
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePtO2/PTSA system catalyzed regioselective hydration of internal arylalkynes bearing electron withdrawing groups†
Hsin-Ping Lin,
Nada Ibrahim,
Olivier Provot ,
Mouad Alami* and
Abdallah Hamze*
,
Mouad Alami* and
Abdallah Hamze*
BioCIS, Univ. Paris-Sud, CNRS, équipe Labellisée Ligue Contre le Cancer, Université Paris-Saclay, 92290, Châtenay-Malabry, France. E-mail: abdallah.hamze@u-psud.fr; mouad.alami@u-psud.fr
First published on 23rd March 2018
Abstract
A highly efficient PtO2/PTSA catalyst system for the hydration of a wide array of alkynes was developed. This method proved to be compatible with a large range of functional groups and the ketone products were obtained in high yields. The scope of this methodology was also extended to the synthesis of 3-aryl-isochromenones, -indoles and -benzofurans.
1 Introduction
Alkyne, which is one of the widely used building blocks in organic synthesis constitutes the toolbox of organic chemists, biochemists, and materials scientists.1 Acetylene chemistry has driven the development of numerous methodologies and various synthetic procedures for many products such as ketones, enynes, and metal acetylides of significant interest to both synthetic and medicinal chemists.2 Among these important applications, the hydration of alkynes is one of the most straightforward and largely studied reactions for the synthesis of carbonyl compounds.3 Since these reactions require a long time to be catalyzed under acidic conditions, they are suitable only for electron-rich alkynes.4 The toxic mercury salt has served as a catalyst for the addition of water and alcohol to unactivated alkynes.5 Alternative and less harmful Au (III) and Pt (II) salts have been used, but they have turned out to be less efficient.6 Regarding the substantial importance of this transformation, the Teles et al. and Tanaka et al. have separately reported the first and very efficient method for hydration of alkynes using cationic gold (I) complexes of [(PR3)Au]+ type.7 Nowadays, a variety of catalytic systems including ruthenium,8 rhodium,9 iridium(III),10 and palladium11 have been well developed for these transformations and have been shown to perform the reaction at different levels of success; among these, gold might be considered by now as the standard catalyst for terminal and internal alkynes hydration with high degree of efficiency.7b,12 However, gold catalyzed hydration of alkynes has some shortcomings, for example, using high acid concentration and relatively high catalyst loadings. Recently, developing bulky N-heterocyclic carbene gold (I) chloride complexes enabled hydration not only under acid-free conditions7b but also at low catalyst loadings (<10 ppm).12a Nevertheless, the reaction required high temperature to proceed. In addition to the aforementioned, regioselectivity is an important feature to be considered; for example, the hydration of terminal alkyne occurred via Markovnikov-type addition, while with unsymmetrical internal alkynes, only moderate regioselectivities were obtained.A recent study showed the regiochemistry in these reactions with a set of examples highlighting the challenges and survey of some of the strategies engaged to address this problem, which has remained a major concern up to now.13 We have no doubt about the great advances achieved in this research area, but we believe that there is still room for improvement, principally in the area of the regioselective hydration and of internal alkynes bearing electron withdrawing groups.
In these regards, our group reported previously the use of PTSA in refluxing alcoholic media for the hydration of electron-rich arylalkynes.14 As expected, no reaction was observed in the presence of electron withdrawing groups (unpublished results).15 With the aim of developing a general catalytic system for carbon–carbon triple bond activation we demonstrated that heterogeneous platinum oxide is a competent catalyst for hydrosilylation of unsymmetrical internal arylalkynes.16 Depending on the source of the solvents used, we observed by gas chromatography some traces of carbonyl compounds in the crude mixture, particularly when solvents were not completely dry, setting off clearly from the water addition to alkyne. We have paid a very close attention to this reaction since hydration of activated alkyne proceeded well with Pt (II)6a salts or Pt(IV)17 under carbon monoxide pressure (200 psi of CO) system but not with the platinum oxide as far as we know.
We wondered whether the catalytic activity of PtO2 would achieve hydration of internal alkynes bearing EWG in a regioselective manner and relatively low catalyst loading. Moreover, we tended to consider the catalysis from an economical point of view with regards to recycling the catalyst and making scalable reactions and finally expanding the reaction to the synthesis of useful heterocycles such as lactones, furans and indoles. In this study, we report that the use of PtO2/PTSA combination in MeOH/H2O serves as a general catalytic system for alkyne hydration with an improvement in activity for diarylalkynes irrespective of the electronic nature of the substituents (electron rich or poor) and regardless of their position on the aromatic ring (ortho, meta, para).
2 Results and discussion
As previously mentioned, we started our optimization studies on the hydration of diphenylacetylene 1a by relying on our previous finding that PTSA promotes water addition-type reactions. For the initial conditions, PTSA (10 mol%) with refluxing in EtOH or MeOH was found to be inactive for the hydration of 1a (Table 1, entry1). The study of the reaction mechanism (vide infra) revealed that water would be required for the hydrolysis of the reaction intermediate; hence, we subsequently investigated the addition of water to the reaction mixture. Indeed, addition of water to the methanolic solution did not improve the yield (entry 2). The reaction between 1a and PtO2 (5 mol%) in MeOH led to the formation of the desired product in a low yield (5%, entry 3). Performing this reaction in the presence of PtO2 (5 mol%) and with catalytic amount of PTSA (10 mol%) in MeOH produced the desired compound 2a in a moderate yield (42%, entry 4). Next, we found that the combination of PtO2/PTSA/MeOH in the presence of H2O had a drastic effect on the reaction efficiency and compound 1a was quantitatively converted to ketone 2a (entry 5). Decreasing the temperature of the reaction from 90 °C to 60 °C led to a slight decrease in the yield (98% vs. 80%). Furthermore, we investigated the use of other Brønsted acid sources such as H2SO4 (90%), which gave a close yield to PTSA (entry 6). However, in the presence of CF3COOH (entry 7) or HCOOH (entry 8), a low yield of 2a was obtained as compared to the most effective Brønsted acid (PTSA). The nature of the solvent significantly affected the hydration reaction since the use of EtOH or dioxane instead of MeOH considerably decreased the yield (entries 9–10). As PtO2 proved to be the most effective catalyst for this transformation, a variety of platinum catalysts were evaluated. Speier's catalyst (H2PtCl6) and platinum(II) acetylacetonate Pt(acac)2 gave moderate and low yields, respectively (entries 11–12). However, a good yield was obtained with PtCl2 (entry 13). Most of the starting material 1a was recovered with palladium, iron, copper, and cobalt catalysts (entries 14–17) and a low yield of ketone 2a was obtained even with a prolonged reaction time when silver-catalyzed (AgBF4) was used (entry 18).18 Next, we examined the hydration of alkyne 1a under commercial AuI-catalyst system (entries 19–20). No reaction occurred when 1,2-diphenylethyne 1a was used as the substrate under cationic gold(I) species [(IPr)AuCl] as described by Li et al.19 in the presence of MeOH/H2O at 110 °C (entry 18). As with stable gold complex (Ph3P)AuCH3, 2a was obtained only in a 53% yield.7b In particular, with longer reaction time (12 h), we were able to decrease the catalyst loading of PtO2 to 1 mol% with minor effect on the yield (Table 1, entry 21).
|
|||||
|---|---|---|---|---|---|
| Entry | [M] (X mol%) | Additive (10 mol%) | Solvent | H2O (equiv.) | Yieldb (%) |
| a Reaction conditions: [catalyst] X mol%, PTSA 10 mol%, alkyne 1 mmol, H2O (3.3 equiv.), MeOH (2.2 mL), 90 °C in a sealed tube.b Isolated yield of 2a.c Performing the reaction at 60 °C furnished ketone 2a in 80% yield.d The reaction was performed overnight.e Conditions from Li et al.,19 the reaction was performed at 110 °C, 6 h.f Conditions from Tanaka et al.,7b reaction was performed at 70 °C, 5 h.g Performing the reaction at a gram scale (1.87 g) gave 2a in 80% isolated yield. | |||||
| 1 | — | PTSA | MeOH | — | 0 |
| 2 | — | PTSA | MeOH | 3.3 | 0 |
| 3 | PtO2 (5) | — | MeOH | 3.3 | 5 |
| 4 | PtO2 (5) | PTSA | MeOH | — | 42 |
| 5c | PtO2 (5) | PTSA | MeOH | 3.3 | 98 |
| 6 | PtO2 (5) | H2SO4 | MeOH | 3.3 | 90 |
| 7 | PtO2 (5) | CF3COOH | MeOH | 3.3 | 30 |
| 8 | PtO2 (5) | HCOOH | MeOH | 3.3 | 15 |
| 9 | PtO2 (5) | PTSA | EtOH | 3.3 | 40 |
| 10 | PtO2 (5) | PTSA | Dioxane | 3.3 | 15 |
| 11 | H2PtCl6 (5) | PTSA | MeOH | 3.3 | 65 |
| 12 | Pt(acac)2 (5) | PTSA | MeOH | 3.3 | 25 |
| 13 | PtCl2 (5) | PTSA | MeOH | 3.3 | 91 |
| 14d | PdCl2 (5) | PTSA | MeOH | 3.3 | 0 |
| 15 | FeCl3 (5) | PTSA | MeOH | 3.3 | 0 |
| 16 | Cu(OTf)2 (5) | PTSA | MeOH | 3.3 | 0 |
| 17 | CoBr2 (5) | PTSA | MeOH | 3.3 | 0 |
| 18e | AgBF4 (5) | PTSA | MeOH | 3.3 | 40 |
| 19 | (IPr)AuCl (1) | — | MeOH/H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
0 | |
| 20f | (Ph3P)AuCH3 (1) | H2SO4 | MeOH | 3.3 | 53 |
| 21d,g | PtO2 (1) | PTSA | MeOH | 3.3 | 88 |

In addition, the robustness of the catalytic conditions is demonstrated through gram scale synthesis of the hydration product 2a and reaction of 1a with the new catalytic system (entry 18) was successfully completed on a 1.87 g scale (10.5 mmol), giving rise to 2a with 80% yield.
As the reusability and the recovery of the catalyst are very important issues, we then studied the recycling of PtO2 in the hydration reaction of diphenylacetylene 1a (Fig. 1). For this, after each run, the MeOH/water solvent was directly evaporated; then, hexane was added at room temperature and the medium was stirred for 20 min. After sedimentation of the solid, the liquid was poured out and the remaining solid catalyst was washed with hexane twice, dried and reused. PtO2 was used four times without any significant decrease of the catalytic activity.
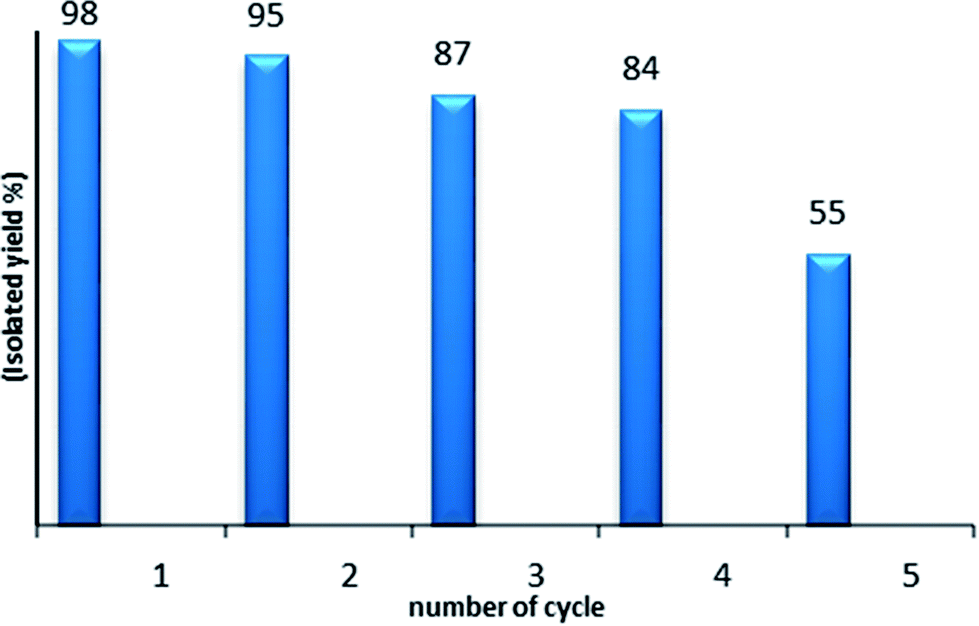 | ||
| Fig. 1 Recycling of PtO2: the hydration of diphenylacetylene 1a catalyzed by PtO2 (5 mol%), PTSA 10 mol% in MeOH (2.2 mL), H2O (3.3 equiv.). | ||
Next, we explored the scope of this reaction with various dissymmetrical alkynes (Table 2). Alkynes having EWGs on para position such as NO2, CN, and CF3 were efficiently converted to their corresponding ketones with complete β-regioselectivity (entries 1–2 and 4). However, in the case of alkyne substituted with a para ester (CO2Me), a mixture of two separable regioisomers (80:20) was obtained, in which the β-regioisomer predominated (entry 3).
|
||
|---|---|---|
| Entry | Conditions | |
| PtO2 X mol%/PTSAa 10 mol%, yieldc | PTSAb 10 mol%, yieldc | |
| a Reactions conditions: PtO2 X mol%, PTSA 10 mol%, alkyne 1 mmol, H2O (3.3 equiv.), MeOH (2.2 mL), 90 °C in a sealed tube.b Same conditions as before but without the addition of PtO2.c Isolated yield of product 2.d NR: no reaction was observed without the addition of PtO2.e Reaction was carried out at 130 °C.f Obtained as separable 80/20 mixture with the other α-regioisomer.g Obtained as separable 95/5 mixture with the other α-regioisomer.h Reaction without PtO2 needs heating under MWI at 150 °C and the addition of 1 equiv. of PTSA. | ||
| 1 | 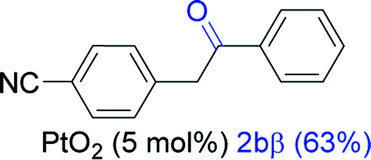 |
NRd |
| 2e | 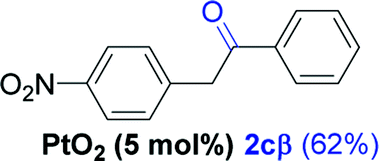 |
NRd |
| 3f | 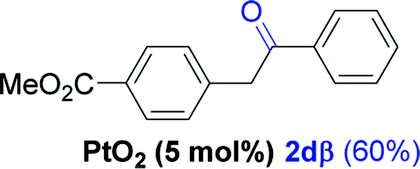 |
NRd |
| 4 | 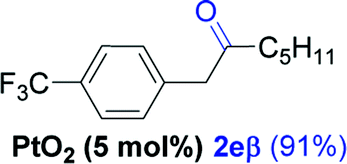 |
NRd |
| 5g,e | 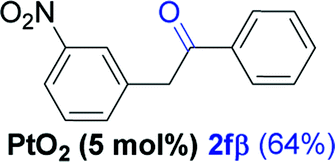 |
NRd |
| 6 | 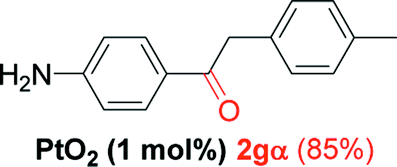 |
 |
| 7 | 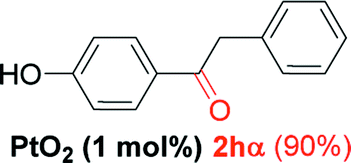 |
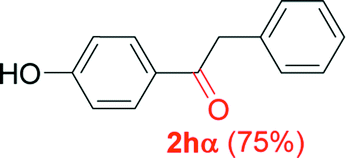 |
| 8e | 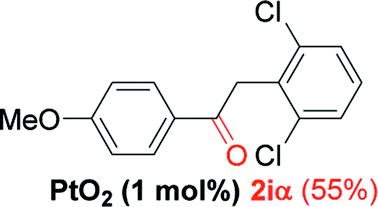 |
NRd |
| 9e | 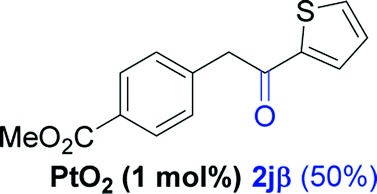 |
NRd |
| 10 | 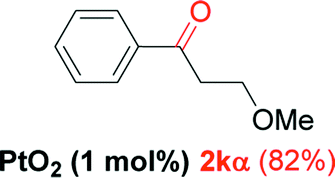 |
NRd |
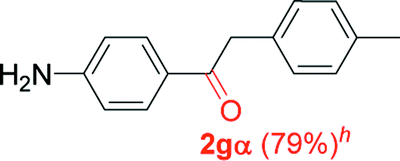
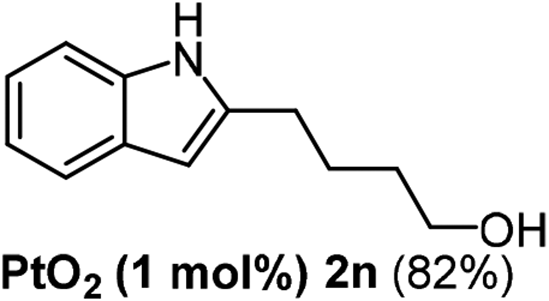
Alkyne having a meta nitro group was successfully regioselectively hydrated into the corresponding ketone 2fβ with good yield (entry 5). In comparison to 1,2-diphenylethyne, the hydration of the electron-poor alkynes (entries 1–5) requires the addition of 5 mol% PtO2 to occur. Next, we applied our conditions with electron-rich internal alkynes. Alkynes having free amino or hydroxyl group in para-position were successfully converted into the corresponding α-ketone derivatives in excellent yields with only (1 mol%) of PtO2 (entries 6–7). It is important to note for compound 2gα that without the use of PtO2, the reaction needs heating under MWI at 150 °C using 1 equiv. of PTSA.15a Performing the hydration of push–pull alkyne led exclusively to the formation of α-regioisomer 2iα (entry 8). In addition, our conditions were found to be efficient for the hydration of heteroaryl-alkyne; the β-ketone 2jβ was obtained with moderate yield (50%, entry 9). Finally, arylalkyl alkyne was easily hydrated under our conditions to give 2kα as a single isomer with 82% yield (entry 10). Furthermore, these results were reproducible as summarized in Table 2, which represents the yields obtained from three parallel experiments.
2.1 Mechanism discussion
The PTSA-catalyzed hydration of alkyne requires the presence of electron-donating substituent in para or in ortho-position (Scheme 1). The methoxy-group in para-position could lead to an allene intermediate, followed by regioselective addition of ethanol to afford an enol form, which rearranges into ketone form after hydrolysis. Thus, terminal alkynes as well as non-activated arylalkynes (EDG) did not react under the reaction conditions using only p-toluenesulfonic acid as the catalyst.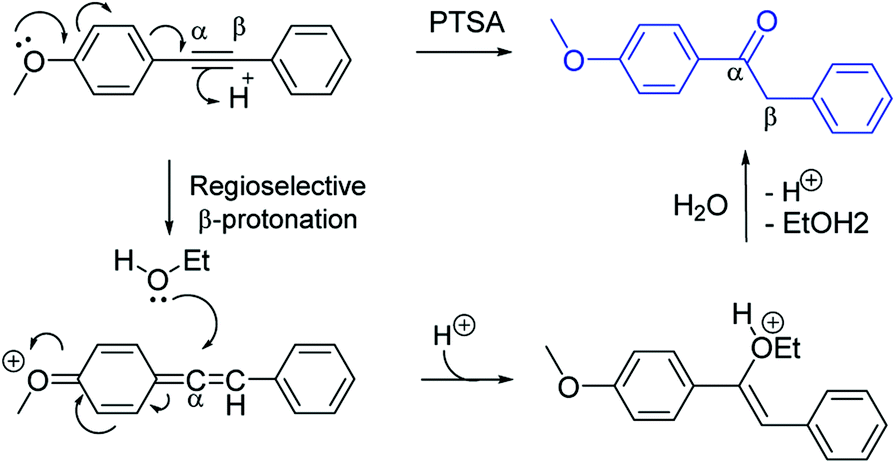 | ||
| Scheme 1 Plausible mechanism for hydration of electron rich 1,2-diphenylethyne with PTSA. | ||
A possible reaction mechanism is proposed to account for the hydration of arylalkynes catalyzed by PtO2/PTSA combination (Scheme 2). Activation of the triple bond can be explained by the formation of π-complex (I) between platinum catalyst and the triple bond, followed by the regioselective addition of MeOH (species II). PTSA/H2O catalyzed proto-demetallation and led to enol form (III), which rearranged into keto form 2.
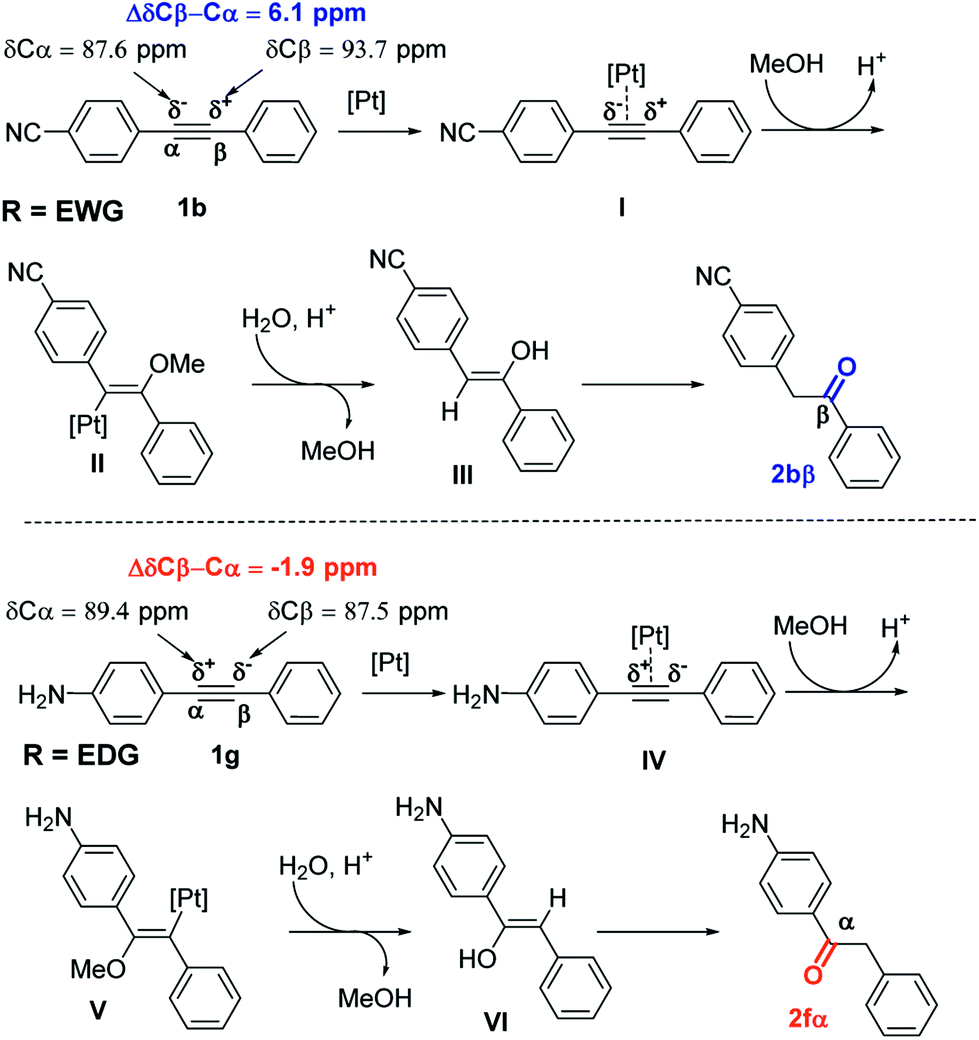 | ||
| Scheme 2 Mechanism of hydration of arylalkynes with platinum/PTSA catalyst system. | ||
The regioselectivity of hydration of dissymmetrical alkynes depends on the nature of the substituent of the aromatic ring, which will induce polarization of the triple bond.
Analysis of 13C NMR chemical shifts of sp-carbon atoms of alkyne 1 can provide a good approximation for electronic polarization of para-alkyne derivatives. Indeed, for estimation of the electronic effects for conjugated systems, analysis of 13C NMR chemical shifts was routinely used.20 The presence of EWG such as CN substituent in para-position increases the difference in the 13C NMR chemical shift of the signal arising from the ΔδCβ–Cα atom from 0 ppm (R = H, diphenylacetylene) to 6.1 ppm (Scheme 2). A similar situation was observed with other EWG substituents such as NO2 or CF3.21 Accordingly, substituents on para-position such as CN, NO2, and CF3 polarize the triple bond in the same way, making the α-sp-carbon more electron-rich and the β-sp-carbon more electron-deficient. The catalytic cycle begins with the formation of Pt-π-alkyne complex I by coordination between alkyne 1b and the platinum catalyst. Nucleophilic attack by PTSA on complex I led to the formation of intermediate II. Then, intermediate II evolved to enol III by protodemetalation in the presence of water in acidic media. Finally, isomerization of enol III produced the ketone 2bβ. As the reaction was performed in MeOH/water mixture, enol III can also be formed by the hydrolysis of vinyl ether intermediate, which can be obtained from the reaction between MeOH and intermediate II.
The presence of EDGs in para-position such as NH2 induced an inversion of the polarization of the carbon–carbon triple bond (the Cα atom becomes more electron-deficient than the Cβ atom). This resulted in the change of sign of ΔδCβ–Cα values, which become negative (ΔδCβ–Cα = −1.9 ppm, Scheme 2). This can explain the inversion of the hydration regioselectivity in the case of para-EDG substituents.
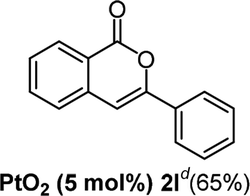
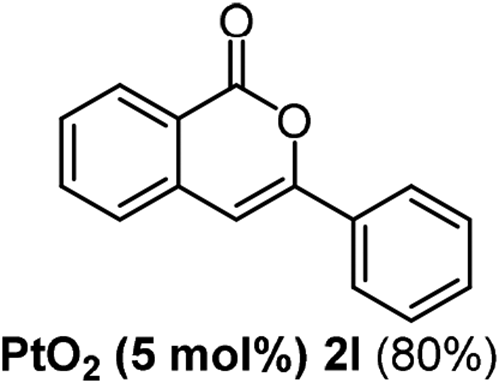
We next examined the synthesis of an important class of heterocyclic compounds under our standard conditions. Thus, cylization of ortho-substituted diarylalkynes proceeded well (Table 3) at 90 °C. Diarylalkynes bearing an ortho-cyano substituent on the aromatic ring 1l provided the cyclized 3-phenyl-isochromen-1-one 2l with low isolated yield (30%). Increasing the temperature of the reaction to 130 °C led to a significant increase in the formation of cyclic product 2l in a good overall yield of 65% (Table 3, entry 1). As expected reaction with alkyne 1m bearing an ortho-ester group (entry 2) gave again the same 3-phenyl-isochromen-1-one 2l in good yield.
|
|||
|---|---|---|---|
| Entry | Alkyne 1 | Conditions | |
| PtO2 Xa mol%, PTSA 10 mol%, yieldc | —b, PTSA 10 mol%, yieldc | ||
| a Reactions conditions: PtO2 X mol%, PTSA 10 mol%, alkyne 1 mmol, H2O (3.3 equiv.), MeOH (2.2 mL), 90 °C in a sealed tube.b Same conditions as before but without the addition of PtO2.c Isolated yield of product 2.d Reaction was realized at 130 °C.e NR: no reaction was observed without the addition of PtO2. | |||
| 1 | 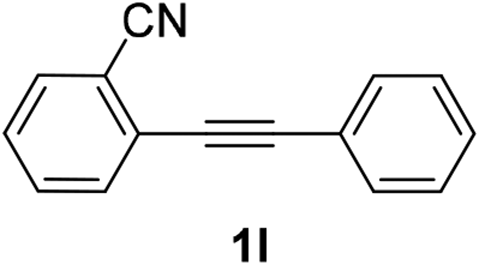 |
 |
NRe |
| 2 | 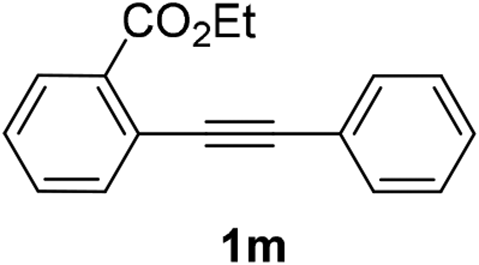 |
 |
NRe |
| 3 | 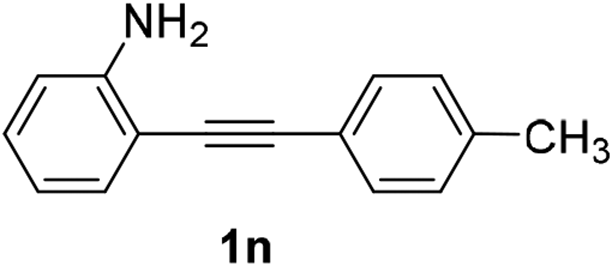 |
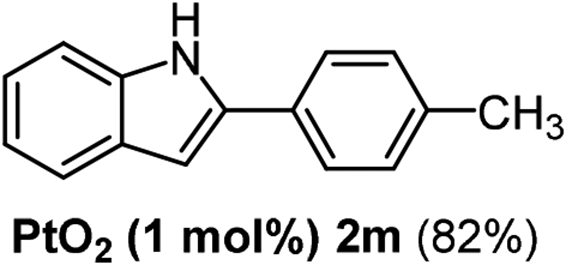 |
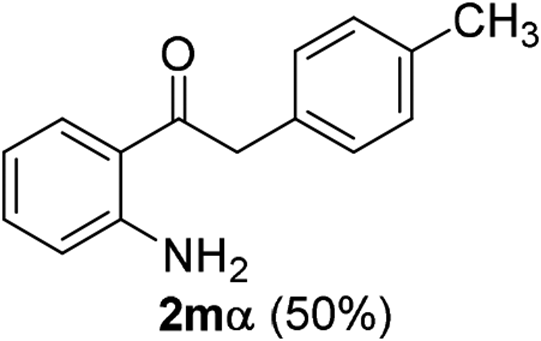 |
| 4 | 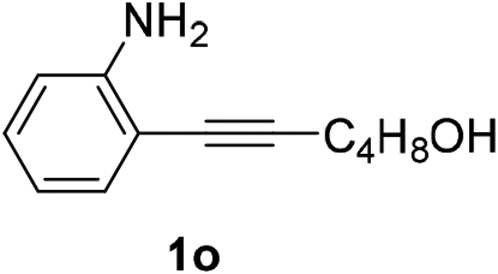 |
 |
 |
| 5 | 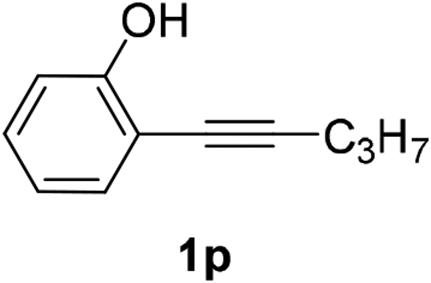 |
 |
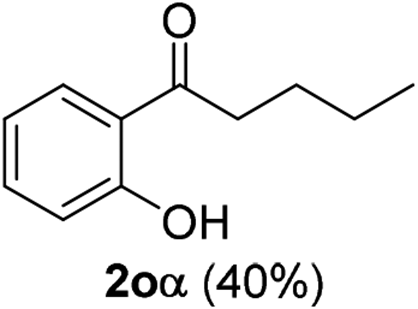 |
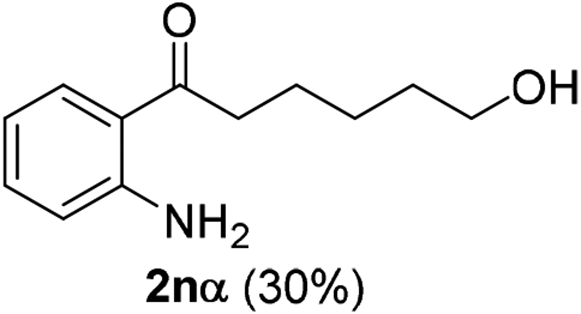
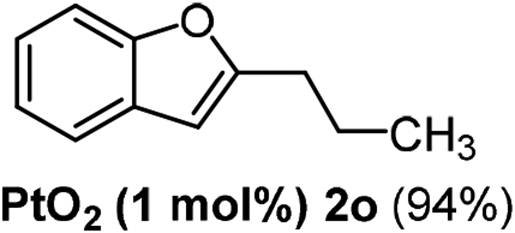
The scope of this cyclization was further examined with a variety of ortho-EDG-substituted diarylalkynes. Substrates bearing an ortho-amino group were successfully transformed to the corresponding indoles derivatives 2m–n in good yields (entries 3–4). In the absence of PtO2, reaction of aniline derivatives (entries 3–4) results in the formation of the hydration products 2mα and 2nα. Starting the reaction from ortho-phenol alkyne 1p leads to the formation of benzofuran derivative 2o in 94% yield (entry 5), while on using only PTSA, product 2oα was obtained.
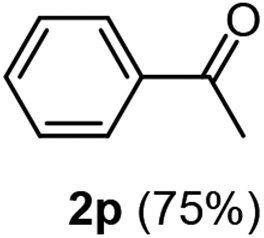
Having succeeded in developing an efficient hydration process of electron deficient diarylalkynes, we next examined this protocol with terminal alkynes so as to compare this system to previously reported catalytic systems (Table 4). We were however delighted to see a successful hydration at 1 mol% of PtO2, regardless of the electronic nature of the terminal alkynes. Thus, hydration of ethynylbenzene derivatives having electron-donating or electron-withdrawing groups efficiently proceeded to afford the corresponding ketones in good to excellent yields. Also, aryl alkynes having a methoxyl group in meta-position of the aryl ring reacted well and furnished the acetophenone derivatives 2q in good yields. Additionally, terminal alkyne having a heterocyclic aromatic substituent such as thiophene reacts well under our standard conditions to afford the hydration product 2t in good yield (62%).
|
|||
|---|---|---|---|
| Entry | Alkyne 1 | Conditions | |
| PtO2 1a mol%, PTSA 10 mol%, yieldc | —b, PTSA 10 mol%, yieldc | ||
| a Reaction conditions: PtO2 1 mol%, PTSA 10 mol%, alkyne 1 mmol, H2O (3.3 equiv.), MeOH (2.2 mL), 90 °C in a sealed tube.b Same conditions as before but without the addition of PtO2.c Isolated yield of product 2.d NR: no reaction was observed without the addition of PtO2. | |||
| 1 |  |
 |
NRd |
| 2 | 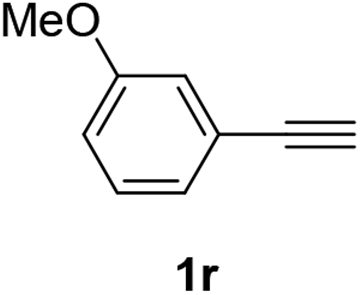 |
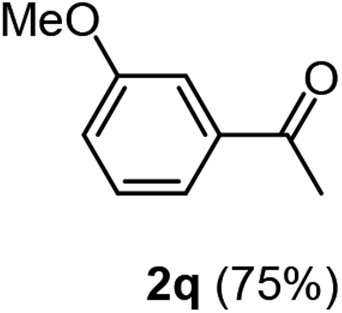 |
NRd |
| 3 | 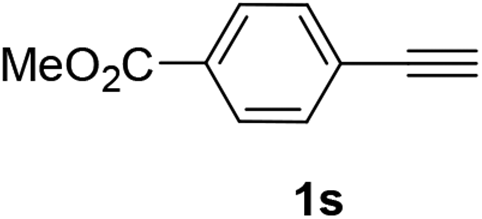 |
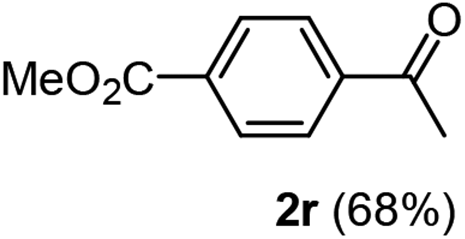 |
NRd |
| 4 | 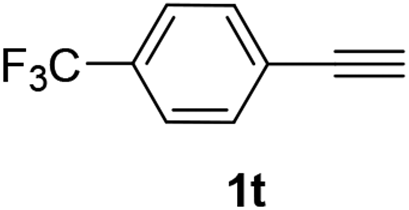 |
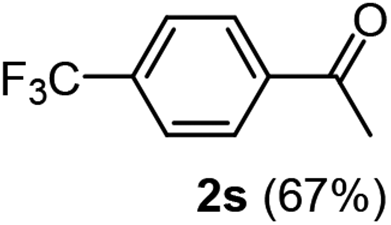 |
NRd |
| 5 | 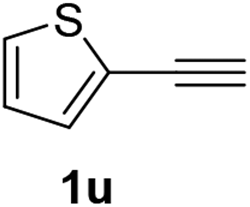 |
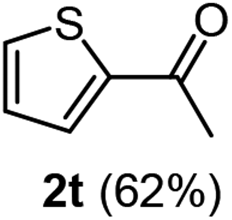 |
NRd |
| 6 |  |
 |
NRd |
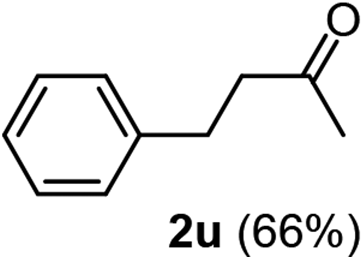
The latter substrate (2t) was studied under PtCl4–CO catalytic system developed by Blum et al.22 However in their study, an unsatisfactory yield of 30% was obtained. Furthermore, our protocol was also efficient for the hydration of aliphatic terminal alkyne and furnished the corresponding ketone 2u with 66% yield.
3 Conclusions
In summary, PtO2/PTSA in MeOH/H2O proved to be a highly potent catalytic system for the transformation of non-activated internal and terminal alkynes to ketones. Performing this reaction in aqueous methanol enables the reaction to proceed smoothly and to afford excellent yields of the resultant ketones 2. Furthermore, the results are highly reproducible and the platinum catalyst is conveniently recovered. This system proved to be compatible with a large range of functionalities including nitrile, nitro, ester, amino and hydroxyl functional groups. Additionally, the application of this methodology to internal ortho-alkynes provides flexible access to phenyl-isochromenones, indoles, and benzofurans.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors gratefully acknowledge the support of this project by CNRS, Univ. Paris-Sud, as well as by La Ligue Nationale Contre le Cancer through an Equipe Labellisée 2014 grant. H.-P. L thanks the Taiwan Paris-Sud scholarship for a Ph.D. funding. Our laboratory is a member of the laboratory of excellence LERMIT supported by a grant from ANR (ANR-10-LABX-33).Notes and references
- F. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry: Chemistry, Biology, and Material Science, Wiley-VCH Verlag GmbH & Co. KGaA, 2005 Search PubMed.
- (a) D. R. Dreyer, H.-P. Jia and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 6813 CAS; (b) M. Rubina, M. Conley and V. Gevorgyan, J. Am. Chem. Soc., 2006, 128, 5818 CrossRef CAS PubMed; (c) V. P. Boyarskiy, D. S. Ryabukhin, N. A. Bokach and A. V. Vasilyev, Chem. Rev., 2016, 116, 5894 CrossRef CAS PubMed.
- (a) L. Hintermann and A. Labonne, Synthesis, 2007, 1121 CrossRef CAS; (b) M. B. Smith and J. March, in March's Advanced Organic Chemistry, John Wiley & Sons, Inc., 2006, p. 999 Search PubMed.
- A. D. Allen, Y. Chiang, A. J. Kresge and T. T. Tidwell, J. Org. Chem., 1982, 47, 775 CrossRef CAS.
- M. Kutscheroff, Ber. Dtsch. Chem. Ges., 1881, 14, 1540 CrossRef.
- (a) J. W. Hartman, W. C. Hiscox and P. W. Jennings, J. Org. Chem., 1993, 58, 7613 CrossRef CAS; (b) J. W. Hartman and L. Sperry, Tetrahedron Lett., 2004, 45, 3787 CrossRef CAS.
- (a) J. H. Teles, S. Brode and M. Chabanas, Angew. Chem., Int. Ed., 1998, 37, 1415 CrossRef CAS; (b) E. Mizushima, K. Sato, T. Hayashi and M. Tanaka, Angew. Chem., Int. Ed., 2002, 41, 4563 CrossRef CAS.
- (a) M. Tokunaga and Y. Wakatsuki, Angew. Chem., Int. Ed., 1998, 37, 2867 CrossRef CAS; (b) P. S. Mainkar, V. Chippala, R. Chegondi and S. Chandrasekhar, Synlett, 2016, 27, 1969 CrossRef CAS.
- J. Blum, H. Huminer and H. Alper, J. Mol. Catal., 1992, 75, 153 CrossRef CAS.
- S. Ogo, K. Uehara, T. Abura, Y. Watanabe and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 16520 CrossRef CAS PubMed.
- (a) Z. Zhang, L. Wu, J. Liao, W. Wu, H. Jiang, J. Li and J. Li, J. Org. Chem., 2015, 80, 7594 CrossRef CAS PubMed; (b) C. Xu, W. Du, Y. Zeng, B. Dai and H. Guo, Org. Lett., 2014, 16, 948 CrossRef CAS PubMed.
- (a) N. Marion, R. S. Ramón and S. P. Nolan, J. Am. Chem. Soc., 2009, 131, 448 CrossRef CAS PubMed; (b) J. H. Teles, in Modern Gold Catalyzed Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, p. 201 Search PubMed.
- J. A. Goodwin and A. Aponick, Chem. Commun., 2015, 51, 8730 RSC.
- (a) N. Olivi, E. Thomas, J. F. Peyrat, M. Alami and J. D. Brion, Synlett, 2004, 2175 CAS; (b) G. Le Bras, O. Provot, J. F. Peyrat, M. Alami and J. D. Brion, Tetrahedron Lett., 2006, 47, 5497 CrossRef CAS.
- (a) M. Jacubert, O. Provot, J.-F. Peyrat, A. Hamze, J.-D. Brion and M. Alami, Tetrahedron, 2010, 66, 3775 CrossRef CAS; (b) G. Le Bras, A. Hamze, S. Messaoudi, O. Provot, P.-B. Le Calvez, J.-D. Brion and M. Alami, Synthesis, 2008, 1607 CAS.
- (a) A. Hamze, O. Provot, M. Alami and J.-D. Brion, Org. Lett., 2005, 7, 5625 CrossRef CAS PubMed; (b) A. Hamze, O. Provot, J.-D. Brion and M. Alami, Synthesis, 2007, 2007, 2025 CrossRef; (c) A. Hamze, O. Provot, J.-D. Brion and M. Alami, J. Organomet. Chem., 2008, 693, 2789 CrossRef CAS.
- W. Baidossi, M. Lahav and J. Blum, J. Org. Chem., 1997, 62, 669 CrossRef CAS PubMed.
- Z.-W. Chen, D.-N. Ye, Y.-P. Qian, M. Ye and L.-X. Liu, Tetrahedron, 2013, 69, 6116 CrossRef CAS.
- F. Li, N. Wang, L. Lu and G. Zhu, J. Org. Chem., 2015, 80, 3538 CrossRef CAS PubMed.
- (a) K. Itami, K. Mitsudo, K. Fujita, Y. Ohashi and J.-i. Yoshida, J. Am. Chem. Soc., 2004, 126, 11058 CrossRef CAS PubMed; (b) B. Happ, T. Bartik, C. Zucchi, M. C. Rossi, F. Ghelfi, G. Palyi, G. Varadi, G. Szalontai, I. T. Horvath and C. Guastini, Organometallics, 1995, 14, 809 CrossRef CAS; (c) M. Alami, F. Liron, M. Gervais, J. F. Peyrat and J. D. Brion, Angew. Chem., Int. Ed., 2002, 41, 1578 CrossRef CAS.
- For more details on 13C NMR chemical shifts of actetylenic carbons of para alkynes please see the ESI part.†.
- O. Israelsohn, K. P. C. Vollhardt and J. Blum, J. Mol. Catal. A: Chem., 2002, 184, 1 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: For experimental details, copies of 1H and 13C NMR spectra for all new compounds. See DOI: 10.1039/c8ra00564h |
| This journal is © The Royal Society of Chemistry 2018 |