
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of conjugation and aromaticity of 3,6 di-substituted carbazoles on triplet energy and the implication of triplet energy in multiple-cyclic aromatic compounds†
Kai Lin Woon *a,
Azhar Ariffin*b,
Kar Wei Hob and
Show-An Chenc
*a,
Azhar Ariffin*b,
Kar Wei Hob and
Show-An Chenc
aLow Dimensional Materials Research Center, Department of Physics, University of Malaya, Malaysia. E-mail: ph7klw76@um.edu.my
bDepartment of Chemistry, University of Malaya, 50603 Kuala Lumpur, Malaysia. E-mail: azhar70@um.edu.my
cDepartment of Chemical Engineering and Frontier Research Center on Fundamental and Applied Sciences of Matters, National Tsing-Hua University, 101, Section 2, Kuang-Fu Road, 30041 Hsinchu, Taiwan, ROC
First published on 9th March 2018
Abstract
It is well-known that short conjugation is needed to obtain a high triplet energy. Carbazole has 3 fused rings and yet it has a high triplet energy. In order to illuminate the reason behind this, we synthesized a range of carbazole derivatives with substitution at the 3,6-positions. All carbazoles with phenyl moieties substituted at the 3,6-positions exhibit a lower triplet energy than that of carbazole itself. We also quantified the aromaticity of carbazole using the nucleus-independent chemical shift tensor. We discovered that the five-membered heterocyclic aromatic ring in carbazole has reduced aromaticity. This results in a reduced conjugation effect between the five-membered heterocyclic aromatic ring and the neighboring benzene rings. Inspired by this finding, the triplet energies of compounds with up to seven benzene units separated by heterocycles (furan, pyrrole, thiophene, silole, and phosphole) and cyclopentadiene were calculated using time-dependent density functional theory. A high triplet energy (>3 eV) can be obtained by alternating high aromaticity and reduced aromaticity in highly extended fused π systems containing furan and pyrrole. In tricyclic aromatic compounds (dibenzofuran, carbazole, fluorene, dibenzothiophene, 5H-benzo[b]phosphinedole and 9H-9-silafluorene) and their extended fused π systems that we have examined so far, the triplet energy is related to the electronegativity of the oxygen, nitrogen, carbon, sulfur, phosphorous and silicon atoms. These findings provide new intuitive insight related to the structures of molecules and the triplet energies, which could be useful in organic optoelectronics.
Introduction
The manipulation of electron delocalization in an extended system allows us to change the electronic and optical properties of materials. By changing the degree of electron delocalization in an organic molecule, the band-gap of the materials can be adjusted. Such a conjugated system can be intuitively modeled as an electron in a quantum box, where the energy difference between two levels is inversely proportional to the length of the box.1 This means that the more extended the electron delocalization, the smaller the material band-gap.This intuitive picture of a quantum box has helped scientists to devise strategies to synthesize a range of novel materials. For organic solar cells and dye sensitized solar cells, more extended conjugation is highly desirable.2 For blue emitting materials, short conjugation is important.3 Carbazole is a wide band-gap material.4 However, the band-gap can be greatly reduced by di-substitution at the 2,7-positions because of the reduced flexibility compared to di-substitution at the 3,6-positions.5 Carbazole and its derivatives are often used as host materials for guest/host systems in organic phosphorescent devices with high efficiency.6 A wide band-gap and high triplet energy are needed to prevent energy back-transfer from the phosphorescent dye.7 Thus, it is of great importance to design and synthesize wide band-gap materials with high triplet energy. It must follow that triplet energy can be reduced by increasing the extent of electron delocalization.8 Since electron localization/delocalization is central in determining various properties of the materials, several methods of quantification have been developed. Most of these descriptors reduce a molecule into a number. These include the noncovalent interaction index, the multicenter delocalization index, the fluctuation index and the harmonic oscillator model of aromaticity index, to name just a few.9 This might be useful for small molecules, but it is hardly useful for a more complex compound where substitution at different positions within the molecule results in totally different behavior. Mapping of how electron delocalization changes within a single molecule might yield useful information. Such methods have not yet been applied in explaining organic optoelectronic materials.
Carbazole is a tricyclic aromatic heterocyclic compound consisting of two benzene rings fused on either side of a five-membered nitrogen ring. Its intriguing structure exhibits high triplet energy. In order to examine how conjugation affects the triplet energy, carbazoles with phenyl derivatives as substituents at the 3,6-positions were synthesized. The aromaticity of a given compound is quantified using isosurface mapping of the magnetic shielding tensor. We not only show that conjugation is important, but also that aromaticity within the compounds must be taken into account in order to explain the high triplet energy of the carbazole molecule.
Results and discussion
Synthesis of the carbazole derivatives
In this study, different groups such as tert-butyl, anisole, fluorophenyl, and carbazole were introduced as side chains onto the tetraphenylsilane-carbazole core. The structures of the derivatives (P1–P5) based on the tetraphenylsilane-carbazole core are shown in Fig. 1.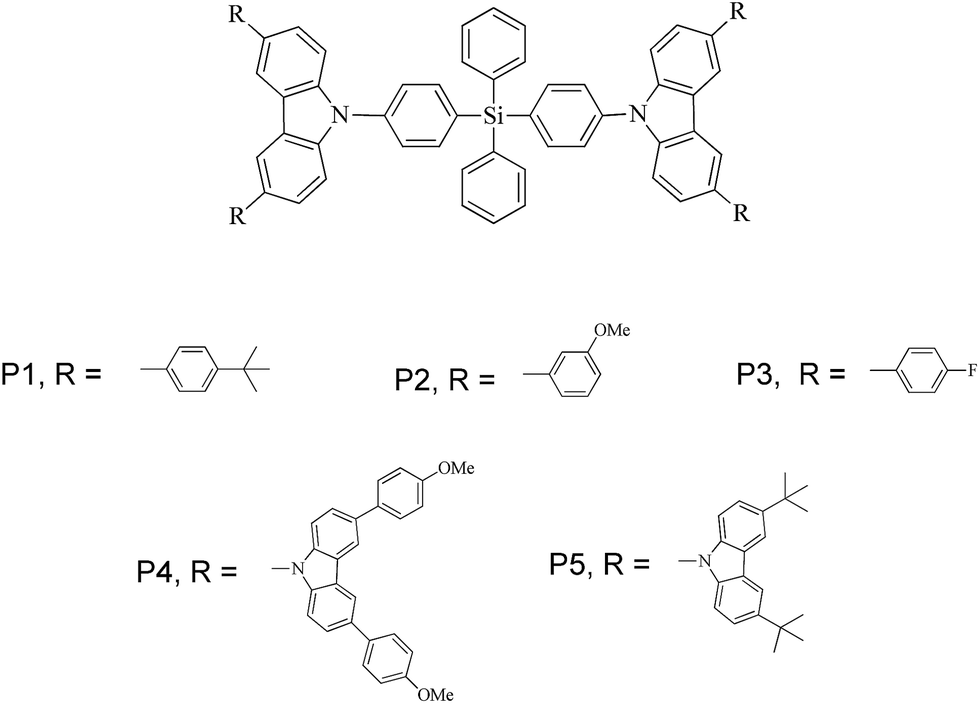 | ||
| Fig. 1 Tetraphenylsilane-carbazole derivatives, P1–P5. | ||
Compounds P1–P5 were obtained through Ullmann coupling reactions of brominated tetraphenylsilane 1 with carbazole side chains (3–5, 11 and 12). Compound 1 was obtained from the reaction between dichlorodiphenylsilane and lithiated 1,4-dibromobenzene at −78 °C, as reported previously.10
Side chains 3–6 were prepared as follows. Suzuki coupling reactions of the iodinated carbazole 2 with boronic acid derivatives afforded the side chains 3–6 (Scheme 1). In the Suzuki coupling reactions, Pd(PPh3)4 was used to catalyze the reactions in the presence of 2 M K2CO3 and toluene solution. Compound 7 was prepared by reacting carbazole with tert-butylchloride in a Friedel–Crafts alkylation reaction.11
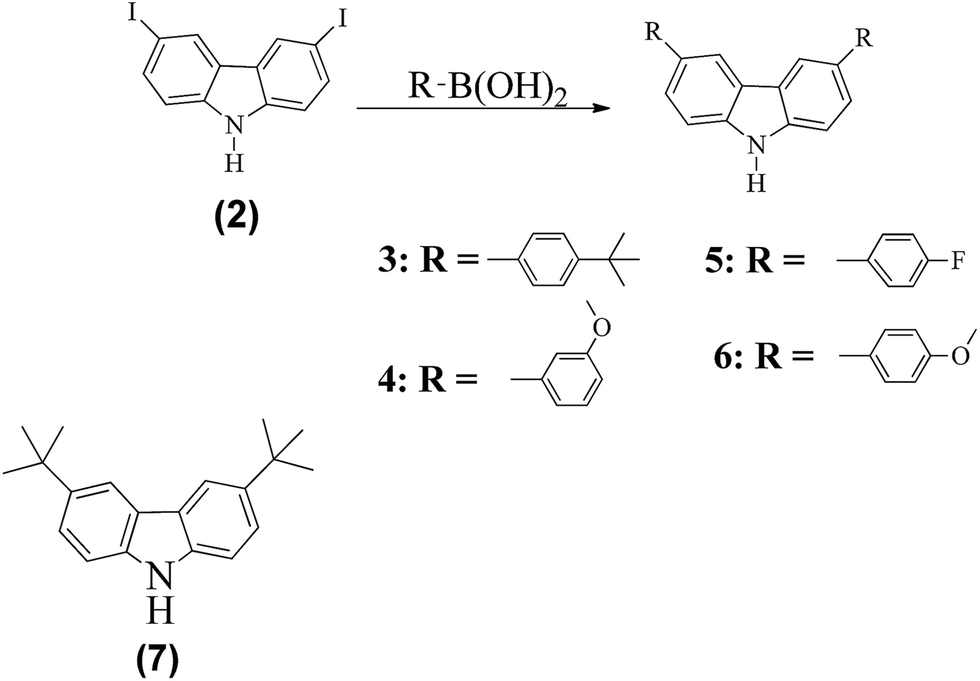 | ||
| Scheme 1 Synthesis routes to compound side chains 3–6. Reagents and conditions: Pd(PPh3)4, 2 M K2CO3, and toluene, at 90 °C overnight. | ||
Compounds P1–P3 were synthesized by reacting compound 1 with the side chains 3–5, respectively, in Ullman coupling reactions as demonstrated in Scheme 2. All three compounds were isolated in very low yields of 20%, 28%, and 17%, respectively.
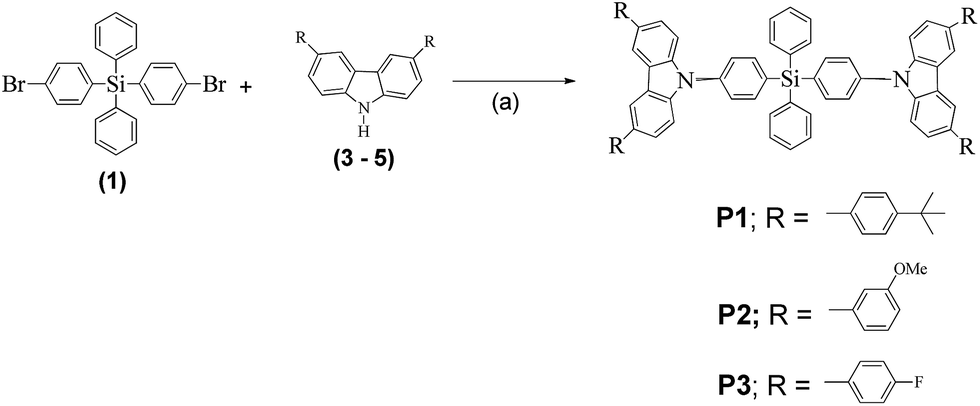 | ||
| Scheme 2 Synthesis of compounds P1–P3. Reaction conditions: (a) copper powder, 18-Crown-6, K2CO3, and o-DCB, at 180 °C for 24–72 h. | ||
The synthesis procedures for compounds P4 and P5 were more complicated than those for compounds P1–P3. Compounds P4 and P5 were synthesized with the intention to extend the molecular backbone by increasing the number of carbazoles at the side chains.
Firstly, the amine proton of the iodinated carbazole (2) was protected using p-toluenesulfonyl chloride according to a literature procedure, to give compound 8 in a 62% isolated yield.12 Compound 8 was then reacted with compound 6 and compound 7 under Ullmann conditions to obtain 9 and 10 in moderate yields (49% and 66%, respectively).12 The tosyl group was then deprotected using base hydrolysis to obtain compounds 11 and 12 in quantitative yields (Scheme 3).
The side chains 11 and 12 were then reacted with compound 1 in Ullman coupling reactions similar to the reactions in Scheme 2, to produce compounds P4 (43%) and P5 (18%). All newly synthesized products were fully characterized using 1H NMR, 13C NMR and MALDI-TOF mass spectroscopy. The results confirmed the proposed structures. The products are readily soluble in common solvents such as dichloromethane, chloroform, tetrahydrofuran and dimethylformamide, which indicates the possibility for solution processing.13
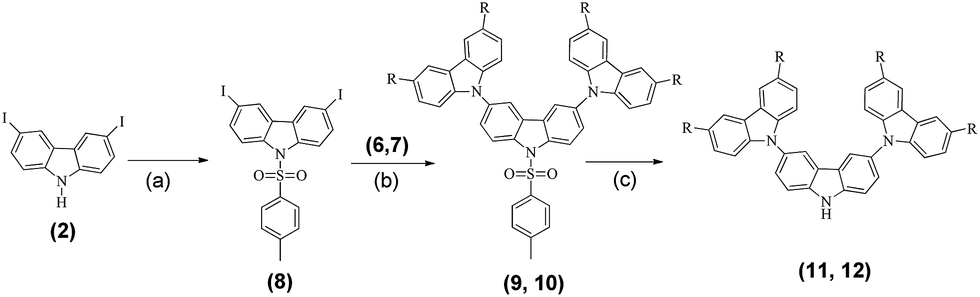 | ||
| Scheme 3 Synthesis routes to compounds 11 and 12. Reagents and conditions: (a) p-toluenesulfonyl chloride, NaH and DMF, at 0 °C for 4 h; (b) compound 6 or 7, Cu2O and N,N-dimethylacetamide, reflux for 48 h; (c) (i) KOH, DMSO, and H2O, reflux for 4 h; (ii) 6 N HCl. | ||
Different substituents result in different degrees of red-shift, as shown in the photoluminescence (PL) spectra in Fig. 2(a), with P3 being the least red-shifted followed by P1, then P2 and lastly P4. The degree of red-shift is often correlated with the degree of conjugation, with P4 being the most conjugated. The peaks of fluorescence for P1, P2, P3 and P4 are at 393.2 nm, 402.5 nm, 388.4 nm and 410.6 nm, respectively. The difference between P2 and P3 is clearly the result of the electron donating ability of the substituent. It seems that the less electron withdrawing the substituent, the more red-shifted the spectrum. Hence, –F is more electron withdrawing than the tert-butyl group while the –OMe group is less electron withdrawing than the tert-butyl group. Comparing P3, P4 and P5 as shown in Fig. 2(b), attaching the 3-methoxyphenyl moiety at the 3,6-positions of carbazole results in significant red-shift compared with functionalizing with a bulky side group at the 3,6-positions. In –OMe, the oxygen donates one of its lone pairs of electrons via resonance into the benzene π system, which in turn increases the electron density in the π system of carbazole. Hence, it experiences the largest red-shift. For the tert-butyl group, the electron-releasing effect occurs via the inductive effect. This effect is weaker than the resonance effect as it involves the strongly held σ-bond electrons. The fluorine atom has 3 lone pairs of electrons, compared to the oxygen atom. The inductive effect of fluorine can occur but it is overwhelmed by the strong electronegativity of the fluorine atom and hence it is the least red-shifted.
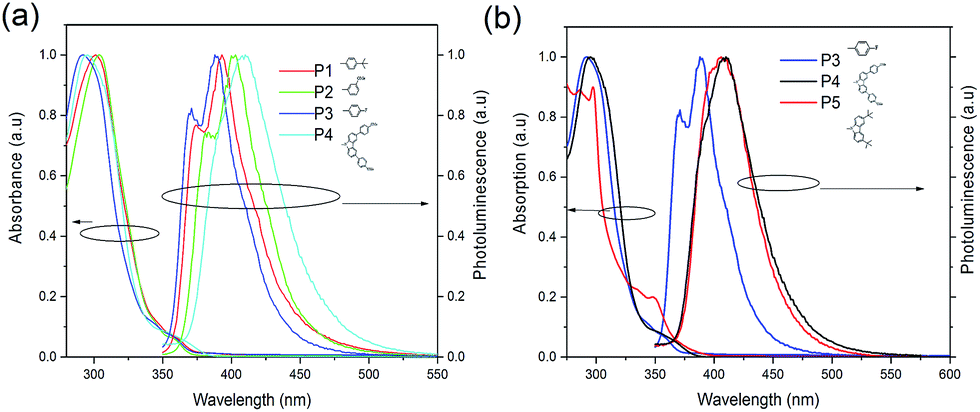 | ||
| Fig. 2 (a) The normalized absorption and photoluminescence of P1, P2, P3 and P4 in solutions of dichloromethane. (b) Comparison of the absorption and photoluminescence of P3, P4 and P5. | ||
Fig. 3 shows the triplet energies for P1, P2, P3, P4 and P5, which are 2.34 eV, 2.49 eV, 2.54 eV, 2.30 eV and 2.72 eV, respectively. For P1, P2, P3 and P4, the triplet energies follow the trends of the PL as shown in Fig. 3(a). The more red-shifted the PL, the lower the triplet energy. Compared with P1 and P3, the fluorine has an unusual effect in reducing the reduction of the triplet energy of the molecules. Despite the fact that P3 has the least red-shifted photoluminescence, it has a lower triplet energy than P5. One would argue that this could be the result of the torsional angle between two aromatic rings, like for the difference in the triplet energy observed between 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) and 4,4′-bis(9-carbazolyl)-2,2′-dimethylbiphenyl (CDBP). The torsional angles and the energies between the carbazole and benzene with different substituent groups, -(4-tert-butylphenyl) (for P1), -(3-methoxyphenyl) (for P2) and -(4-fluorophenyl) (for P3), are similar to those of P5 (see ESI Fig. S49†). In this case, the torsional angle shouldn’t have a significant effect on the difference in the triplet energy observed between P1, P2, P3 and P5. Carbazole has 3 fused rings, while the derivative groups such as P1, P2 and P3 have only one ring. The intuitive picture of conjugation seems to fail. The presence of fluorine in P3 gives us a clue that the intuitive picture of conjugation from the chemical structure might be incorrect. Fluorine is a highly electron withdrawing group and there is a very strong likelihood that within the molecule, the π-delocalized electrons are not evenly distributed. It may be that aromaticity gives a better quantification.
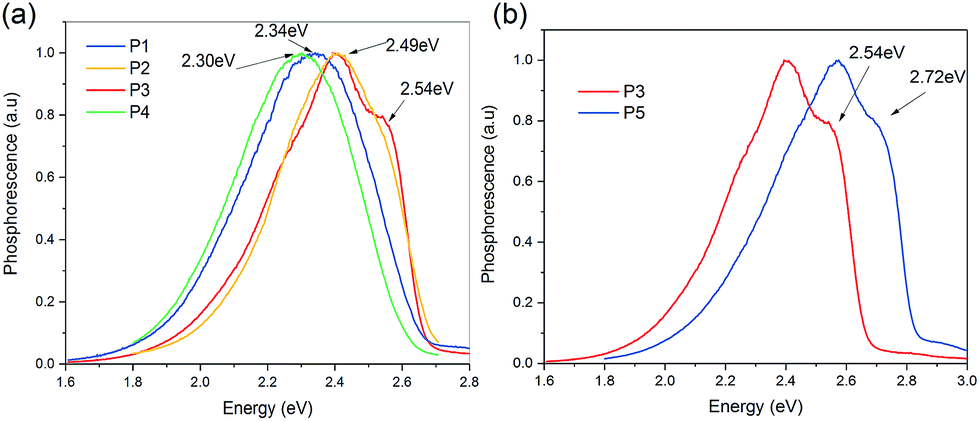 | ||
| Fig. 3 (a) The normalized low temperature phosphorescence of P1, P2, P3 and P4 in solutions of dichloromethane. (b) Comparison of the low temperature phosphorescence of P3 and P5. | ||
Aromaticity is not a directly measurable quantity. However, magnetic properties are the most closely related to aromaticity, as the induced ring currents are associated with cyclic electron delocalization.14 These can be used to intuitively visualize the aromaticity. Since ring currents due to the cyclic π electron delocalization are induced primarily by the external magnetic field applied perpendicular to the ring (the z direction), the out-of-plane component of the nucleus-independent chemical shift (NICS) tensor should contain the information most relevant for aromaticity evaluations.15 Using the NICS concept, the Nuclear Magnetic Resonance (NMR) shieldings can be visualized as iso-chemical-shielding surfaces (ICSSs).16
Fig. 4(a) represents the isosurface mapping of the magnetic shielding tensor perpendicular to and 1 Å above the plane of the molecule. Here, we refer to it as ICSSZZ(1). The different colour schemes are used to indicate different magnetic shielding isosurface values in ppm. The orange-red region at the two benzene rings of carbazole indicates that the z-direction magnetic field is largely shielded due to the induced ring current originating from the delocalized π-electrons. This observation implies the strong aromaticity of the two benzene rings of carbazole. However, the five membered heterocyclic ring fused at the middle by the two benzene rings has significantly less aromaticity.
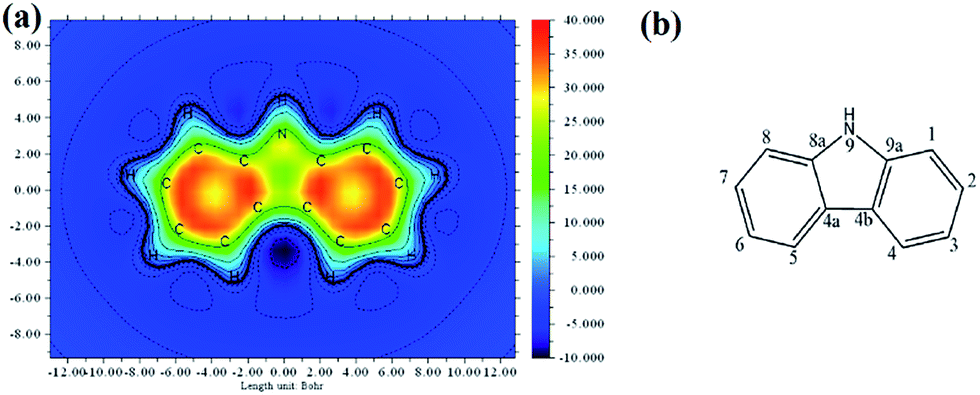 | ||
| Fig. 4 (a) ICSSZZ(1) mapping of the carbazole molecule. Positive and negative Z-components indicate shielding and de-shielding values when a magnetic field along the z-direction is applied. (b) The labelling of positions of the atoms in the carbazole molecule. | ||
Carbazole has a lone pair of electrons in the pz orbital of the nitrogen atom. However, there is a small orbital overlap between the lone pair of electrons occupying the pz orbital with the electrons in the pz orbitals of the neighboring carbon atoms (8a and 9a as shown in Fig. 4(b)). This is probably due to the high electronegativity of the nitrogen atom resulting in contraction of the pz orbital and hence a smaller wavefunction overlap, despite the bond length being 1.39 Å as indicated by single crystal X-ray diffraction.17 This is supported by natural bond orbital (NBO) analysis, where the occupancy of the lone pair of electrons is 1.67 at the nitrogen atom. NBO analysis also indicated that C8a–C4a and C9a–C4a are electron rich, as supported by the ICSSZZ(1) in Fig. 4(a). The bond length between the carbon atoms at C4a–C4b is 1.47 Å.16 The ICSSZZ(1) indicates a sigma bond character. This is in contrast with pyrrole, where the carbon–carbon double bond shows a strong π-character.18 Carbazole is analogous to phenanthrene, where according to Clar’s rule, the two side rings are expected to have a larger local aromaticity than the central ring.19 Clar’s rule only applies to aromatic π-sextets, however, carbazole is not truly an aromatic π-sextet due to the presence of a five membered ring at the center. The large bond length between C4a and C4b could significantly reduce the overlapping of π-orbitals. This is in contrast to molecular orbital (MO) theory where the highest occupied molecular orbital is spread out between C4a and C4b, suggesting the existence of extended π-electrons across the two benzenes.20 This is probably the main weakness of MO theory, where the electrons are treated such that they are spread out under the influence of the nuclei in the whole molecule. The partial break of conjugation between C8a–N9 and N9–C9a allows the nitrogen atom to be connected with another aromatic ring with a small reduction of the triplet energy. This is clearly indicated by the P5 molecule. The two benzene rings of carbazole are electron rich. Attaching the benzene rings at the 3,6-positions of carbazole will effectively extend the conjugation. Hence, a significant reduction of triplet energy occurs. This also explains why naphthalene has such a low triplet energy compared to carbazole, as two benzene rings are conjugated together. However, for a high triplet energy, both short aromatic rings and a high torsional angle between aromatic moieties are important.21
Fluorene and dibenzofuran, as tricyclic aromatic compounds, might share some of the characteristics of carbazole to various degrees. Inspired by this idea, we carried out triplet energy calculations with up to seven benzene rings separated by different types of 5-membered aromatic ring and fused together. The general structure of the molecules is shown in Fig. 5(a), with oxygen, nitrogen, carbon, sulfur, phosphorous or silicon as the X atoms. Fig. 5(b) shows the ICSSZZ(1) mapping of four benzene units separated by pyrrole rings. We can see that there is alternating high aromaticity and reduced aromaticity. Dibenzofuran and carbazole have the highest triplet energy, followed by dibenzothiophene, fluorene, then 5H-benzo[b]phosphinedole and lastly 9H-9-silafluorene, as shown in Fig. 5(c). There is a reduction of triplet energy when more and more repeated units are fused together. The degree of reduction seems to follow the trend of how electronegative the X atom is, as shown in Fig. 5(d). Using the Pauling scale, oxygen has an electronegativity of 3.44 followed by nitrogen (3.04), sulfur (2.58), carbon (2.55), phosphorus (2.19) and silicon (1.90). However, carbon has a slightly smaller reduction than sulfur despite the fact that sulfur is 0.04 higher in electronegativity on the Pauling scale than carbon. It may be that the number of electronic shells (size of the atom) is the underlying secondary factor. For the seven benzene units separated by furan, the triplet energy only reduced by 0.14 eV compared to the dibenzofuran, while for the seven benzene rings separated by silole, the triplet energy is reduced by as much as 0.32 eV compared to 9H-9-silafluorene. These extended fused π-systems might improve the charge mobility as the intermolecular π–π overlap can be higher between two extended fused π-systems.22 The 4-membered ring in the biphenylene molecule is well-known to exhibit an anti-aromatic character. However, such a molecule has a very low calculated triplet energy (1.16 eV). Due to the extended aromaticity between two benzene rings in naphthalene and three benzene rings in anthracene, their triplet energies are lower than that of carbazole, with naphthalene having a triplet energy of 2.65 eV (ref. 23) and anthracene having a triplet energy of 1.85 eV.24
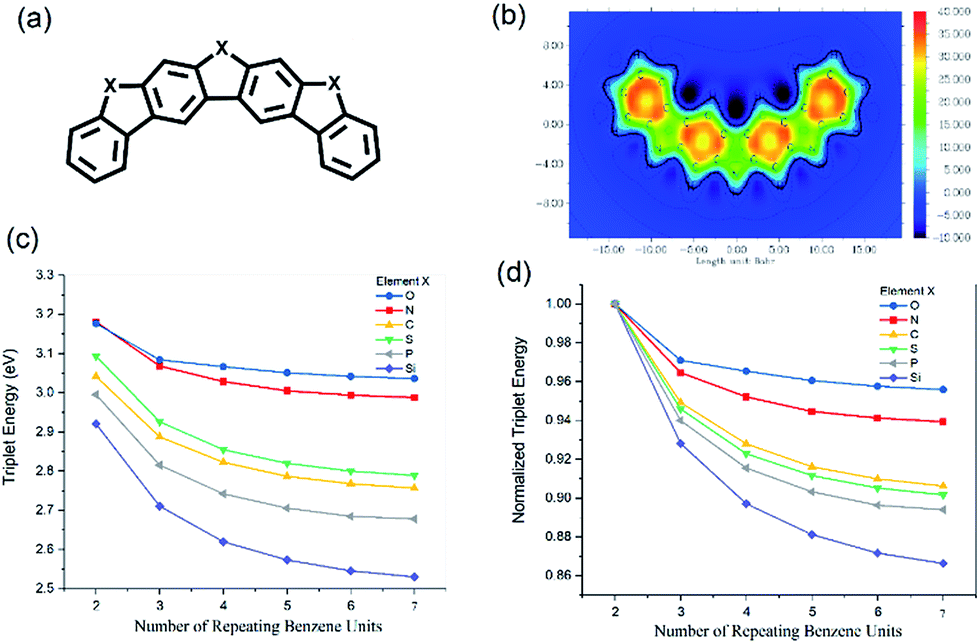 | ||
| Fig. 5 (a) The general molecular structure of alternating benzene and heterocycle rings where X can be O, N, C, S, P or Si. (b) The ICSSZZ(1) mapping of four benzene rings separated by pyrrole rings. (c) The triplet energies of extended π systems with different numbers of benzene units separated by different heterocycles. (d) The normalized triplet energies with different numbers of repeating benzene units. | ||
Conclusions
The decrease of aromaticity of the five membered rings in carbazole results in insensitivity of triplet energy change when the 9-position of carbazole is functionalized with a benzene ring. A range of tricyclic aromatic compounds were examined. It seems that by alternating high aromaticity and reduced aromaticity in an extended fused π-system, high triplet energy can be somewhat preserved, particularly when furan and pyrrole are used. In future, extended fused π-systems with high triplet energy can be synthesized and the charge mobility of such materials tested.Experimental
General information
Proton and carbon nuclear magnetic resonance spectra (1H NMR and 13C NMR spectra) were recorded on a Bruker AVANCE III 400 MHz FT-NMR spectrometer using deuterated chloroform as the solvent with tetramethylsilane (TMS) as an internal standard. Matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) measurements were carried out using a Bruker Microflex series spectrometer in order to measure the molecular masses. 2,5-Dihydroxybenzoic acid (DHB) was used as a matrix.3,6-Bis(4-(tert-butyl)phenyl)-9H-carbazole. 4-Tert-butylphenylboronic acid (1.1 g, 6.2 mmol) and compound 2 (1.25 g, 3 mmol) were dissolved in 25 ml toluene in a round bottom flask under nitrogen. A solution of potassium carbonate (2 M, 1.7 ml) was added to the solution. Tetrakis(triphenyl-phosphine)palladium(0) (0.14 g, 0.12 mmol) was added. The reaction mixture was refluxed for 24 h under nitrogen. After cooling to room temperature, the reaction mixture was evaporated to remove solvent and extracted with chloroform and water. The organic layer was removed under reduced pressure, and the residue was subjected to column chromatography using hexane/ethyl acetate as the eluent to obtain the known compound as a white solid (yield 85.6%, 1.11 g). Mp: decomposes >200 °C. 1H NMR (400 MHz, CDCl3) δ [ppm]: 8.34 (d, J = 1.71 Hz, 2H), 8.13 (s, 1H), 7.66–7.73 (m, 6H), 7.49–7.56 (m, 6H), 1.42 (s, 18H). 13C NMR (100 MHz, CDCl3) δ [ppm]: 149.46, 139.30, 139.16, 132.99, 126.92, 125.72, 125.52, 124.04, 118.71, 110.85, 34.51, 31.44. MALDI-TOF MS (m/z): 431.165.
3,6-Bis(3-methoxyphenyl)-9H-carbazole. 3-Methoxyphenylboronic acid (3.17 g, 20.9 mmol) and compound 2 (3.98 g, 9.49 mmol) were dissolved in 40 ml toluene in a round bottom flask under nitrogen. A solution of potassium carbonate (2 M, 10 ml) was added to the solution. Tetrakis(triphenyl-phosphine)palladium(0) (0.47 g, 0.41 mmol) was added. The reaction mixture was refluxed for 24 h under nitrogen. After cooling to room temperature, the reaction mixture was evaporated to remove solvent and extracted with chloroform and water. The organic layer was removed under reduced pressure, and the residue was subjected to column chromatography using hexane/ethyl acetate as the eluent to obtain the known compound as a white solid (yield 17.4%, 0.63 g). Mp: decomposes >200 °C. 1H NMR (400 MHz, CDCl3) δ [ppm]: 8.36 (d, J = 1.83 Hz, 2H), 8.17 (s, 1H), 7.72 (dd, J = 8.24, 1.83 Hz, 2H), 7.53, (d, J = 8.24 Hz, 2H), 7.38–7.48 (m, 2H), 7.31–7.37 (m, 2H), 7.24–7.31 (m, 2H), 6.87–7.00 (m, 2H), 3.94 (s, 6H). 13C NMR (100 MHz, CDCl3) δ [ppm]: 160.07, 143.63, 139.58, 133.12, 129.83, 125.75, 124.05, 119.93, 119.04, 113.06, 112.09, 110.99, 55.44. MALDI-TOF MS (m/z): 378.868.
3,6-Bis(4-fluorophenyl)-9H-carbazole. 4-Fluorophenylboronic acid (0.93 g, 6.7 mmol) and compound 2 (1.27 g, 3.03 mmol) were dissolved in 24 ml toluene in a round bottom flask under nitrogen. A solution of potassium carbonate (2 M, 6 ml) was added to the solution. Tetrakis(triphenyl-phosphine)palladium(0) (0.13 g, 0.11 mmol) was added. The reaction mixture was refluxed for 24 h under nitrogen. After cooling to room temperature, the reaction mixture was evaporated to remove solvent and extracted with chloroform and water. The organic layer was removed under reduced pressure, and the residue was subjected to column chromatography using hexane/ethyl acetate as the eluent to obtain the known compound as a white solid (yield 30.4%, 0.33 g). Mp: decomposes >200 °C. 1H NMR (400 MHz, CDCl3) δ [ppm]: 8.29 (s, 2H), 8.17 (br s, 1H), 7.62–7.72 (m, 6H), 7.53 (d, J = 8.44 Hz, 2H), 7.19 (t, J = 8.68 Hz, 4H). 13C NMR (100 MHz, CDCl3) δ [ppm]: 163.34, 160.90, 139.36, 138.15, 132.32, 128.80, 128.71, 125.58, 123.97, 118.79, 115.69, 115.48, 111.01. MALDI-TOF MS (m/z): 354.818.
3,6-Bis(4-methoxyphenyl)-9H-carbazole. 4-Methoxyphenylboronic acid (1.31 g, 8.6 mmol) and compound 2 (1.51 g, 3.6 mmol) were dissolved in 30 ml toluene in a round bottom flask under nitrogen. A solution of potassium carbonate (2 M, 3.6 ml) was added to the solution. Tetrakis(triphenyl-phosphine)palladium(0) (0.17 g, 0.14 mmol) was added. The reaction mixture was refluxed for 24 h under nitrogen. After cooling to room temperature, the reaction mixture was evaporated to remove solvent and extracted with chloroform and water. The organic layer was removed under reduced pressure, and the residue was subjected to column chromatography using hexane/ethyl acetate as the eluent to obtain the known compound as a white solid (yield 43.5%, 1.13 g). Mp: 198–200 °C. 1H NMR (600 MHz, CDCl3) δ [ppm]: 8.30 (s, 2H), 8.10 (br s, 1H), 7.60–7.78 (m, 6H), 7.50 (d, J = 8.25 Hz, 2H), 6.97–7.09 (m, 4H), 3.90 (s, 6H). 13C NMR (100 MHz, CDCl3) δ [ppm]: 158.65, 139.10, 134.74, 132.82, 128.27, 125.32, 124.05, 118.43, 114.24, 110.85, 55.40. MALDI-TOF MS (m/z): 379.511.
3,3′′,6,6′′-Tetrakis(4-methoxyphenyl)-9′-tosyl-9′H-9,3’:6′,9′′-tercarbazole. A solution of compound 8 (1.16 g, 2.03 mmol) and compound 6 (1.68 g, 4.42 mmol) in N,N-dimethylacetamide (10 ml) was prepared. Copper oxide (0.59 g, 4.1 mmol) was added to the solution. The mixture was then refluxed for 48 h and cooled to room temperature. After cooling to room temperature, the reaction mixture was evaporated under reduced pressure to remove solvent and extracted with chloroform and water. The organic layer was evaporated under reduced pressure, and the residue was subjected to column chromatography using hexane/ethyl acetate as the eluent to obtain the new compound as a white solid (yield 49.1%, 1.07 g). Mp: decomposes >300 °C. 1H NMR (400 MHz, CDCl3) δ [ppm]: 8.66 (d, J = 8.80 Hz, 2H), 8.36 (d, J = 1.35 Hz, 4H), 8.18 (d, J = 1.83 Hz, 2H), 7.98 (d, J = 8.44 Hz, 2H), 7.83 (dd, J = 8.93, 2.08 Hz, 2H), 7.66 (d, J = 8.80 Hz, 8H), 7.63 (dd, J = 8.25, 1.83 Hz, 4H), 7.46 (d, J = 8.56 Hz, 4H), 7.35 (d, J = 8.07 Hz, 2H), 7.04 (d, J = 8.80 Hz, 8H), 3.90 (s, 12H), 2.44 (s, 3H). 13C NMR (100 MHz, CDCl3) δ [ppm]: 158.75, 140.74, 137.71, 135.05, 134.51, 134.08, 133.51, 130.17, 128.35, 128.29, 127.06, 126.82, 125.49, 125.46, 124.09, 118.88, 118.50, 116.47, 114.28, 109.87, 55.41, 21.73. MALDI-TOF MS (m/z): 1075.875.
3,3′′,6,6′′-Tetra-tert-butyl-9′-tosyl-9′H-9,3’:6′,9′′-tercarbazole. A solution of compound 8 (1.44 g, 2.51 mmol) and compound 7 (1.54 g, 5.5 mmol) in N,N-dimethylacetamide (10 ml) was prepared. Copper oxide (0.73 g, 5.1 mmol) was added to the solution. The mixture was then refluxed for 48 h and cooled to room temperature. After cooling to room temperature, the reaction mixture was evaporated under reduced pressure to remove solvent and extracted with chloroform and water. The organic layer was evaporated under reduced pressure, and the residue was subjected to column chromatography using hexane/ethyl acetate as the eluent to obtain the known compound as a white solid with a yield of 65.5% (1.44 g). Mp: decomposes >300 °C. 1H NMR (600 M Hz, CDCl3) δ [ppm]: 8.59 (d, J = 8.80 Hz, 2H), 8.16 (d, J = 1.65 Hz, 4H), 8.07 (d, J = 2.02 Hz, 2H), 7.94 (d, J = 8.25 Hz, 2H), 7.75 (dd, J = 8.80, 2.20 Hz, 2H), 7.47 (dd, J = 8.62, 2.02 Hz, 4H), 7.35 (d, J = 8.25 Hz, 4H), 7.32 (d, J = 8.25 Hz, 2H), 2.42 (s, 3H), 1.48 (s, 36H). 13C NMR (100 MHz, CDCl3) δ [ppm]: 145.49, 143.02, 139.51, 137.38, 135.02, 134.54, 130.07, 127.09, 126.77, 123.70, 123.38, 118.50, 116.32, 108.97, 34.74, 32.00, 21.69. MALDI-TOF MS (m/z): 875.756.
3,3′′,6,6′′-Tetrakis(4-methoxyphenyl)-9′H-9,3’:6′,9′′-tercarbazole. To a solution of compound 9 (1.04 g, 0.965 mmol) in 9 ml tetrahydrofuran, dimethyl sulfoxide (2.45 ml) and water (1.0 ml) were added, and the mixture was stirred for 10 min. Subsequently, potassium hydroxide (1.36 g, 0.024 mol) was added to this mixture, and the reaction mixture was refluxed for 4 hours, cooled to room temperature and diluted with water. After neutralization with HCl solution, the crude product was filtered and recrystallized from dichloromethane–hexane (1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give the new compound as a white solid (yield 99.4%, 0.884 g). Mp: decomposes >300 °C. 1H NMR (600 MHz, CDCl3) δ [ppm]: 8.62 (br s, 1H), 8.25–8.44 (m, 6H), 7.40–7.82 (m, 20H), 7.04 (d, J = 8.80 Hz, 8H), 3.89 (s, 12H). 13C NMR (100 MHz, CDCl3) δ [ppm]: 158.66, 146.60, 141.42, 139.33, 134.73, 130.06, 128.30, 126.11, 125.35, 123.82, 120.44, 119.70, 118.41, 114.25, 112.13, 110.03, 55.40. MALDI-TOF MS (m/z): 921.795.
:1) to give the new compound as a white solid (yield 99.4%, 0.884 g). Mp: decomposes >300 °C. 1H NMR (600 MHz, CDCl3) δ [ppm]: 8.62 (br s, 1H), 8.25–8.44 (m, 6H), 7.40–7.82 (m, 20H), 7.04 (d, J = 8.80 Hz, 8H), 3.89 (s, 12H). 13C NMR (100 MHz, CDCl3) δ [ppm]: 158.66, 146.60, 141.42, 139.33, 134.73, 130.06, 128.30, 126.11, 125.35, 123.82, 120.44, 119.70, 118.41, 114.25, 112.13, 110.03, 55.40. MALDI-TOF MS (m/z): 921.795.
3,3′′,6,6′′-Tetra-tert-butyl-9′H-9,3’:6′,9′′-tercarbazole. To a solution of compound 10 (1.39 g, 1.59 mmol) in 10 ml tetrahydrofuran, dimethyl sulfoxide (4.8 ml) and water (1.6 ml) were added, and the mixture was stirred for 10 min. Subsequently, potassium hydroxide (1.59 g, 0.03 mol) was added to this mixture, and the reaction mixture was refluxed for 4 hours, cooled to room temperature and diluted with water. After neutralization with HCl solution, the crude product was filtered and recrystallized from dichloromethane–hexane (1
:1) to give the known compound 12 as a white solid (yield 99.5%, 1.14 g). Mp: decomposes >300 °C. 1H NMR (600 MHz, CDCl3) δ [ppm]: 8.52 (br s, 1H), 8.18 (d, J = 1.65 Hz, 6H), 7.70 (d, J = 8.16 Hz, 2H), 7.63 (dd, J = 8.48, 1.88 Hz, 2H), 7.47 (dd, J = 8.57, 1.65 Hz, 4H), 7.33 (d, J = 8.07 Hz, 4H), 1.48 (s, 36H). 13C NMR (100 MHz, CDCl3) δ [ppm]: 142.50, 140.19, 139.07, 125.97, 124.11, 123.54, 123.07, 119.43, 116.18, 111.84, 109.11, 102.16, 34.73, 32.05. MALDI-TOF MS (m/z): 721.654.
Photophysical properties
The UV-visible and photoluminescence emission spectra of the compounds in solutions of dichloromethane (CH2Cl2) at room temperature were obtained using a Shimadzu UV-Vis spectrophotometer (UV-2600) and a fluorescence spectrophotometer (Cary, Eclipse), respectively. For triplet energy measurement, thin film samples were prepared via drop-casting onto a glass substrate by heating at 60 °C from chloroform solution containing hosts at a concentration of 40 mg ml−1. The samples were attached to the sample holder in a nitrogen cryostat (Janis). The samples were excited at 350 nm by a 150 fs pulsed Ti:sapphire laser (Spectra-Physics Hurricane i) at a 1 kHz repetition rate in conjunction with an ultrafast optical parametric amplifier (Quantronix TOPAS). The luminescence from the film was allowed to pass through a long pass filter (395 nm) and a monochromator (Princeton Instruments Acton SP 2150) and then to a gated intensified CCD camera (Andor ICCD 334T). The intensified CCD was operated synchronously with a gate delay of 100 μs for all samples and a detection window width of 850 μs. Measurements were carried out in a cryostat under a vacuum of 10−7 Torr at 77 K for all samples.Computational calculations
Geometry optimizations were performed at the B3LYP/6-31G** level of theory. Iso-chemical-shielding surfaces (ICSSs) were easily constructed by employing the Nucleus-Independent Chemical Shift (NICS) concept of Paul von Rague Schleyer. In this process, a lattice of “ghost atoms” surrounding the π electron system located in the center of a virtual cube were calculated by employing the Gauge-Independent Atomic Orbital (GIAO) perturbation method25 as implemented in Gaussian 09 using the same basis set. The resulting data sets were transformed into a contour file of ICSSs using a wavefunction analysis program, Multiwfn.26 The GIAO perturbation was performed using an Intel core i7 6700K system overclocked at 4.4 GHz with 16GB RAM. For triplet energy calculations, density functional theory (DFT) as implemented in the Terachem 1.9 software package27 was used. The Becke three parameter hybrid exchange-correlation functional (B3LYP) with increasingly large basis sets was used until the molecule was optimized at the 6-311G** basis set. Unrestricted Hartree Fock theory was used to obtain the triplet energy of the molecules, since spin coupling in time-dependent DFT has not yet been implemented in Terachem 1.9. The ground state and triplet energy calculations were performed using a GPU server that had 64 GB RAM installed to support eight Tesla K10 graphic cards.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the University of Malaya Chancellery High Impact Research Grant (UM.C/625/1/HIR/208) and the Postgraduate Research Grant (PPP PG181-2014B). We thank ItraMAS Corporation. Sdn. Bhd for partial support of the research. We would like to thank Dr Liew Chee Sun from the Faculty of Computer Science and Information for the support with the computing facility.Notes and references
- G. Malloci, G. Cappellini, G. Mulas and A. Mattoni, Chem. Phys., 2011, 384, 19–27 CrossRef CAS.
- (a) A. Ganguly, J. F. Zhu and T. L. Kelly, J. Phys. Chem. C, 2017, 121, 9110–9119 CrossRef CAS; (b) L. L. Wang, Y. Zhang, N. Yin, Y. Lin, W. Gao, Q. Luo, H. W. Tan, H. B. Yang and C. Q. Ma, Sol. Energy Mater. Sol. Cells, 2016, 157, 831–843 CrossRef CAS; (c) Z. Q. Wan, C. Y. Jia, Y. Wang and X. J. Yao, ACS Appl. Mater. Interfaces, 2017, 9, 25225–25231 CrossRef CAS PubMed; (d) J. M. Huang, K. Nakano, K. Suzuki, Y. J. Chen, F. J. Wang, T. Koganezawa and K. Tajima, Macromolecules, 2017, 50, 3557–3564 CrossRef CAS.
- (a) A. Tsurusaki, A. Kobayashi and S. Kyushin, Asian J. Org. Chem., 2017, 6, 737–745 CrossRef CAS; (b) H. H. Li, L. J. Xu, Y. T. Tang, Y. Tao, S. Xu, C. Zheng, G. C. Xing, X. H. Zhou, W. Huang and R. F. Chen, J. Mater. Chem. C, 2016, 4, 10047–10052 RSC; (c) X. Y. Liu, F. Liang, L. S. Cui, X. D. Yuan, Z. Q. Jiang and L. S. Liao, Chem.–Asian J., 2015, 10, 1402–1409 CrossRef CAS PubMed.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletete, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS PubMed.
- (a) N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2995 CrossRef; (b) N. Blouin and M. Leclerc, Acc. Chem. Res., 2008, 41, 1110–1119 CrossRef CAS PubMed; (c) Y. P. Zou, D. Gendron, R. Badrou-Aich, A. Najari, Y. Tao and M. Leclerc, Macromolecules, 2009, 42, 2891–2894 CrossRef CAS; (d) H. Liu, G. Cheng, D. H. Hu, F. Z. Shen, Y. Lv, G. N. Sun, B. Yang, P. Lu and Y. G. Ma, Adv. Funct. Mater., 2012, 22, 2830–2836 CrossRef CAS.
- (a) C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048 CrossRef CAS; (b) X. Ren, J. Li, R. J. Holmes, P. I. Djurovich, S. R. Forrest and M. E. Thompson, Ultrahigh Energy Gap Hosts in Deep Blue Organic Electrophosphorescent Devices, Chem. Mater., 2004, 16, 4743–4747 CrossRef CAS; (c) M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS; (d) R. J. Holmes, S. R. Forrest, Y. J. Tung, R. C. Kwong, J. J. Brown, S. Garon and M. E. Thompson, Appl. Phys. Lett., 2003, 82, 2422–2424 CrossRef CAS; (e) S. R. Forrest, Org. Electron., 2003, 4, 45–48 CrossRef.
- S. O. Jung, Y. H. Kim, S. K. Kwon, H. Y. Oh and J. H. Yang, Org. Electron., 2007, 8, 349–356 CrossRef CAS.
- (a) C. Fan, Y. H. Chen, P. Gan, C. L. Yang, C. Zhong, J. G. Qin and D. G. Ma, Org. Lett., 2010, 12, 5648–5651 CrossRef CAS PubMed; (b) M. Kim, S. K. Jeon and J. Y. Lee, RSC Adv., 2015, 5, 100378–100383 RSC.
- F. Feixas, E. Matito, J. Poater and M. Sola, Chem. Soc. Rev., 2015, 44, 6434–6451 RSC.
- H. K. Wei and A. Ariffin, Russ. J. Phys. Chem. A, 2016, 90, 2590–2599 CrossRef.
- Z. A. Hasan, K. L. Woon, W. S. Wong, A. Ariffin and S. A. Chen, J. Lumin., 2017, 183, 150–158 CrossRef CAS.
- M. E. El-Khouly, S. H. Lee, K. Y. Kay and S. Fukuzumi, New J. Chem., 2013, 37, 3252 RSC.
- (a) K. Y. Wang, C. Chen, J. F. Liu, Q. Wang, J. Chang, H. J. Zhu and C. Li, Org. Biomol. Chem., 2012, 10, 6693–6704 RSC; (b) A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402–428 CrossRef.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS PubMed.
- Z. F. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- K. Erich, K. Sabrina and K. Andreas, THEOCHEM, 2007, 811, 45–60 CrossRef.
- M. Kurahashi, M. Fukuyo, A. Shimada and I. Nitta, Bull. Chem. Soc. Jpn., 1969, 42, 2174–2179 CrossRef CAS.
- K. E. Horner and P. B. Karadakov, J. Org. Chem., 2013, 78, 8037–8043 CrossRef CAS PubMed.
- M. Solà, Front. Chem., 2013, 1, UNSP 22 Search PubMed.
- K. L. Woon, Z. N. Nadiah, Z. A. Hasan, A. Ariffin and S. A. Chen, Dyes Pigm., 2016, 132, 1–6 CrossRef CAS.
- K. L. Woon, A. H. Zainal, B. K. Ong, A. Ariffin, R. Griniene, S. Grigalevicius and S. A. Chen, RSC Adv., 2015, 5, 59960–59969 RSC.
- (a) L. Li, R. G. Clevenger, L. Jin, K. V. Kilway and Z. H. Peng, J. Phys. Chem. C, 2016, 120, 841–852 CrossRef; (b) K. L. Woon, M. P. Aldred, P. Vlachos, G. H. Mehl, T. Stirner, S. M. Kelly and M. O’Neill, Chem. Mater., 2006, 18, 2311–2317 CrossRef CAS.
- P. Mathias, S. Dage and K. Andreas, J. Phys. Chem. C, 2012, 116, 15203–15217 Search PubMed.
- R. P. Madhu, P. M. Sean and K. Michael, J. Chem. Phys., 1956, 24, 588–594 CrossRef.
- (a) J. R. Ditchfield, Mol. Phys., 1974, 27, 789 CrossRef; (b) J. P. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, J. Chem. Phys., 1996, 104, 5497 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- I. S. Ufimtsev and T. J. Martinez, J. Chem. Theory Comput., 2009, 5, 2619–2628 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00674a |
| This journal is © The Royal Society of Chemistry 2018 |