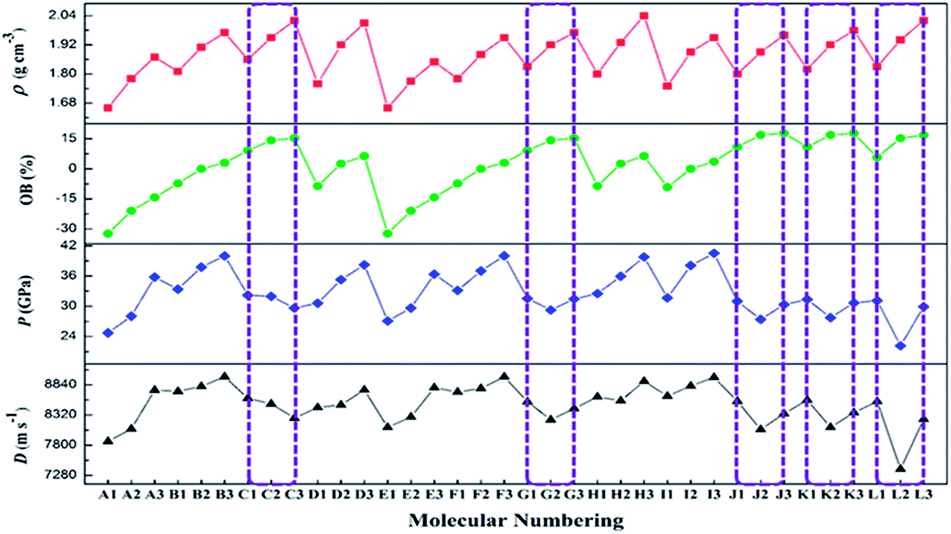 | ||
| Fig. 3 Detonation properties, oxygen balance and densities for the compounds A1–L3. | ||
Conclusion
In summary, we systematically investigated the molecular structures, electronic structures, gas-phase and solid-phase HOFs, energetic properties, and thermal stabilities of three series of CH(NO2)2-, CF(NO2)2-, and C(NF2)(NO2)2-substituted triazole and tetrazole derivatives. As a function of the substituent, the densities of each series of 1,2,4-triazole, 1,2,3-triazole, and tetrazole-based compounds decreased according to –C(NF2)(NO2)2 > –CF(NO2)2 > –CH(NO2)2. This trend indicates that the introduction of highly dense –F or –NF2 groups is a very effective method for increasing the densities of energetic molecules. The introduction of –CF(NO2)2 groups in these materials was both advantageous, as the densities were increased, and disadvantageous, as highly positive HOFs were not obtained. In addition, substitution with a –CF(NO2)2 group was very useful for increasing the thermal stability of 1,2,4-triazole, 1,2,3-triazole, and tetrazole derivatives. In terms of detonation properties, B3, F3, H3, and I3 displayed excellent integrated performance, with higher ρ (≥1.95 g cm−3), OB (≥2.97%), D (>8900 m s−1), and P values (>40.0 GPa) than RDX. Thus, these compounds are promising HEDM candidates. In addition, our design strategy, involving the use of C- or N-functionalization to construct nitrogen-rich molecular skeletons with highly dense substituents and highly positive HOFs, is a valuable approach for developing novel HEDMs with excellent performance and stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (Grant No. 21576026).Notes and references
- H. Gao and J. M. Shreeve, Chem. Rev., 2011, 111, 7377–7436 CrossRef CAS PubMed.
- R. P. Singh, R. D. Verma, D. T. Meshri and J. M. Shreeve, Angew. Chem., Int. Ed., 2006, 45, 3584–3601 CrossRef CAS PubMed.
- D. Fischer, T. M. Klapötke and J. Stierstorfer, Angew. Chem., Int. Ed., 2014, 53, 8172–8175 CrossRef CAS PubMed.
- Y. Tang, J. Zhang, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 15984–15987 CrossRef CAS PubMed.
- J. P. Agrawal, Prog. Energy Combust. Sci., 1998, 24, 1–30 CrossRef CAS.
- C. Qi, S. Li, Y. Li, Y. Wang, X. Zhao and S. Pang, Chem. - Eur. J., 2012, 18, 16562–16570 CrossRef CAS PubMed.
- A. A. Dippold and T. M. Klapötke, J. Am. Chem. Soc., 2013, 135, 9931–9938 CrossRef CAS PubMed.
- D. E. Chavez, S. K. Hanson, J. M. Veauthier and D. A. Parrish, Angew. Chem., Int. Ed., 2013, 52, 6876–6879 CrossRef CAS PubMed.
- J. Zhang and J. M. Shreeve, J. Am. Chem. Soc., 2014, 136, 4437–4445 CrossRef CAS PubMed.
- Q. Liu, B. Jin, R. Peng, Z. Guo, J. Zhao, Q. Zhang and Y. Shang, J. Mater. Chem. A, 2016, 4, 4971–4981 CAS.
- D. E. Chavez, D. A. Parrish and L. Mitchell, Angew. Chem., Int. Ed., 2016, 55, 8666–8669 CrossRef CAS PubMed.
- D. E. Chavez, J. C. Bottaro, M. Petrie and D. A. Parrish, Angew. Chem., Int. Ed., 2015, 54, 12973–12975 CrossRef CAS PubMed.
- L. Liu, Y. Zhang, Z. Li and S. Zhang, J. Mater. Chem. A, 2015, 3, 14768–14778 CAS.
- N. Fischer, D. Izsák, T. M. Klapötke, S. Rappenglück and J. Stierstorfer, Chem. - Eur. J., 2012, 18, 4051–4062 CrossRef CAS PubMed.
- P. He, L. Wu, J. Wu, Q. Wang, Z. Li, M. Gozin and J. Zhang, Chem. - Eur. J., 2017, 23, 11159–11168 CrossRef CAS PubMed.
- D. R. Miller, D. C. Swenson and E. G. Gillan, J. Am. Chem. Soc., 2004, 126, 5372–5373 CrossRef CAS PubMed.
- P. Yin, J. Zhang, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2015, 3, 8606–8612 CAS.
- X. Zhao, S. Li, Y. Wang, Y. C. Li, F. Zhao and S. Pang, J. Mater. Chem. A, 2016, 4, 5495–5504 CAS.
- Y. Tang, H. Gao, G. H. Imler, D. A. Parrish and J. M. Shreeve, RSC Adv., 2016, 6, 91477–91482 RSC.
- K. O. Christe, W. W. Wilson, G. Bélanger-Chabot, R. Haiges, J. A. Boatz, M. Rahm, G. K. S. Prakash, T. Saal and M. Hopfinger, Angew. Chem., Int. Ed., 2015, 54, 1316–1320 CrossRef CAS PubMed.
- S. Huang, J. Tian, X. Qi, K. Wang and Q. Zhang, Chem. - Eur. J., 2017, 23, 12787–12794 CrossRef CAS PubMed.
- M. A. Kettner, K. Karaghiosoff, T. M. Klapötke, M. Sućeska and S. Wunder, Chem. - Eur. J., 2014, 20, 7622–7631 CrossRef CAS PubMed.
- M. A. Kettner, T. M. Klapötke, T. G. Witkowski and F. von Hundling, Chem. - Eur. J., 2015, 21, 4238–4241 CrossRef CAS PubMed.
- L. Zhai, B. Wang, K. Xu, H. Huo, N. Liu, Y. Li, H. Li, P. Lian and X. Fan, J. Energ. Mater., 2016, 34, 92–102 CrossRef CAS.
- H. Li, F. Zhao, B. Wang, L. Zhai, W. Lai and N. Liu, RSC Adv., 2015, 5, 21422–21429 RSC.
- A. S. Batsanov, Y. T. Struchkov, A. A. Gakh and A. A. Fainzil'berg, Russ. Chem. Bull., 1994, 43, 588–590 CrossRef.
- I. L. Dalinger, A. K. Shakhnes, K. A. Monogarov, K. Y. Suponitsky and A. B. Sheremetev, Mendeleev Commun., 2015, 25, 429–431 CrossRef CAS.
- R. Haiges and K. O. Christe, Dalton Trans., 2015, 44, 10166–10176 RSC.
- B. S. Fedorov, N. I. Golovina, S. P. Smirnov, I. S. Abdrakhmanov, A. I. Firkin and L. O. Atovmyan, Chem. Heterocycl. Compd., 2002, 38, 385–389 CrossRef CAS.
- A. B. Sheremetev, D. E. Dmitriev and S. M. Konkina, Russ. Chem. Bull., 2004, 53, 1130–1132 CrossRef CAS.
- Q. Ma, Z. Lu, L. Liao, J. Huang, D. Liu, J. Li and G. Fan, RSC Adv., 2017, 7, 38844–38852 RSC.
- T. M. Klapötke, B. Krumm and R. Moll, Chem. - Eur. J., 2013, 19, 12113–12123 CrossRef PubMed.
- M. Lustig and G. H. Cady, Inorg. Chem., 1963, 2, 388–390 CrossRef CAS.
- A. V. Fokin, Y. N. Studnev, A. I. Rapkin and L. D. Kuznetsova, Russ. Chem. Bull., 1996, 45, 2547–2550 CrossRef.
- Y. Pan and W. Zhu, J. Phys. Chem. A, 2017, 121, 9163–9171 CrossRef CAS PubMed.
- G. Wang, X. Gong, Y. Liu, H. Du, X. Xu and H. Xiao, J. Hazard. Mater., 2010, 177, 703–710 CrossRef CAS PubMed.
- W. Zhu, T. Wei, W. Zhu and H. Xiao, J. Phys. Chem. A, 2008, 112, 4688–4693 CrossRef CAS PubMed.
- G. Wang, X. Gong, Y. Liu and H. Xiao, Int. J. Quantum Chem., 2010, 110, 1691–1701 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Peterssonand et al., Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- X. Fan and X. Ju, J. Comput. Chem., 2008, 29, 505–513 CrossRef CAS PubMed.
- Q. Zhang, J. Zhang, X. Qi and J. M. Shreeve, J. Phys. Chem. A, 2014, 118, 10857–10865 CrossRef CAS PubMed.
- Y. Pan, J. Li, B. Cheng, W. Zhu and H. Xiao, Comput. Theor. Chem., 2012, 992, 110–119 CrossRef CAS.
- V. D. Ghule, P. M. Jadhav, R. S. Patil, S. Radhakrishnan and T. Soman, J. Phys. Chem. A, 2010, 114, 498–503 CrossRef CAS PubMed.
- Q. Wu, W. Zhu and H. Xiao, J. Chem. Eng. Data, 2013, 58, 2748–2762 CrossRef CAS.
- T. Wei, W. Zhu, X. Zhang, Y. Li and H. Xiao, J. Phys. Chem. A, 2009, 113, 9404–9412 CrossRef CAS PubMed.
- P. C. Redfern, J. P. Blaudeau and L. A. Curtiss, J. Phys. Chem. A, 1997, 101, 8701–8705 CrossRef CAS.
- P. Politzer and J. S. Murray, Cent. Eur. J. Energ. Mater., 2011, 8, 209–220 CAS.
- P. W. Atkins, J. Phys. Chem., Oxford University Press, Oxford, U.K., 1982 Search PubMed.
- P. Politzer, P. Lane and J. S. Murray, Cent. Eur. J. Energ. Mater., 2011, 8, 39–52 CAS.
- E. F. C. Byrd and B. M. Rice, J. Phys. Chem. A, 2006, 110, 1005–1013 CrossRef CAS PubMed.
- M. Jaidann, S. Roy, H. Abou-Rachid and L. S. Lussier, J. Hazard. Mater., 2010, 176, 165–173 CrossRef CAS PubMed.
- F. Wang, H. Du, J. Zhang and X. Gong, J. Phys. Chem. A, 2011, 115, 11852–11860 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- P. Politzer, J. Martinez, J. S. Murray, M. C. Concha and A. Toro-Labbé, Mol. Phys., 2009, 107, 2095–2101 CrossRef CAS.
- B. M. Rice and E. F. C. Byrd, J. Comput. Chem., 2013, 34, 2146–2151 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- M. Pospíšil, P. Vávra, M. C. Concha, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 895–901 CrossRef PubMed.
- M. Sucćeska, EXPLO5, version 6.01, Brodarski Institute, Zagreb, Croatia, 2013 Search PubMed.
- B. M. Rice and J. J. Hare, J. Phys. Chem. A, 2002, 106, 1770–1783 CrossRef CAS.
- T. Lu and S. Manzetti, Struct. Chem., 2014, 25, 1521–1533 CrossRef CAS.
- T. Wei, W. Zhu, J. Zhang and H. Xiao, J. Hazard. Mater., 2010, 179, 581–590 CrossRef CAS PubMed.
- T. Fei, H. Cai, Y. Zhang, L. Liu and S. Zhang, J. Energ. Mater., 2016, 34, 138–151 CrossRef CAS.
- Y. Li, Y. Shu, S. Zhang, B. Wang, L. Zhai and H. Huo, Chin. J. Energ. Mater., 2017, 25, 612–616 Search PubMed.
- T. M. Klapötke, B. Krumm, R. Moll, S. F. Rest, W. Schnick and M. Seibald, J. Fluorine Chem., 2013, 156, 253–261 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Summary of surface analysis of compounds A1–L3. See DOI: 10.1039/c8ra00699g |
| This journal is © The Royal Society of Chemistry 2018 |