
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDominant formation of disulfidic linkages in the sulfur cross-linking reaction of isoprene rubber by using zinc stearate as an activator
Yuta Sakakia,
Ryota Usamia,
Atitaya Tohsanb,
Preeyanuch Junkongcd and
Yuko Ikeda *ce
*ce
aGraduate School of Science and Technology, Kyoto Institute of Technology, Matsugasaki, Sakyo, Kyoto 606-8585, Japan
bDepartment of Materials and Production Technology Engineering, Faculty of Engineering, King Mongkut's University of Technology North Bangkok, Wongsawang, Bangsue, Bangkok 10800, Thailand
cCenter for Rubber Science and Technology, Kyoto Institute of Technology, Matsugasaki, Sakyo, Kyoto 606-8585, Japan
dResearch Strategy Promotion Center, Kyoto Institute of Technology, Matsugasaki, Sakyo, Kyoto 606-8585, Japan
eFaculty of Molecular Chemistry and Engineering, Kyoto Institute of Technology, Matsugasaki, Sakyo, Kyoto 606-8585, Japan. E-mail: yuko@kit.ac.jp
First published on 19th March 2018
Abstract
A linear combination fitting in sulfur K-edge X-ray absorption near edge structure (S-XANES) measurements reveals each fraction of monosulfidic, disulfidic and polysulfidic linkages in solvent extracted sulfur cross-linked isoprene rubbers. The sulfidic linkage of a disulfidic type is found for the first time to be dominant when zinc stearate and N-(1,3-benzothiazol-2-ylsulfanyl)cyclohexanamine are used as the activator and accelerator, respectively, for the sulfur cross-linking reaction at 140 °C. The presence of the bridging bidentate zinc/stearate complex as an intermediate for the sulfur cross-linking reaction is supposed to induce the generation of abundant disulfidic linkages in the rubber networks. This unexpected observation is of use for the material design of high performance rubber products with anti-aging and thermal stabilities. S-XANES is a powerful tool that was used to reveal the characteristics of the sulfur cross-linking of rubber. These results will contribute to the development of rubber science and technology.
Introduction
It is well-known that various kinds of rubber products have been widely used for numerous applications.1–4 Since the first use of sulfur (S8) by Goodyear in 1839, vulcanization has been acknowledged as one of the traditional important reactive processes in polymer science and technology. The mechanical properties of rubber can be enhanced by introducing vulcanization, which provides the three-dimensional cross-linked network structure of the final product.5–8Regarding the current progress in analytical techniques, it has been accepted that vulcanization leads to the inhomogeneous network structure. Previously, our group reported the effects of the combination and composition of the vulcanizing reagents on the formation of the rubber network structure.9 The results obtained from the small-angle neutron scattering technique indicated that zinc oxide (ZnO) and other reagents are crucial in the control of the two-phase inhomogeneous network structure of vulcanized isoprene rubber, as illustrated in Fig. 1,9 where the network domains of high network-chain density are speculated to be embedded in the mesh network.
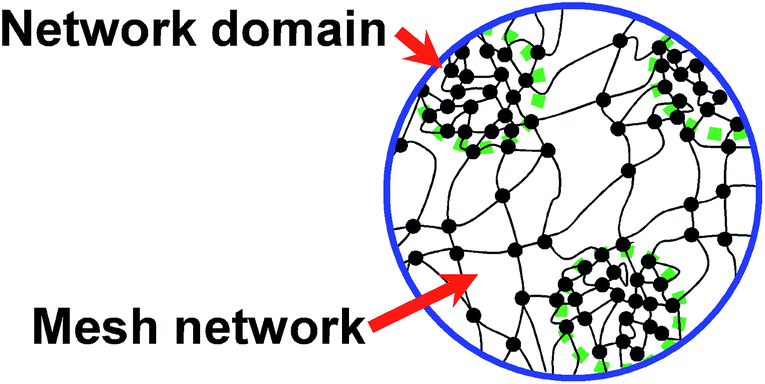 | ||
| Fig. 1 Proposed two-phase inhomogeneous network structure in sulfur cross-linked isoprene rubber. Reproduced with permission.9 Copyright 2009, American Chemical Society. | ||
The two-phase network structure was proposed to be initially formed by zinc stearate (ZnSt2) generated by a reaction between ZnO and stearic acid. The ZnSt2 was proposed to further react with the accelerator and sulfur to result in sulfur cross-links and was therefore supposed to be a key factor in controlling the size of the mesh network in the matrix.9 In contrast, sulfur and accelerator were also considered to be adsorbed on the remaining ZnO in the rubber matrix, followed by the sulfur cross-linking reaction around the ZnO particles. Therefore, the amounts of accelerator and sulfur adsorbed on the ZnO were also proposed to control the size of the network domain.9
The formation of a two-phase inhomogeneous network structure was confirmed by employing time-resolved zinc K-edge X-ray absorption fine structure (Zn-XAFS) spectroscopy and differential scanning calorimetry.10 It was detected that the formation of the mesh network occurred a little earlier than that of the network domains and its fraction was unexpectedly observed to be larger than that calculated on the basis of the concentration of ZnSt2. The result was ascribed to the amount of zinc/stearate complex intermediates forming the mesh network.10 These previous studies clearly emphasized that the compositions of vulcanizing reagents play an important role in controlling the two-phase network structure and hence the mechanical properties of vulcanized rubber.
On the other hand, it is also well-known that the compositions of vulcanizing reagents change the sequence of sulfidic linkages in the cross-linking sites in a vulcanization reaction. Particularly, it has been recognized that sulfidic linkages in vulcanizates drastically affect the mechanical properties and thermal resistance of rubber products.5,11–13 For example, a polysulfidic linkage is more easily decomposed by heat and stress than monosulfidic and disulfidic linkages, due to the lower bonding energy of the polysulfidic segment. Generally, sulfur cross-linked rubber has been believed to be composed of a mixture of monosulfidic, disulfidic and polysulfidic cross-linkages with C–C cross-linkings.11,13,14 However, the effect of curing reagents on the sulfidic linkages has not yet been satisfactorily clarified, due to the difficulty of analysis of the linkages in the vulcanizates. So far, a chemical method for the cleavage of sulfidic linkages by lithium aluminium hydride has been utilized for investigating the sulfidic structure in vulcanized rubbers.15 This method is known to not distinguish between cross-links and other sulfurated network features such as pendant accelerator residues and cyclic structures.16 Chemical analysis using reagents of thiols and amines has been used to selectively cleave disulfidic and polysulfidic cross-linked segments.16 However, the handling of these reagents is problematic due to their strong smell and dangerousness. For both chemical methods, the additional effect of the trapped entanglement has not been taken into account for the determination of each concentration of sulfidic linkages. Alternatively, sulfur K-edge X-ray absorption near edge structure (S-XANES) spectroscopy is a tool that can be used to evaluate the sulfidic linkages in vulcanizates, which allows insight into the stereochemistry and electronic state of the absorbing atom.17–22 In addition, this technique gives good structural information as a fingerprint, together with being non-destructive to the sample and it can be used with various types of samples. Synchrotron-based S-XANES measurement under high vacuum is broadly employed to investigate the sulfidic linkage for rubber materials,23–30 where the energy states at the absorption edges are determined.31 Brendebach et al. observed a variation in the cleavage of cyclic sulfur during the vulcanization of carbon black filled conventional rubber.21 Pattanasiriwisawa et al. clarified that sulfidic linkages were controlled by the ratio between the contents of sulfur and accelerator in the conventional vulcanization, effective vulcanization (EV), and semi-EV systems of natural rubber.25 Taweepreda et al. researched the effect of accelerators such as thiuram, thiazole, and dithiocarbamic acid on the sulfidic linkages in the vulcanized squalene.27 However, there has been a request for this S-XANES method to be improved for the quantitative analyses of sulfidic linkages in the vulcanizates.
In this paper, a variation of sulfidic linkages in the solvent-extracted isoprene rubber vulcanizates is intensively studied using S-XANES in order to quantitatively investigate the type of sulfidic linkage in the mesh network in Fig. 1. As a model network for the mesh, a benzothiazolesulfenamide-accelerated curing system with ZnSt2 was utilized for the investigation. An unexpected result is reported in this article, which indicates the unique characteristics of the vulcanizate. The findings are useful for producing important rubber materials with anti-aging and thermal stabilities, as predicted in the ACS News Service Weekly PressPac in 2015.32
Experimental
Sample preparation
The isoprene rubber vulcanizates (IR–ZnSt2–CBS–S8) were prepared by the heat pressing at 140 °C of their compounds obtained by the conventional milling of isoprene rubber (IR, the commercial grade: IR2200, JSR Co.) with 3.88 phr of ZnSt2 (Wako Pure Chemical Industries, Ltd.), 1.0 phr of N-(1,3-benzothiazol-2-ylsulfanyl)cyclohexanamine (Sanshin Chemical Industry Co. Ltd., Sanceler CM-G) and 1.5 phr of elemental sulfur (Hosoi Chemical Industry Co. Ltd., powder, 150 mesh) on a two-roll mill at room temperature (r.t.). All reagents were commercial grade for rubber processing and were used as received. Herein, “phr” is a conventional unit in rubber science and technology, which is the abbreviation of “parts per one hundred rubbers by weight”. In this study, the same compounding recipe used in previous studies10,33 was used to represent a model of the mesh network in the two-phase network structure shown in Fig. 1. Six flat sheets of about 0.2 mm thickness were prepared with different heat-pressing times, determined on the basis of a variation of the torque in the cure curve shown in Fig. 2. All sample codes are presented as a combination of the reagents, using their abbreviations and the heat-pressing time in minutes. Here, each reagent is abbreviated as follows; zinc stearate, N-(1,3-benzothiazol-2-ylsulfanyl)cyclohexanamine and sulfur are ZnSt2, CBS and S8, respectively.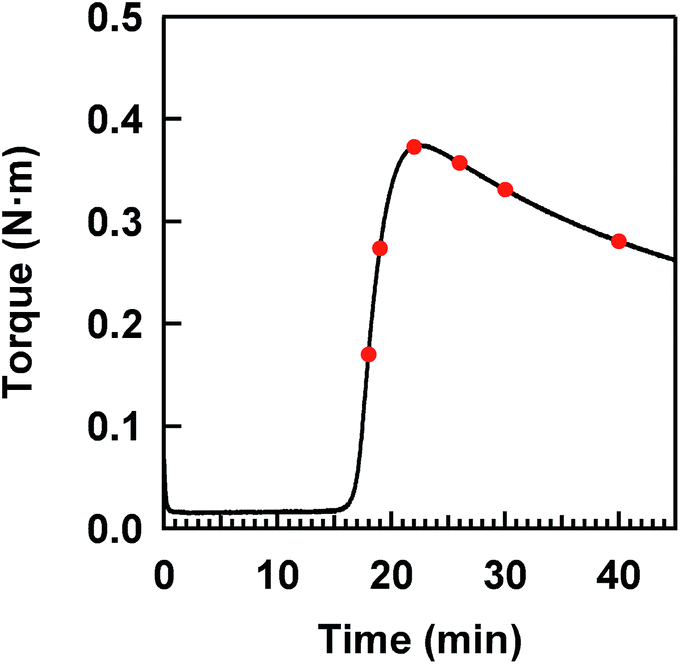 | ||
| Fig. 2 A cure curve at 140 °C for the compound of IR–ZnSt2–CBS–S8. | ||
Measurement of curing degree
The cure characteristics of IR–ZnSt2–CBS–S8 were evaluated using a Curelastometer III (JSR Co.) at 140 °C. A cure curve is illustrated in Fig. 2. According to red points in the cure curve, the adequate heat-pressing times were 18, 19, 22, 26, 30 and 40 min for preparing flat sheets of about 0.2 mm thickness.Measurement of network-chain density
The network-chain densities of the sulfur cross-linked rubbers prepared using different pressing times were calculated by the modified Flory–Rehner equation34 using the swelling results in toluene at 25 °C for 24 hours, and volume change was measured using a CCD camera (VC1000 Digital Fine Scope, OMRON Co.). The sample size for the swelling test was about 3 mm in width × about 4 mm in length × about 0.2 mm in thickness. The details of the swelling experiment were described in our previous paper.30 The network-chain densities, degrees of swelling by volume and gel fractions of IR–ZnSt2–CBS–S8 samples at different heat-pressing times are summarized in Table 1.| Sample code | Pressing time (min) | Network-chain densitya × 104 (mol cm−3) | Degree of swelling by volumeb | Gel fractionc |
|---|---|---|---|---|
| a Determined by the swelling measurement in toluene at 25 °C.b Determined by the change of volume in toluene before and after swelling.c Determined by the change of weight in toluene before and after swelling followed by drying. | ||||
| IR–ZnSt2–CBS–S8-18 | 18 | 0.66 | 7.0 | 0.92 |
| IR–ZnSt2–CBS–S8-19 | 19 | 0.69 | 6.9 | 0.97 |
| IR–ZnSt2–CBS–S8-22 | 22 | 1.28 | 5.2 | 0.95 |
| IR–ZnSt2–CBS–S8-26 | 26 | 1.16 | 5.5 | 0.96 |
| IR–ZnSt2–CBS–S8-30 | 30 | 0.84 | 6.3 | 0.94 |
| IR–ZnSt2–CBS–S8-40 | 40 | 0.71 | 6.8 | 0.91 |
Sulfur K-edge X-ray absorption near edge structure (S-XANES) measurement
S-XANES measurements were carried out at the BL-10, SR center of the Ritsumeikan University at r.t. using synchrotron radiation at 575 MeV electron beam energy and an electron beam current of 120–190 mA. An InSb (111) crystal was used as a monochromator in the synchrotron energy range from 2400 to 2600 eV. The X-ray absorption spectra for the samples were recorded in the transmission mode using a photodiode detector, in which the energy of K2SO4 was calibrated as a secondary standard at the maximum absorption peak (white line) of 2481.7 eV.The XANES spectra were linear-background subtracted and normalized before analysis by the ATHENA (version 0.8.056) software package.35 The quantitative sulfidic linkage analysis, i.e., mono-, di- and polysulfidic linkages, were evaluated by a linear combination fitting method in the energy range of 2461 to 2491 eV. The R-factor is defined as a sum-of-squares measure of fractional misfit showing a degree of best fit; the smaller the R-factor, the better the fitting accuracy.
For S-XANES measurements, the IR vulcanizates of about 10 mm in width × about 20 mm in length × about 0.2 mm in thickness were subjected to the solvent extractions in advance using 30 mL of chloroform/acetone mixture (7/3 vol%, special grade solvents of Wako Pure Chemical Industries, Ltd.), followed by 30 mL of special grade tetrahydrofuran (THF, Wako Pure Chemical Industries, Ltd.). Both extraction steps were performed twice at 25 °C for 24 hours. After the extractions, the sample sheets were rinsed several times using the same solvent for each step. The extracted samples were then dried under air and vacuum for 1 day to remove the solvent. The amounts of extracts (wt%) are summarized in Table 2. No heating was applied for the extraction in order to prevent post-vulcanization at high temperature. These extraction conditions were adequate for removing unreacted S8 and CBS because the solubilities of the reagents at 25 °C were confirmed before the extractions, as follows:30 for S8 and CBS in 100 g of the chloroform/acetone mixture (7/3 vol%) the solubilities were 0.6 g and 5.0 g, respectively; for S8 and CBS in 100 g of THF the solubilities were 2.0 g and 18.2 g, respectively. In the fourth extraction, the extracts of each vulcanizate were subjected to mass spectroscopy with EI mode at 220 °C, where THF was used as the solvent. As shown in Table 2, the amounts of extracts from each sample decreased by repeating the extractions, and trace amounts were observed in each sample after the fourth extraction by THF. Therefore, all unreacted CBS and S8 were judged to be removed almost completely.
| Sample code | 1st extractiona | 2nd extractiona | 3rd extractionb | 4th extractionb | ||
|---|---|---|---|---|---|---|
| Q (wt%) | Q (wt%) | Q (wt%) | Q (wt%) | Mass spectroscopy | ||
| CBSc | S8 | |||||
| a Solvent extraction using chloroform/acetone mixture (7/3 vol%).b Solvent extraction using THF.c N-(1,3-Benzothiazol-2-ylsulfanyl) cyclohexanamine.d ×: none, T: trace. | ||||||
| IR–ZnSt2–CBS–S8-18 | 7.7 | 2.8 | 0.6 | 0.9 | Td | T |
| IR–ZnSt2–CBS–S8-19 | 5.2 | 1.5 | 0.5 | 0.6 | ×d | T |
| IR–ZnSt2–CBS–S8-22 | 4.5 | 1.3 | 0.1 | 0.4 | × | T |
| IR–ZnSt2–CBS–S8-26 | 4.9 | 1.4 | 0.0 | 0.2 | T | T |
| IR–ZnSt2–CBS–S8-30 | 5.9 | 1.8 | 0.4 | 0.7 | T | T |
| IR–ZnSt2–CBS–S8-40 | 6.5 | 2.2 | 0.3 | 0.5 | T | T |
Reference samples for S-XANES
Prior to the evaluation of sulfidic linkages, the characteristic absorption peaks of the reference samples were examined. Thirteen phr of octadecyl sulfide (ODS), 7 phr of dibenzyl disulfide (DBS) obtained from Tokyo Chemical Industry Co., Ltd. and 1.5 phr of S8 were mixed with IR, representing the reference compounds of mono-, di- and polysulfidic linkages, respectively. Moreover, 5 phr of zinc sulfide (ZnS, Rare Metallic Co., Ltd.), 8 phr of zinc sulfate (ZnSO4, Wako Pure Chemical Industries, Ltd.) and 8 phr of di-n-butyl sulfoxide (DBSO, Ouchi Shinko Chemical Industrial Co., Ltd.) were also mixed with IR as other reference compounds for the evaluation of the sulfur atom state in the thermal-oxidative process, e.g., –SO− and –SO42−.Fig. 3 shows the XANES spectra of all reference samples plotted for the energy range of 2465–2490 eV with normalized intensity. Their characteristic absorption peaks correspond to their energies at absorption edges and shapes, indicating each local environment of the sulfur atom. The energies at the tops of peaks can be used to assign the sulfidic linkages in the IR vulcanizates. The absorption peaks of the IR–ODS, IR–DBS and IR–S8 were observed at 2473.02, 2472.42 and 2472.24 eV, indicating the mono-, di- and polysulfidic linkages, respectively. The characteristic peaks and shapes of the reference compounds were in good agreement with previous reports.19,31,36 In practice, ZnS is formed during the progress of the vulcanization reaction together with the thermal degradation and oxidation reaction, i.e., the generation of –SO− and –SO42− species; therefore, in this study, ZnS, DBSO and ZnSO4 were selected to assign the characteristic peaks of Zn–S, –SO− and –SO42−, respectively. From Fig. 3, the energies at the tops of the peaks of these sulfur reference species were observed at 2473.58, 2475.70 and 2481.99 eV, respectively.
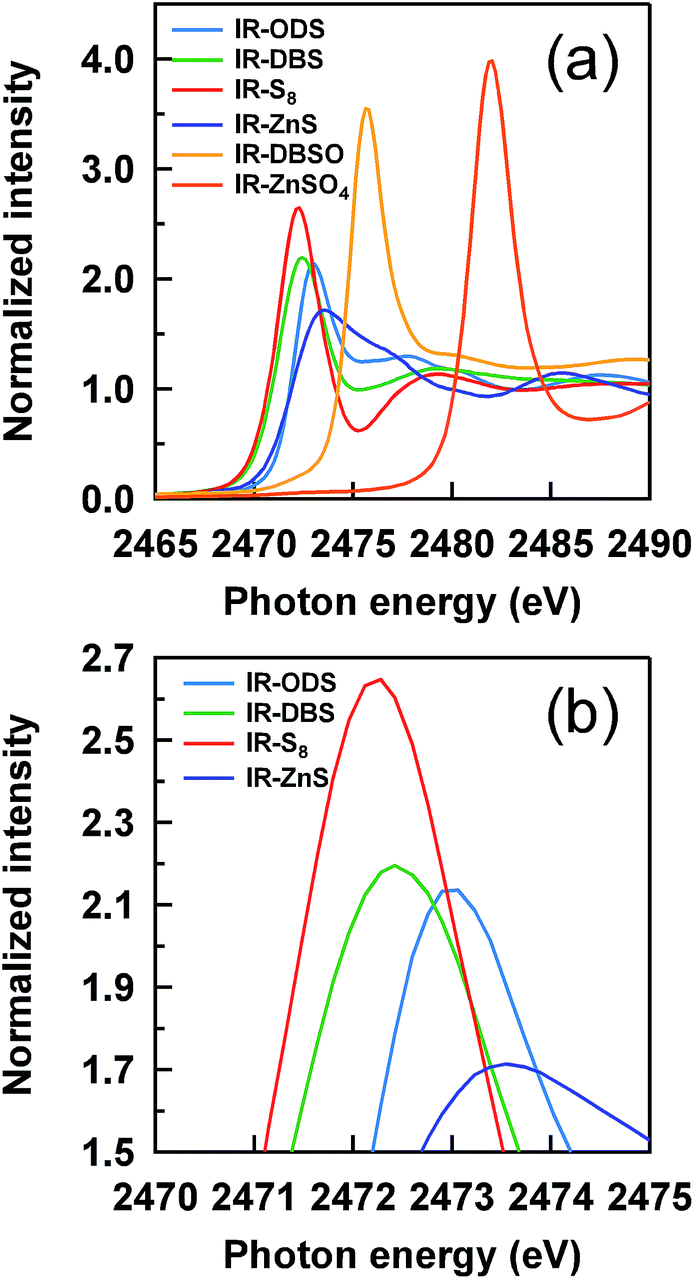 | ||
| Fig. 3 S-XANES spectra of the reference samples; (a) white lines for all reference samples and (b) their magnified region. | ||
Tensile measurement
For evaluation of the tensile properties of the vulcanizates, tensile measurements were conducted using a tailor-made tensile tester (ISUT-2201, Aiesu Giken, Co., Kyoto, Japan) at 25 °C for ring-shaped samples, whose inner and outer diameters were about 11.7 and 13.7 mm, respectively. The thickness was about 0.2 mm. The stretching speed was 100 mm min−1, i.e., the strain speed was about 4.98 min−1.Results and discussion
Variation of S-XANES spectra with the progress of curing
Fig. 2 shows a cure curve for IR–ZnSt2–CBS–S8, which was used as a model sample representing the formation of the mesh network in the vulcanizate in this study. The cure curve showed a drastic upturn of its torque from about 16 min, a maximum torque at about 22 min, followed by over-curing behavior. During the curing, we investigated what kind of sulfidic linkage was produced. In order to follow the change in sulfidic linkages at different cross-linking degrees in the cure curve, S-XANES measurements were conducted for the samples prepared at various curing times as illustrated by red circles on the cure curve in Fig. 2. Fig. 4(a) shows the S-XANES spectra under the transmission mode for the samples that were prepared by changing the heat-pressing time. It was found that a white line was detected in each spectrum in the energy range of 2472–2473 eV. The positions slightly increased to the higher photon energy region with increasing the heat-pressing time for the samples, which are obviously seen from the enlarged view of this region in Fig. 4(b). This observation suggests the gradual change of polysulfidic linkages to a mixed state of disulfidic and monosulfidic linkages with polysulfidic linkage by a progress of vulcanization for up to 40 min of heat pressing. However, the results were far from those obtained by Taweepreda et al.26 in the CBS-accelerated sulfur-based vulcanization of squalene. They reported that the monosulfidic linkages were dominantly detected in their study. The difference is ascribed to our solvent extraction method for the vulcanizates. By using adequate solvent extractions, the unreacted reagents, e.g., free S8 and free CBS, were removed from the vulcanizates as shown in the experimental section.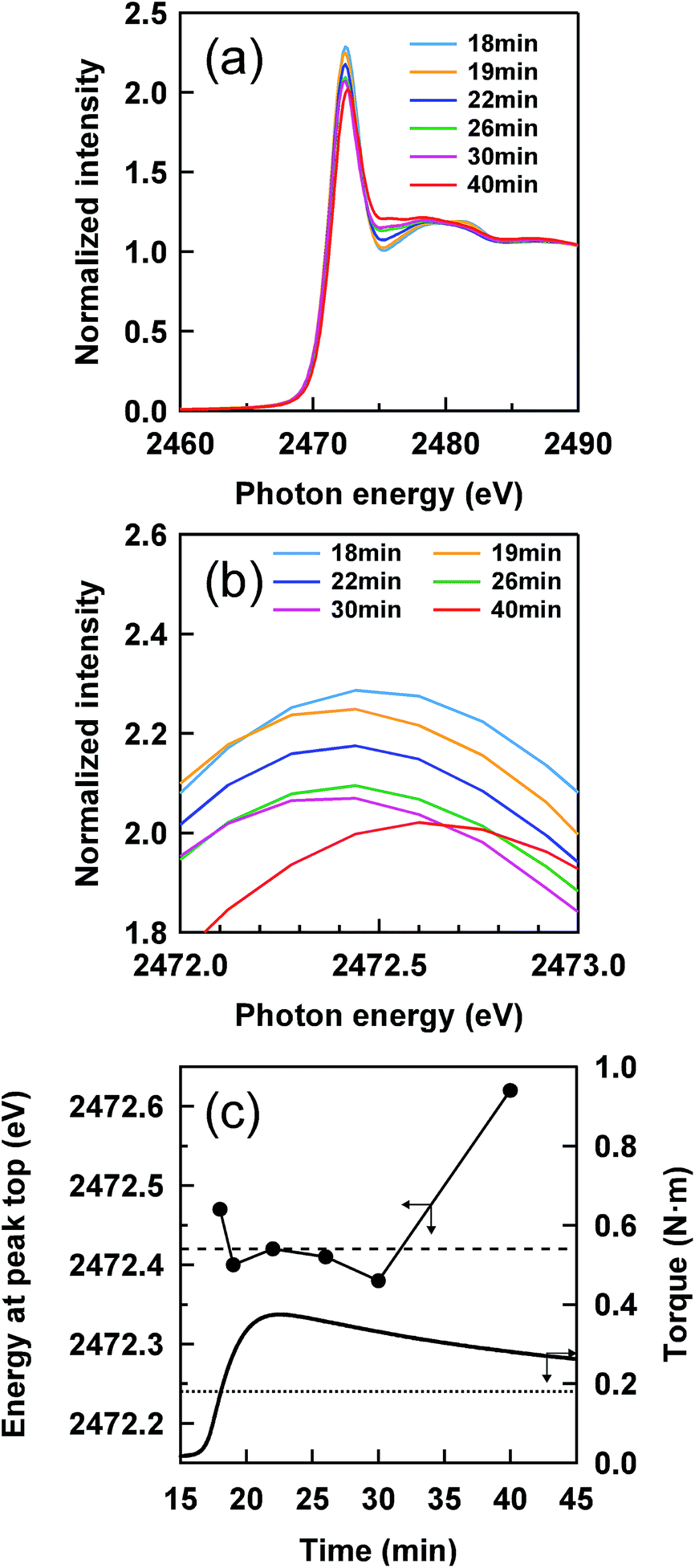 | ||
| Fig. 4 (a) S-XANES spectra of IR–ZnSt2–CBS–S8 samples at different heat-pressing times, (b) the enlarged view of the white lines, and (c) the plots of the energies of white lines against the heat-pressing time. The energies of the white lines of disulfidic and polysulfidic linkages of reference samples are also indicated by the dashed and dotted lines, respectively. | ||
A relationship between the energy of the white line and the heat-pressing time of each sample is shown in Fig. 4(c), in which the dashed and dotted lines represent the energies at the white lines of two reference samples, IR–DBS and IR–S8, suggesting disulfidic and polysulfidic linkages at 2472.42 and 2472.24 eV, respectively. The torque obtained from the cure curve is also plotted as a secondary y-axis with the heat-pressing time. The IR–ZnSt2–CBS–S8 samples prepared by heat pressing for 18, 19, 22, 26 and 30 min show the white lines at about 2472.42 eV, at a similar level to that of the disulfidic linkage. However, the white line of IR–ZnSt2–CBS–S8 prepared by heat pressing for 40 min was substantially higher than 2472.42 eV. These results indicate that disulfidic linkages may be dominantly formed in the mesh network during both the vulcanization and reversion processes. Additionally, the cleavage of polysulfidic linkages to di- and/or monosulfidic linkages is proposed to be significantly accelerated under a long heating time up to 40 min. However, it must be emphasized here that the white lines are represented as a summation of all linkages. Therefore, it is difficult to directly discuss the variation in the length of sulfidic linkages during vulcanization from only the white lines.
Variation of sulfidic linkages with the progress of curing
In order to accurately follow the change in sulfidic linkages upon vulcanization, linear combination fittings using the reference S-XANES spectra were carried out for each S-XANES spectrum of the vulcanizates. Fig. 5(a–f) shows the fitting results of the samples at various heat-pressing times. The S-XANES spectra were sufficiently well decomposed into each component, and thus R-factors were quite small (Fig. 6). It is worth noting that disulfidic linkages were detected to serve as the main sulfidic linkage during both the vulcanization and reversion processes of IR–ZnSt2–CBS–S8, and this is the first report, as far as we know, of the CBS-accelerated sulfur curing system with zinc stearate. Of course, we have to keep in mind that presently it is impossible for S-XANES studies to separately characterize the cross-linking segments and pendant segments. However, the cure curve in Fig. 2 may support the cross-linking segments in IR–ZnSt2–CBS–S8-18, IR–ZnSt2–CBS–S8-19 and IR–ZnSt2–CBS–S8-22, which were mainly composed of disulfidic cross-linkages because the cross-linking reaction occurred predominantly until the heat-pressing time of 22 min. In addition, the tensile properties of these samples were found to be high, as shown in Fig. 7, which suggests that the cross-linked segments to load the stress were abundant.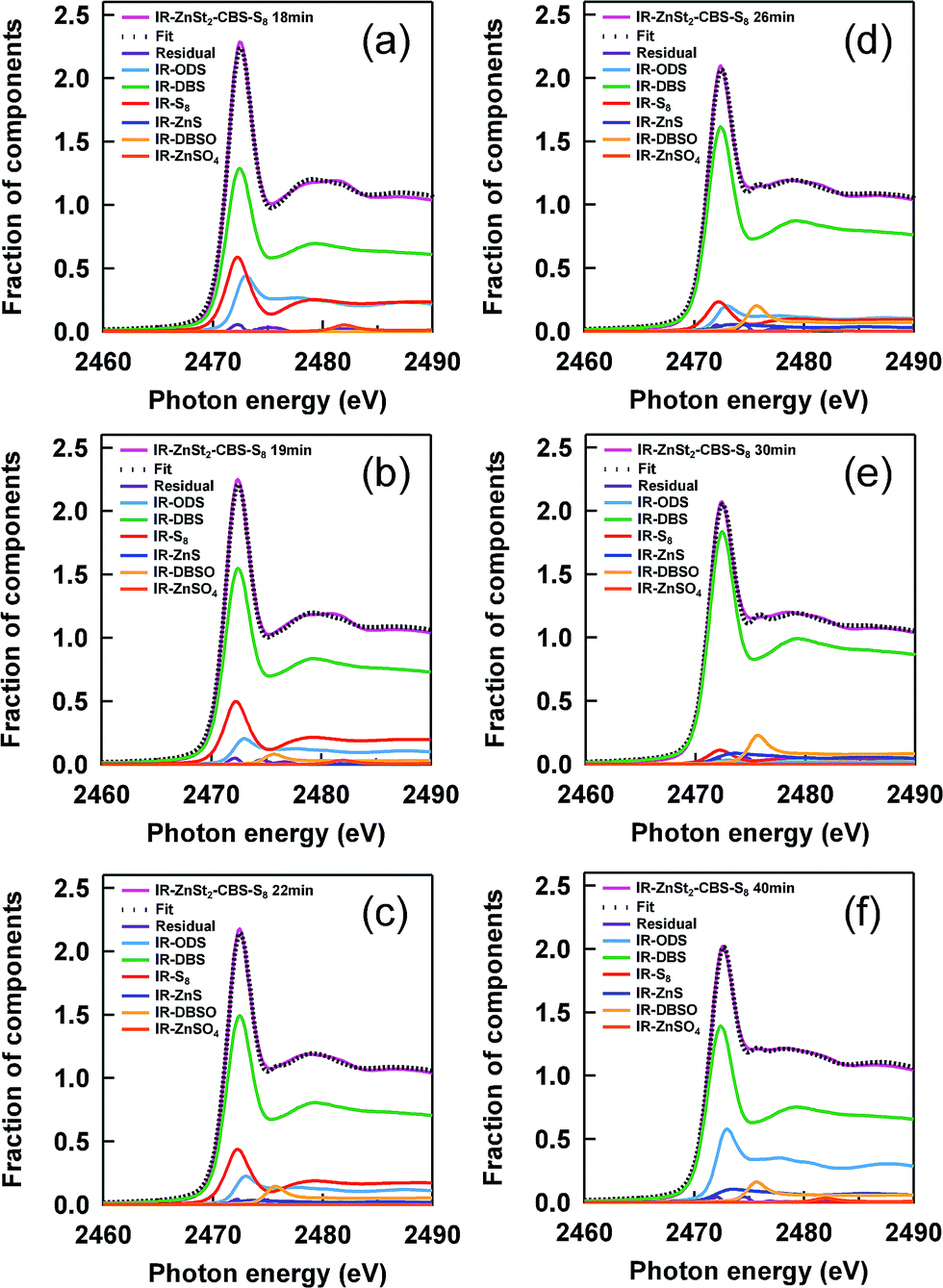 | ||
| Fig. 5 Fitting results for the S-XANES spectra of IR–ZnSt2–CBS–S8 samples at various heat-pressing times: (a) 18 min, (b) 19 min, (c) 22 min, (d) 26 min, (e) 30 min and (f) 40 min. The pink lines show the raw data and the dotted black lines show the results of linear combination fitting using each reference S-XANES spectrum. | ||
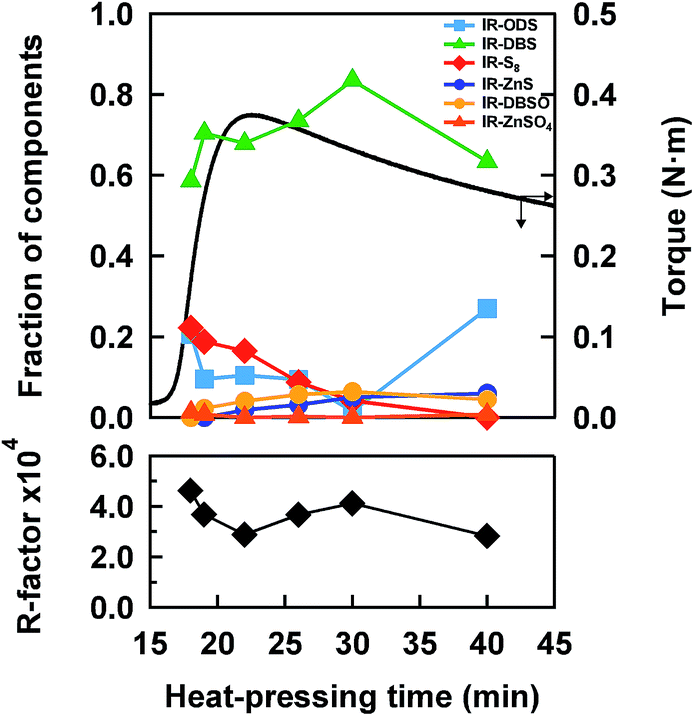 | ||
| Fig. 6 The variation of sulfidic linkages and oxidative species in IR–ZnSt2–CBS–S8 with the heat-pressing time. Each fraction was obtained by linear combination fitting. The cure curve is also shown. The R-factor is separately plotted below. | ||
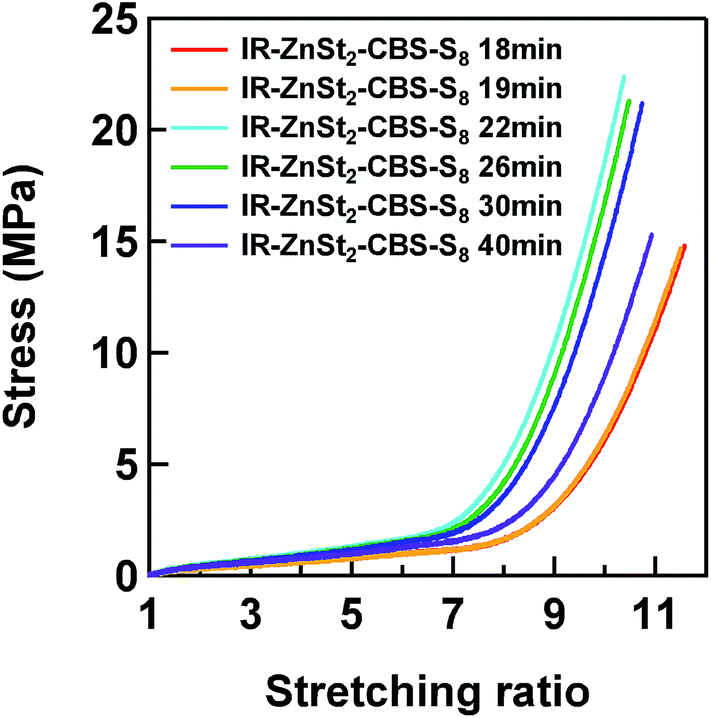 | ||
| Fig. 7 Tensile stress–strain curves of IR–ZnSt2–CBS–S8 samples. | ||
On the other hand, the vulcanizate of IR–ZnSt2–CBS–S8-40 showed the presence of di- and monosulfidic linkages without any polysulfidic linkage. As is well understood, the reversion process is generally ascribed to the cleavage of sulfur segments of the cross-linked sites in the network by thermal aging. It is also well-known that the cleavage of the network structure occurs most easily in polysulfidic cross-linking segments, followed by the cleavage of disulfidic linkages. In this study, Fig. 6 clearly and quantitatively supports the decrease in the fraction of polysulfidic linkages with the time of heat pressing and the increase in the fraction of disulfidic linkages from 22 to 30 min. When the cleavage of polysulfidic linkages was mostly finished at about 30 min, there was a decrease in disulfidic linkages, as shown in Fig. 6. Since the thermal stability of the disulfidic linkage is a little weaker than that of the monosulfidic linkage,37 the decrease in disulfidic linkages and increase in monosulfidic linkages were reasonable for heat pressing at 140 °C. The variation in network-chain densities of IR–ZnSt2–CBS–S8 samples supported this consideration. As shown in Table 1, the network-chain densities clearly increased from 18 to 22 min, but decreased after 22 min, where the maximum torque of the cure curve was detected.
In general, zinc sulfide has been recognized as one of the by-products in the sulfur cross-linking reaction.38 In this study, our results support the common opinion that the fraction of zinc sulfide increased with the increase in heat-pressing time. Thus, both the sulfur cross-linking reaction and the decross-linking reaction are concluded to occur even in the reversion process. It is also noted in Fig. 4(a) that the absorption intensities at about 2475 and 2482 eV increased with time, which may be ascribed to the generation of sulfoxide and/or sulfate groups by oxidation.18,21 By fitting, the oxidative products composed of –SO− and –SO42− groups were evidently detected in the samples as trace amounts. Since our samples do not contain any antioxidant reagents, the thin films may have been a little oxidized during the handling and storage of the samples. From these results, it is obvious that the linear combination fitting using the appropriate reference spectra gives us reasonable and quantitative results to monitor the changes in sulfidic linkages as a function of heat-pressing time.
A possible factor to generate disulfidic linkages
As shown above, it was found that the CBS-accelerated sulfur curing system predominantly formed the disulfidic linkages when using zinc stearate as an activator at 140 °C. A key to producing a disulfidic linkage is discussed in this section on the basis of our previous data.39 Generally, it is thought that stearic acid (StH) reacts with ZnO to form the activator (ZnSt2) in the vulcanization reaction. Zinc 1,3-benzothiazole-2-thiolate with a stearate group as a ligand has been accepted as an active intermediate generated from ZnO, StH, and CBS in a rubber matrix of the CBS-accelerated sulfur curing system with ZnO and StH. However, the details regarding the zinc salt of StH for sulfur cross-linking have not been well understood, because most of the past studies have only focused on the intermediates isolated from vulcanization reactions. Thus, the role of the zinc salt of StH in the vulcanization reaction was not sufficiently clear until our study in 2015.39 While studying the role of the zinc salt of StH, we observed the formation of a new complex generated from ZnO and StH at a high temperature by using a combination of in situ time-resolved Zn K-edge XAFS spectroscopy, in situ time-resolved infrared spectroscopy, and the density functional theory calculation.39 The bridging bidentate zinc/stearate complex (intermediate as shown in Fig. 8) was found to be a novel dominant intermediate. Interestingly, the zinc ion-to-stearate molar ratio of the complex was unexpectedly found to be 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The most likely structure was proposed to be [Zn2(μ-O2CC17H35)2]2+(OH−)2·XY like enzymes, where X and Y are water and/or a rubber segment, as shown in Fig. 8. In addition, this bridging bidentate zinc/stearate complex was also detected in the isoprene rubber matrix containing ZnSt2.39 Therefore, this intermediate may be one of the important factors to dominantly generate the disulfidic linkages during the vulcanization reaction of IR–ZnSt2–CBS–S8 in this study.
:2. The most likely structure was proposed to be [Zn2(μ-O2CC17H35)2]2+(OH−)2·XY like enzymes, where X and Y are water and/or a rubber segment, as shown in Fig. 8. In addition, this bridging bidentate zinc/stearate complex was also detected in the isoprene rubber matrix containing ZnSt2.39 Therefore, this intermediate may be one of the important factors to dominantly generate the disulfidic linkages during the vulcanization reaction of IR–ZnSt2–CBS–S8 in this study.
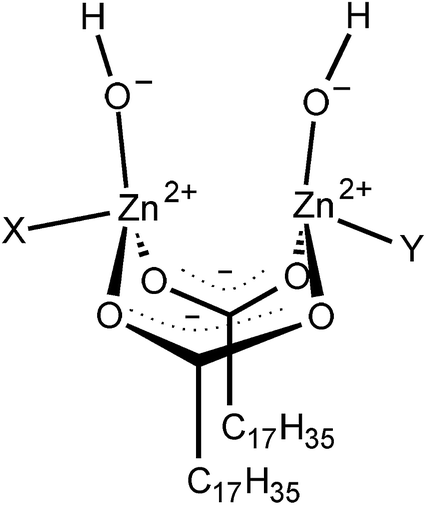 | ||
| Fig. 8 Dinuclear type bridging bidentate zinc/stearate complex composed of (Zn2(μ-O2CC17H35)2)2+(OH−)2·XY, where X and Y are water and/or a rubber segment. Reproduced with permission.39 Copyright 2015, American Chemical Society. | ||
As shown in Fig. 6, the fraction of disulfidic linkages of IR–ZnSt2–CBS–S8-18 was about 59%, although the torque in the cure curve was about 0.17 N m, which was about 43% of the maximum torque of the sample prepared at 22 min in the cure curve shown in Fig. 2. This clearly suggests that the disulfidic linkage was already formed in the first half of the sulfur cross-linking reaction in IR–ZnSt2–CBS–S8. Therefore, the results in this S-XANES study suggest that the intermediate of the bridging bidentate zinc/stearate complex may induce the formation of disulfidic linkages during the vulcanization reaction. As is well-known, the disulfidic linkages are more stable against heat and oxidation compared to the polysulfidic linkages. Therefore, the observed results for the disulfidic linkage in the mesh network formation of this study must be a strategic key for producing high performance rubber products. The other experimental evidence to support this proposal will be reported in the near future.
The effect of sulfidic linkages on tensile properties
Fig. 7 shows the stress–strain curves of IR–ZnSt2–CBS–S8 samples. The nominal stress drastically increased with the heat-pressing time for the vulcanization up to 22 min, but decreased in the sample prepared by heat pressing for 30 and 40 min. Here, α is defined as α = l/l0, in which l0 is the initial length and l is the length after deformation. The effect of the variation of sulfidic linkages formed in the mesh network models on their mechanical properties is discussed using these results. The tensile stress–strain curves of IR–ZnSt2–CBS–S8-19 and IR–ZnSt2–CBS–S8-40, which have similar network-chain densities, are compared for the discussion.It was found that IR–ZnSt2–CBS–S8-40 showed higher tensile stresses than IR–ZnSt2–CBS–S8-19, even though their fractions of disulfidic linkages were comparable as shown in Fig. 6. This difference is ascribable to the fractions of polysulfidic and monosulfidic linkages: the fraction of polysulfidic linkages of IR–ZnSt2–CBS–S8-40 was zero and monosulfidic linkages were much higher than that of IR–ZnSt2–CBS–S8-19. The results suggest that the monosulfidic linkage (C–S–C) is much stronger than the polysulfidic linkage (C–Sx–C), due to the higher bonding energy of the C–S linkage. The vulcanizate mainly cross-linked by monosulfidic and disulfidic linkages can load the higher stresses, but give the lower flexibility, compared to the vulcanizate composed of the polysulfidic linkages. This phenomenon was clearly shown in this study by using the S-XANES analyses for the vulcanizates. Due to the absence of ZnO in IR–ZnSt2–CBS–S8 in this study, only the mesh network must have been formed without any network domains that could act as a reinforcing filler. Therefore, the effect of different kinds of sulfidic linkages was apparently detected in the tensile properties of vulcanizates.
Conclusions
Sulfur K-edge absorption near edge structure (S-XANES) measurements were conducted to evaluate the variation of the sulfidic linkage of the network structure with the progress of vulcanization of IR, where the IR vulcanizates were subjected to solvent extraction. The disulfidic linkage was found for the first time to be dominantly generated in the rubber network formation when zinc stearate and N-(1,3-benzothiazol-2-ylsulfanyl)cyclohexanamine were used as activator and accelerator for the sulfur cross-linking reaction at 140 °C, respectively. This was supposed to be induced by the intermediate of the bridging bidentate zinc/stearate complex generated from zinc stearate, together with the cleavage of polysulfidic linkages during heating. Therefore, the control of the generation of the intermediate complex must be one of the factors governing the properties of rubber vulcanizates. This observation is useful for the preparation of high performance rubber products because anti-aging and thermal stabilities are demanded for these rubber products. As demonstrated herein, the S-XANES measurement was found to be a very powerful tool to identify the structural details of the sulfur cross-linked rubber networks. These results are definitely valuable for the development of rubber science.40 Further study for the elucidation of the mechanism of the generation of disulfidic linkage by the intermediate of the bridging bidentate zinc/stearate complex is necessary, and will be reported elsewhere in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST ALCA program (2015–) and Sumitomo Science and Technology Foundation. The XANES experiment was performed at the BL-10 in the SR center of Ritsumeikan University with the approval of the Ritsumeikan University (Proposal No. R1247, R1308, R1312). The authors thank to Prof. Dr Toshiaki Ohta and Mr Masahiro Ogawa in SR Center of Ritumeikan University, and Dr Treethip Phakkeree and Dr Nawaphorn Kuhakongkiat in Kyoto Institute of Technology.Notes and references
- J. L. Bras, Rubber: Fundamentals of Its Science and Technology, Chemical Publishing Company, New York, 1957 Search PubMed.
- A. K. Bhowmick, Rubber Products Manufacturing Technology, CRC Press, USA, 1994 Search PubMed.
- J. L. White, Rubber Processing: Technology, Materials, Hanser Publishers, Germany, 1995 Search PubMed.
- A. Ciesielski, An Introduction to Rubber Technology, iSmithers Rapra Publishing, USA, 1999 Search PubMed.
- A. Y. Coran, The Science and Technology of Rubber, Academic Press, San Diego, 2nd edn, 1994 Search PubMed.
- D. Craig, Rubber Chem. Technol., 1957, 30, 1291–1346 CrossRef CAS.
- M. Porter, The Chemistry of Sulfides, Interscience Publishers, New York, 1968 Search PubMed.
- M. M. Coleman, J. R. Shelton and J. L. Koenig, Ind. Eng. Chem. Prod. Res. Dev., 1974, 13, 154–166 CAS.
- Y. Ikeda, N. Higashitani, K. Hijikata, Y. Kokubo, Y. Morita, M. Shibayama, N. Osaka, T. Suzuki, H. Endo and S. Kohjiya, Macromolecules, 2009, 42, 2741–2748 CrossRef CAS.
- Y. Yasuda, S. Minoda, T. Ohashi, H. Yokohama and Y. Ikeda, Macromol. Chem. Phys., 2014, 215, 971–977 CrossRef CAS.
- E. Morita, Rubber Chem. Technol., 1980, 53, 393–436 CrossRef CAS.
- S. K. Mandal, R. N. Datta and D. K. Basu, Rubber Chem. Technol., 1989, 62, 569–584 CrossRef CAS.
- P. Ghosh, S. Katare, P. Patkar, J. M. Caruthers, V. Venkatasubramanian and K. A. Walker, Rubber Chem. Technol., 2003, 76, 592–693 CrossRef CAS.
- G. Heideman, R. N. Datta, J. W. M. Noordermeer and V. B. Baarle, Rubber Chem. Technol., 2004, 77, 512–541 CrossRef CAS.
- M. L. Studebaker and L. G. Nabors, Rubber Chem. Technol., 1959, 32, 941–961 CrossRef CAS.
- D. S. Campbell, J. Appl. Polym. Sci., 1969, 13, 1201–1214 CrossRef CAS.
- S. Paste, V. Gotte, C. Goulon-Ginet, A. Rogalev, J. Goulon, P. Georget and J. Marcilloux, J. Phys. IV, 1997, 7, 665–666 CrossRef CAS.
- R. Chauvistré, J. Hormes, D. Brück, K. Sommer and H.-W. Engels, Kautsch. Gummi Kunstst., 1992, 47, 808–813 Search PubMed.
- R. Chauvistré, J. Hormes and K. Sommer, Kautsch. Gummi Kunstst., 1994, 47, 481–484 Search PubMed.
- R. Chauvistré, J. Hormes, E. Hartmann, N. Etzenbach, R. Hosch and J. Hahn, Chem. Phys., 1997, 223, 293–302 CrossRef.
- B. Brendebach and H. Modrow, Kautsch. Gummi Kunstst., 2002, 55, 157–163 CAS.
- A. Prange and H. Modrow, Rev. Environ. Sci. Bio/Technol., 2002, 1, 259–276 CrossRef CAS.
- H. Modrow, R. Zimmer, F. Visel and J. Hormes, Kautsch. Gummi Kunstst., 2000, 53, 328–337 CAS.
- H. Modrow, J. Hormes, F. Visel and R. Zimmer, Rubber Chem. Technol., 2001, 74, 281–294 CrossRef CAS.
- W. Pattanasiriwisawa, J. Siritapetawee, O. Patarapaiboolchai and W. Klysubun, J. Synchrotron Radiat., 2008, 15, 510–513 CrossRef CAS PubMed.
- W. Taweepreda, R. Nu-Mard, W. Pattanasiriwisawa and P. Songsiriritthigul, J. Phys.: Conf. Ser., 2009, 190, 8–12 CrossRef.
- W. Taweepreda, W. Limphirat and P. Songsiriritthigul, Adv. Mater. Res., 2013, 620, 440–445 CrossRef.
- K. Boonkerd and W. Limphirat, Adv. Mater. Res., 2014, 905, 128–131 CrossRef.
- K. Fukumori, International Polymer Science and. Technology, 2014, 41, T23–T29 Search PubMed.
- Y. Yasuda, A. Tohsan, W. Limphirat and Y. Ikeda, Kobunshi Ronbunshu, 2015, 72, 16–21 CrossRef CAS.
- A. Funatsuki, K. Shiota, M. Takaoka and Y. Tamenori, Forensic Sci. Int., 2015, 250, 53–56 CrossRef CAS PubMed.
- ACS News Service Weely PressPac, New insight into how rubber is made could improve tires, reduce air pollution, https://www.acs.org/content/acs/en/pressroom/presspacs/2015/acs-presspac-february-11-2015/new-insight-into-how-rubber-is-made-could-improve-tires-reduce-air-pollution.html, accessed 8 November 2017.
- Y. Ikeda, N. Higashitani, K. Hijikata, Y. Kokubo, Y. Morita, M. Shibayama, N. Osaka, T. Suzuki, H. Endo and S. Kohjiya, Macromolecules, 2009, 42, 2741–2748 CrossRef CAS.
- P. J. Flory, J. Chem. Phys., 1950, 18, 108–111 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- P. Pangamol, C. Sirisinha, Y. Hu and S. G. Urquhart, Ind. Eng. Chem. Res., 2013, 52, 17179–17183 CrossRef CAS.
- D. J. Elliott, in Developments in Rubber Technology—1, ed. A. Whelan and K. S. Lee, Applied science publishers Ltd., London, 1979, pp. 1–44 Search PubMed.
- A. V. Chapman and M. Porter, Natural Rubber Science and Technology, Oxford University Press, Oxford, 1988 Search PubMed.
- Y. Ikeda, Y. Yasuda, T. Ohashi, H. Yokohama, S. Minoda, H. Kobayashi and T. Honma, Macromolecules, 2015, 48, 462–475 CrossRef CAS.
- Y. Ikeda, A. Kato, S. Kohjiya and Y. Nakajima, Rubber Science: A Modern Approach, Springer, Singapore, 2017 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |