
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel strategy for the manufacture of idelalisib: controlling the formation of an enantiomer†
Nagaraju Mekalaab,
Srinivasa Rao Buddepuab,
Sanjay K. Dehurya,
Krishna Murthy V. R. Moturu *a,
Sunil Kumar V. Indukuria,
Umamaheswara Rao Vasireddia and
Atchuta R. Parimib
*a,
Sunil Kumar V. Indukuria,
Umamaheswara Rao Vasireddia and
Atchuta R. Parimib
aOncology Division, Process Development Laboratories, Laurus Laboratories Limited, ICICI Knowledge Park, Turkapally, Shameerpet, Hyderabad-500 078, Telangana, India. E-mail: murthy.moturu@lauruslabs.com; Fax: +91 40 23480481; Tel: +91 40 30413393
bDepartment of Organic Chemistry, Food, Drugs & Water, School of Chemistry, Andhra University, Visakhapatnam-530 003, Andhra Pradesh, India
First published on 27th April 2018
Abstract
A novel and scalable synthesis of 5-fluoro-3-phenyl-2-[(1S)-1-(9H-purin-6-ylamino)propyl]-4(3H)-quinazolinone, idelalisib 1, has been developed. This strategy controls the desfluoro impurity of 13 during reduction of nitro intermediate 4, and also arrests the formation of the enantiomer during cyclisation of diamide 17, without affecting the neighbouring chiral centre. This process is demonstrated on a larger scale in the laboratory and achieved good chemical and chiral purities coupled with good yields.
Introduction
Quinazolinones are fused heterocyclic alkaloids and have attracted many scientists working in organic and medicinal chemistry due to their substantial and extensive range of therapeutic activities. The remarkable therapeutic activities1 such as antiinflammatory, anticonvulsant, antidiabetic, anticancer, antitussive, antibacterial, analgesic, hypnotic, and sedative activities coupled with exotic structural features have impelled a lot of activity in synthetic chemistry research groups towards the development of new synthetic strategies and methodologies for the synthesis of quinazolinone alkaloids. Till now approximately 200 naturally occurring quinazolinone alkaloids have been isolated from various sources.2Idelalisib 1, (trade name; Zydelig), 5-fluoro-3-phenyl-2-[(1S)-1-(9H-purin-6-ylamino)propyl]-4(3H)-quinazolinone is a quinazolinone drug used for the treatment of chronic lymphocytic leukemia. The substance acts as a phosphoinositide 3-kinase inhibitor; More specifically, it blocks P110δ, the delta isoform of the enzyme phosphoinositide 3-kinase. The U.S. Food and Drug Administration approved idelalisib,3 in 2014, for the treatment of relapsed Follicular B-cell non-Hodgkin Lymphoma (FL), relapsed Chronic Lymphocytic Leukemia (CLL) and relapsed Small Lymphocytic Lymphoma (SLL). The European Medicines Agency (EMA) granted idelalisib4 approval for the treatment of adult patients with Chronic Lymphocytic Leukaemia (CLL) who have received at least one prior therapy and as first line treatment in the presence of 17p depletion.
As part of our research programme on the development of a series of anticancer drugs, herein we disclose a novel strategy for the synthesis of idelalisib, 1. Our approach has resulted not only in achieving almost total control of undesired enantiomer of 1, but also enhanced the yields tremendously during the cyclisation step when compared to known related reports in the literature.5–7
Results and discussion
Idelalisib was first developed by Icos corporation.5 The synthesis of idelalisib reported by Icos corporation was started with 2-fluoro-6-nitro benzoic acid (Scheme 1). Nitrobenzoic acid on treatment with oxalylchloride followed by condensation with aniline in DCM affords the corresponding 2-fluoro-6-nitro-N-phenylbenzamide 4. Coupling of this N-phenylbenzamide, 4 with N-Boc-L-2-amino butyric acid in presence of oxalylchloride yields N-boc-2-amino butyrate intermediate 6 which on reduction with Zn/AcOH affords the corresponding quinazolinone intermediate. Deprotection of Boc group followed by coupling with 6-bromopurine affords idelalisib 1. In this route reaction of N-Boc-L-2-amino butyric acid, 5 with nitro amide intermediate 4 is a tedious reaction. In this process only 50% conversion observed with 60% purity, due to back ward reaction. Changing the solvent medium from DCM to acetonitrile or toluene also did not give any advantage in terms of conversion and purity. In the subsequent stage, nitro group is reduced but further cyclisation is not completed to form the quinazolinone (Scheme 1).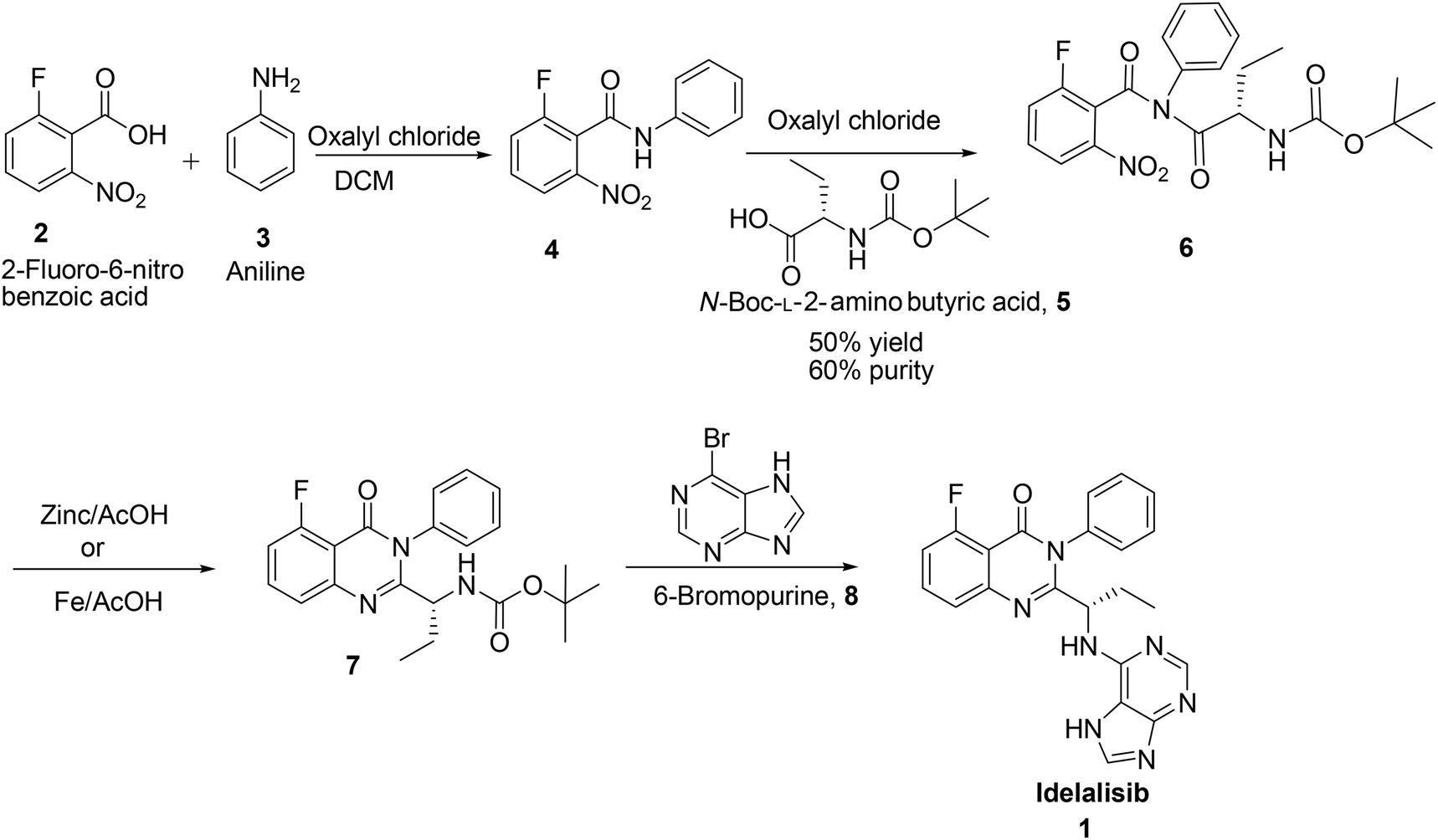 | ||
| Scheme 1 Synthesis of idelalisib, 1 reported by Icos Corporation& Shandong Kangmeile Pharmaceutical Technology Co Ltd. | ||
In the second synthesis reported by Geng Fengluan et al.6 of Shandong Kangmeile Pharmaceutical Technology Co Ltd. avoided acid chloride reagent for the activation of 2-fluoro-6-nitro benzoic acid, 2 instead used CDI (Scheme 1). In the reductive cyclisation of 6, Fe/AcOH was used in place of Zn/AcOH. However, CDI is expensive to use and Fe/AcOH reagent also not viable in commercial scale.
The third synthesis reported by Suzhou Mirac Pharma Technology Co. Ltd7 is started with coupling of adenine 10 with 2-hydroxy butyrate 9 to afford the corresponding adenine derivative 11. Treatment of 11 with 2-amino-6-fluoro benzoic acid 2 in trimethylaluminium yields the corresponding acid intermediate 12. This acid intermediate on treatment with aniline, 3 in presence of acetic anhydride and toluene at high temperatures (80–120 °C) followed by cyclisation afforded idelalisib, 1. In this route coupling of aniline 3 with acid intermediate, 12 followed by cyclisation is a cumbersome reaction forming many products. The removal of aniline from the reaction mass to desired level as per ICH limit is also not achieved (Scheme 2).
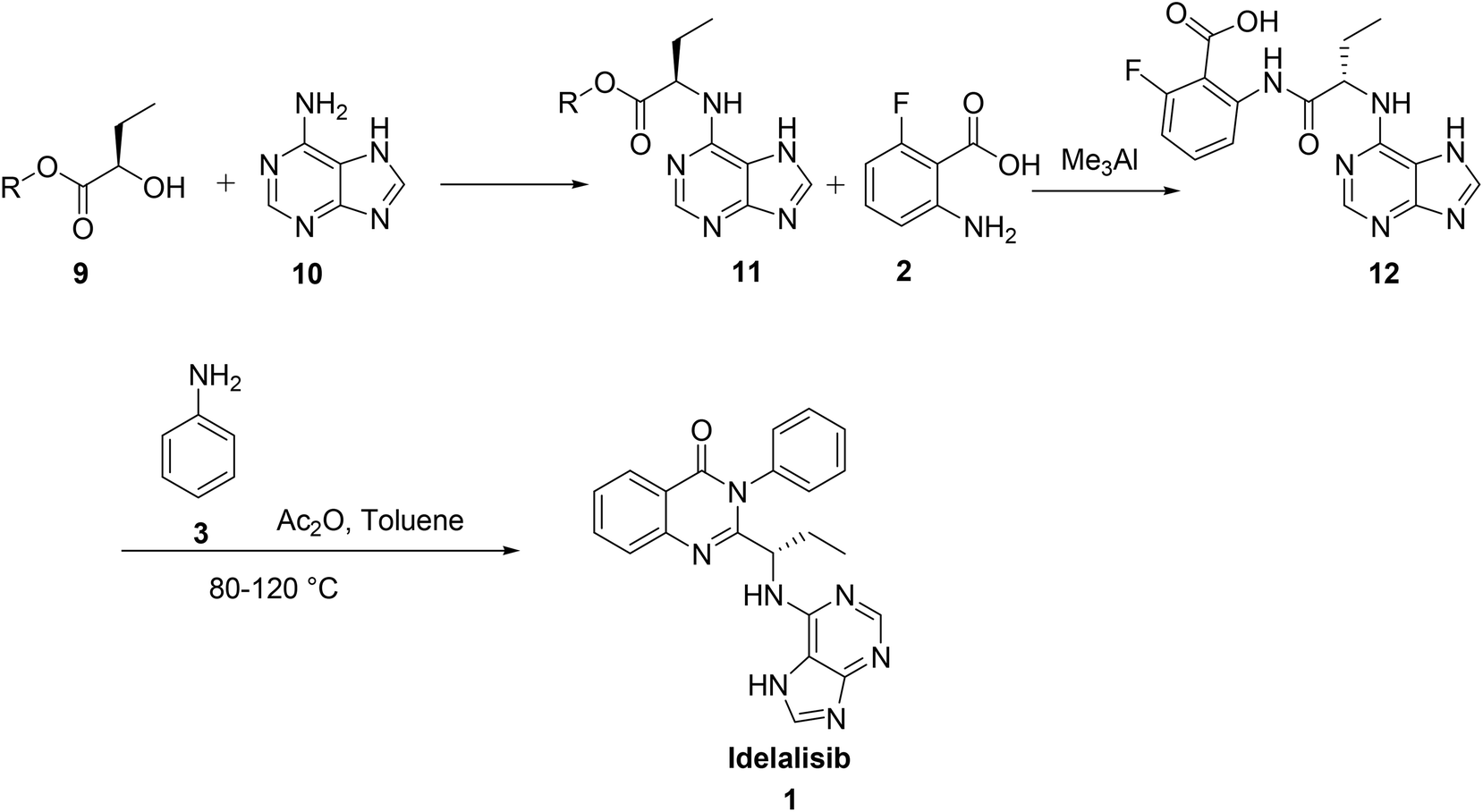 | ||
| Scheme 2 Synthesis of idelalisib, 1 reported by Suzhou Mirac Pharma Technology Co. Ltd. | ||
Hence, development of idelalisib, 1 process for the manufacture requires an alternate approach to achieve ICH purity and a scalable process. As part of our process development to prepare 1 in desired cost, we have designed a novel route (Scheme 3). Unlike the innovator process (Scheme 1), N-Boc-L-2-amino butyric acid, 5 was coupled with 2-amino-6-fluoro-N-phenylbenzamide at ambient temperature and achieved >98% purities with reasonably good yield (Table 1). The reduction of nitro group for the preparation of amino intermediate is carried out at ambient temperature with conventional catalysts like 10% Pd–C (Table 1; entry 1), 5% Pd–C (Table 1; entry 2), Pt–C (Table 1; entry 3) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of methanol and dichloromethane. With these catalysts desfluoro impurity is formed up to 5% level which is very difficult to separate by means of any crystallisation from the product once it is formed. Finally, this impurity is completely controlled in the reaction itself by changing the catalyst to zinc and ammonium formate8 to afford the 2-amino-6-fluoro-N-phenylbenzamide in 90% yield coupled with 99% purity (Table 1; entry 4).
:1 mixture of methanol and dichloromethane. With these catalysts desfluoro impurity is formed up to 5% level which is very difficult to separate by means of any crystallisation from the product once it is formed. Finally, this impurity is completely controlled in the reaction itself by changing the catalyst to zinc and ammonium formate8 to afford the 2-amino-6-fluoro-N-phenylbenzamide in 90% yield coupled with 99% purity (Table 1; entry 4).
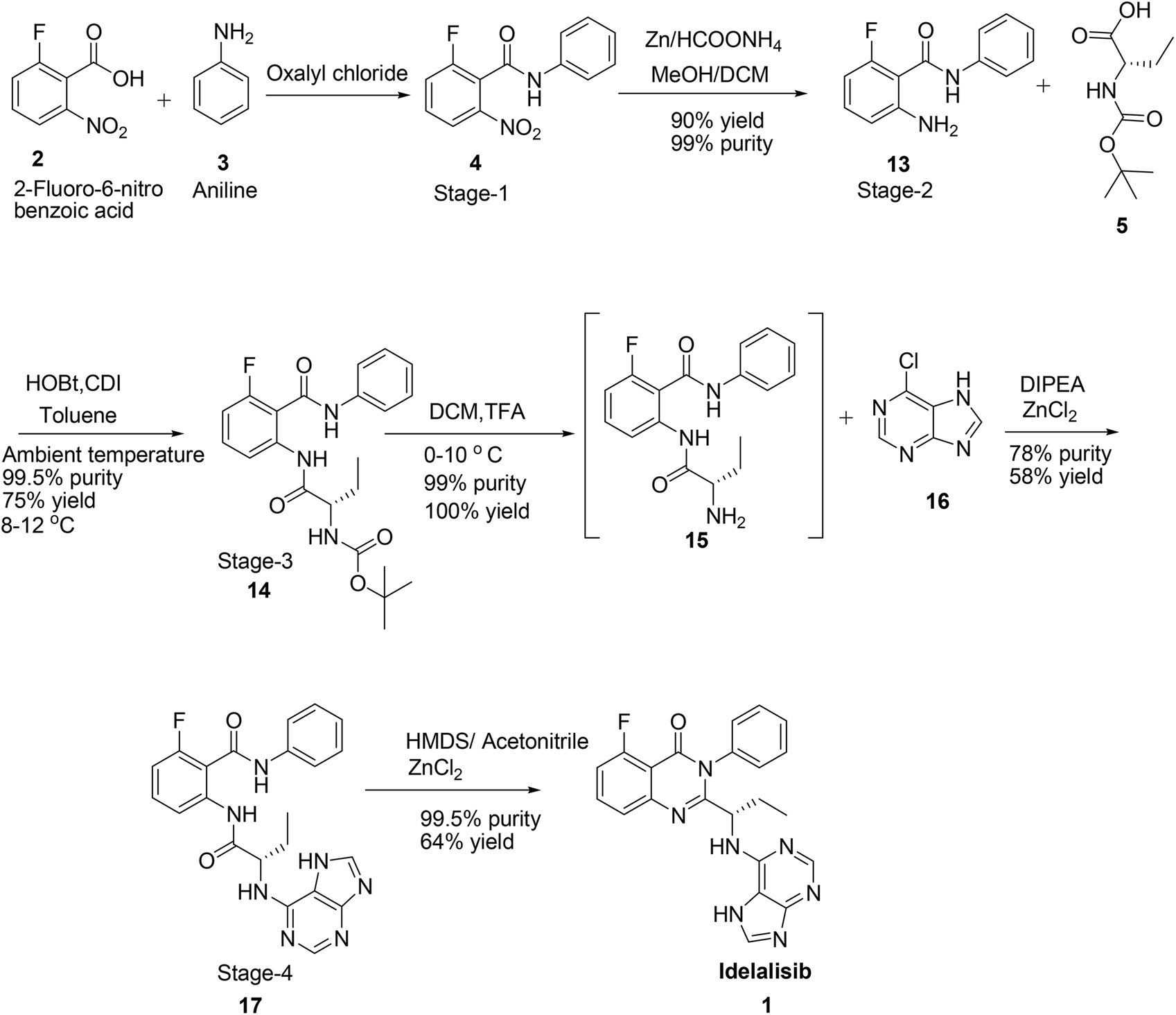 | ||
| Scheme 3 Synthesis of idelalisib, 1 by Laurus Labs Ltd. | ||
| S. No. | Catalyst | Input (g) | Output (g) | Yield (%) | Purity by HPLC (% area) | |
|---|---|---|---|---|---|---|
| Product | Desfluoro impurity | |||||
| a Catalyst used: 50% wet.b Catalyst used: 0.2 w/w to the input.c Catalyst used: Zn: 6.0 eq. and HCOONH4 7.0 eq. | ||||||
| 1a | 10% Pd–C | 110 | 84 | 87 | 93.5 | 5.6 |
| 2a | 5% Pd–C | 100 | 82 | 93 | 98.0 | 0.28 |
| 3b | Pt/C | 10 | 8 | 91 | — | 0.2 |
| 4c | Zn/HCOONH4 | 180 | 142 | 90 | 99.0 | Not detected |
In the next step for the formation of amide bond, we have attempted different coupling reagents such as benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP), N,N′-dicyclohexylcarbodiimide (DCC), carbonyldiimidazole (CDI), CDI/HOBt (1-hydroxybenzotriazole) and PyBOP/HOBt for the coupling reaction of N-Boc-L-2-amino butyric acid with 2-amino-6-fluoro-N-phenylbenzamide with different solvents to achieve good yield and purity (Table 2).
| S. No. | Coupling reagent | Input (g) | Solvent | Output (g) | Yield (%) | Purity by HPLC (% area) | Remarks | ||
|---|---|---|---|---|---|---|---|---|---|
| Chemical | Chiral | ||||||||
| Product | Product | Enantiomer | |||||||
| a Reagents used: 2.0 eq. of reagents used in all experiments except HOBt 1.0 eq. | |||||||||
| 1 | PyBOp | 10 | DMF | 5 | 10 | 99.0 | 99.95 | 0.05 | Reaction not completed (∼50% conversion observed by TLC) |
| 2 | PyBOp | 10 | Acetonitrile | 5 | 32 | 99.0 | 95.0 | 5.0 | |
| 3 | PyBOp | 10 | Toluene | 5 | 28 | 99.0 | 99.9 | 0.1 | |
| 4 | PyBOp/HOBt | 10 | Toluene | 5 | 28 | 99.0 | 100 | 0.0 | |
| 5 | DCC | 10 | Toluene | 8 | 44 | 97.0 | 98.0 | 2.0 | Reaction not completed (∼70% conversion observed by TLC) |
| 6 | CDI | 10 | Toluene | 8 | 44 | 97.0 | 99.5 | 0.5 | |
| 7 | CDI/HOBt | 140 | Toluene | 182 | 75 | 99.5 | 99.99 | 0.01 | Reaction completed by TLC |
As shown in Table 2, the conversion of 13 was observed ∼50% by TLC when the reaction was carried out in DMF at room temperature using PyBOP as coupling agent. However, after work up it was found that only 10% of the product was isolated with 99% purity along with control of other isomer formation (Table 2; entry 1). Changing the solvent to acetonitrile or toluene there was not much improvement in the yield but up to 5% racemisation is observed (Table 2; entry 2) in acetonitrile whereas the isomerisation was controlled to 0.10% in the latter solvent (Table 2; entry 3). Thus we decided to use toluene as solvent in the further experiments. To our good fortune racemisation is controlled completely with similar conversion and yield when 1.0 eq. of HOBt is added to PyBOP (Table 2; entry 4). Conversion is improved in both DCC (Table 2; entry 5) and CDI (Table 2; entry 6) with improved yield 44% in the same solvent but up to 2% racemisation was observed with DCC and 0.5% with CDI. Complete conversion with reasonably good yield was observed when 1.0 eq. of HOBt is added to CDI (2.0 eq.) and also totally controlled the racemisation to 0.01% (Table 2; entry 7) at lower temperature (8–12 °C). The same reaction at room temperature gives other isomer up to 0.15% level and at higher temperatures racemisation is observed up to 25%.
Deprotection of the Boc group in DCM/trifluoroacetic acid at 0–10 °C is achieved smoothly to afford the corresponding amine, 15 in quantitative yield coupled with 99% purity. This amine, 15 when coupled with 6-chloropurine, 16 in acetonitrile with diisopropylethylamine (DIPEA; 1.5 eq.) and ZnCl2 (1.5 eq.) at 80–90 °C for 10–12 h afforded the corresponding diamide in 58% yield with 60% purity (Table 3; entry 1). Conversion is observed only 50% by TLC when the base is changed to triethylamine but only 39% yield coupled with 98% purity with 5% of enantiomer is observed after isolation (Table 3; entry 2). Reaction accelerates when zinc chloride loading is doubled (3.0 eq.) with DIPEA base and controlled the racemisation to below 0.5% (Table 3; entry 3). Reaction does not go to completion without zinc chloride and up to 20% of other isomer is formed (Table 3; entry 4). Reaction not moved at all in other bases, such as pyridine and ammonia (Table 3; entries 5 & 6). Conversion is also observed in water but 50% racemisation is observed.
| S. No. | Base/reagent | Input (g) | Output (g) | Yield (%) | Chemical purity by HPLC (% area) | Chiral purity by HPLC (% area) | Conversion by TLC (%) | |
|---|---|---|---|---|---|---|---|---|
| Product | Enantiomer | |||||||
| 1 | DIPEA/ZnCl2 | 10.0 | 6.0 | 58 | 60 | 98 | 2 | 70 |
| 2 | TEA/ZnCl2 | 5.0 | 2.0 | 39 | 98 | 95 | 5 | 50 |
| 3 | DIPEA/ZnCl2 | 10.0 | 6.0 | 58 | 78 | 99.5 | 0.5 | 75 |
| 4 | DIPEA | 10.0 | 3.0 | 29 | 60 | 80 | 20 | 30 |
| 5 | Pyridine/ZnCl2 | 3.0 | — | — | — | — | — | No conversion |
| 6 | NH4OH/ZnCl2 | 3.0 | — | — | — | — | — | |
Cyclisation of 17 to form the quinazoline ring closing by retaining the neighbouring chiral centre is a crucial step in the preparation of idelalisib, 1. To accomplish the cyclisation of diamide, 17 various reagents such as HMDS, HMDS/I2 and piperdine/I2 (Table 4) and solvents (Table 5) have been studied. Cyclization of diamide attempted in Zn/AcOH as per innovator process5a at room temperature but did not result in any conversion (Table 4; entry 1). Reaction did not progress at all with different reagent/solvent systems such as PTSA/toluene, DMF, formamide and trimethyl aluminium/toluene (Table 4; entries 2–5). Reaction also attempted with ZnCl2/acetonitrile9 but found no advantage (Table 4; entry 6). When the reagent system is changed to TPP/I2/piperidine in acetonitrile the product was obtained in 60% purity but racemisation could not be controlled (Table 4; entry 7). When the reaction is carried out in HMDS/I2 in DCM only 42% yield was observed with complete racemisation (Table 4; entry 8). Complete conversion is observed but racemisation is not controlled when the DCM replaced with toluene, though 60% purity of the product is obtained (Table 4; entry 9). Reaction also attempted by replacing iodine with ZnBr2 but product formation was not observed at all (Table 4; entry 10).10 When neat reaction was carried out in HMDS/I211 (Table 4; entry 11) 58% yield coupled with 98.9% purity was observed but racemisation could not be prevented. Finally, racemisation is completely arrested by changing the reagent to HMDS/ZnCl212 (Table 4; entry 12) in acetonitrile and obtained the compound with good purity 99.5% in 53% yield.
| S. No. | Base/reagent | Input (g) | Output (g) | Yield (%) | Chemical purity (%) | Chiral purity by HPLC (% area) | |
|---|---|---|---|---|---|---|---|
| Product | Enantiomer | ||||||
| 1 | Zn/AcOH | 0.5 | — | — | — | — | — |
| 2 | pTSA/toluene | 0.5 | — | — | — | — | — |
| 3 | DMF | 0.5 | — | — | — | — | — |
| 4 | Formamide | 0.5 | — | — | — | — | — |
| 5 | Trimethyl aluminium/toluene | 2.0 | — | — | — | — | — |
| 6 | ZnCl2/acetonitrile | 5.0 | — | — | — | — | — |
| 7 | TPP/I2/piperidine | 10.0 | 4.5 | 47 | 60 | 50 | 50 |
| 8 | HMDS/I2/DCM | 0.5 | 0.2 | 42 | 10 | 50 | 50 |
| 9 | HMDS/I2/toluene | 0.5 | 0.3 | 63 | 60 | 50 | 50 |
| 10 | HMDS/ZnBr2/toluene | 0.5 | — | — | — | — | — |
| 11 | HMDS/I2 | 20.0 | 11.0 | 58 | 98.9 | 50 | 50 |
| 12 | HMDS/ZnCl2/acetonitrile | 20.0 | 10.0 | 53 | 99.5 | 99.95 | 0.05 |
| S. No. | Solvent | Input (g) | Output (g) | Yield (%) | Chemical purity (%) | Chiral purity by HPLC (%) | |
|---|---|---|---|---|---|---|---|
| Product | Enantiomer | ||||||
| 1 | Toluene | 40 | 20 | 48 | 78 | 50 | 50 |
| 2 | t-Butanol | 20 | 10 | 48 | 90 | 50 | 50 |
| 3 | n-Butanol | 3 | 2 | 64 | 80 | 50 | 50 |
| 4 | 2-Methyl THF | 300 | 208 | 66 | 75 | 98.0 | 2.0 |
| 5 | Acetonitrile | 30 | 20 | 64 | 99.5 | 99.95 | 0.05 |
Having this result in hand further efforts were directed towards finding a suitable solvent to achieve ICH quality material coupled with good yield. Reaction in toluene afforded the product in 48% yield and 78% purity but racemised completely (Table 5; entry 1). Changing the solvent to t-butanol (Table 5; entry 2) or n-butanol (Table 5; entry 3) did not arrest the racemisation but improved yield in n-butanol. Reaction in 2-methyl THF afforded the similar yield as observed in n-butanol, however, racemisation controlled to 2% with reasonable purity (Table 5; entry 4). No conversion was observed when the reaction was carried out in water. Finally when reaction was carried out in acetonitrile racemisation was controlled to 0.05% level along with achieving high purity of 99.95% in 64% yield (Table 5; entry 5).
Finally, with all these results in hand the optimised process was executed in 150–190 g scale in the Laboratory. We observed good yield and purities in all the steps (Table 6).13
Experimental procedures
Preparation of 2-fluoro-6-nitro-N-phenylbenzamide, 4
To a DCM solution of 2-fluoro-5-nitro benzoic acid (100 g; 540 mmol), was added catalytic amount of DMF (7.3 g, 100 mmol) and oxalylchloride (102.8 g, 810 mmol) at ambient temperature and maintained for 3 h. After completion of the reaction which was monitored by TLC, the solvent was stripped off and the obtained residue was dissolved in 1,4-dioxane (80 mL) and kept a side. In another RBF having 250 mL 1,4-dioxane was added aniline (50.3 g; 541 mmol) and aq. sodium bicarbonate (90 g in 250 water) solution. To this aniline solution was added the above 1,4-dioxane solution at 0–5 °C, maintained at ambient temperature for 90 min. After completion of the reaction which was monitored by TLC, the reaction mass was quenched into water (1500 mL) and stirred for 2 h at ambient temperature. The obtained pale yellow colour solids were filtered, washed with water to afford 2-fluoro-6-nitro-N-phenylbenzamide in 86% yield and 99.0% purity which was proceed to next step without further purification. 1H NMR data were identical with those reported in the literature.5a,bPreparation of 2-amino-6-fluoro-N-phenylbenzamide, 13
To a mixture of methanol (500 mL) and dichloromethane (500 mL), was added 2-fluoro-6-nitro-N-phenylbenzamide (100 g; 385 mmol), zinc powder (140 g; 2154 mmol) and then slowly added aq. ammonium formate solution (dissolved 170 g ammonium formate in 300 mL water) and stirred the reaction at ambient temperature for 4–5 h. The progress of the reaction was monitored by TLC. After completion of the reaction zinc salts were filtered, washed with dichloromethane (500 mL). The solvent from filtrate was stripped off completely under vacuum and added water (1000 mL) to the obtained residue. The emitted white-pale pink colour solids were filtered washed with water to afford 2-amino-6-fluoro-N-phenylbenzamide in 90% yield. The product used as such to next step without further purification. Mp by DSC: 112.58 °C; 1HNMR (300 MHz, DMSO-d6) δ 10.27 (s, 1H), 7.63 (m, 2H), 7.33 (t, J = 7.5 Hz, 2H), 7.14 (m, 2H), 6.75 (d, J = 8.1 Hz, 1H), 6.42 (m, 2H), 5.7 (s, 2H); 13CNMR (75 MHz, DMSO-d6) δ 167.87, 163.03, 161.80, 158.58, 149.36, 139.15, 131.41, 128.50, 123.50, 120.25, 111.38, 108.60, 101.90; MS: m/z 231(M+ + H).Preparation of 2-{[(2S)-2-amino(butoxycarbonyl)butanoyl]amino}-6-fluoro-N-phenylbenzamide, 14
To a toluene (500 mL) solution of N-Boc-L-2-amino butyric acid (100 g; 493 mmol), was added 2-fluoro-6-amino-N-phenylbenzamide (100 g; 435 mmol) cooled to 8–15 °C and added carbonyldiimidazole (140.8 g, 869 mmol) followed by 1-hydroxybenzotriazole (117.4 g, 868.6 mmol) and stirred for 30–32 h. After completion of the reaction which was monitored by TLC, the mass was quenched into water (500 mL). The obtained precipitate was filtered, washed with water. The wet material was leached with acetonitrile (300 mL), filtered and dried to afford off-white coloured 2-{[(2S)-2-amino(butoxycarbonyl)butanoyl]amino}-6-fluoro-N-phenylbenzamide, 14 in 67% yield. Mp by DSC: 167.48 °C; 1HNMR (300 MHz, DMSO-d6) δ 10.54 (s, 1H), 9.93 (s, 1H), 7.99 (d, 1H, J = 8.4 Hz), 7.74 (m, 2H), 7.52 (m, 1H), 7.32 (m, 2H), 7.12 (m, 2H), 3.89 (broad s, 1H), 1.74 (m, 1H), 1.55 (m, 1H), 1.30 (s, 9H), 0.86 (t, J = 7.2 Hz, 3H); 13CNMR (75 MHz, DMSO-d6) δ 171.50, 161.41, 160.69, 157.43, 155.70, 138.60, 137.70, 131.75, 128.50, 124.07, 120.93, 119.96, 117.56, 115.75, 111.12, 78.33, 56.99, 28.00, 24.38, 10.54; MS: m/z 414(M+ − H).Preparation of 2-{[(2S)-2-amino(7H-purin)butanoyl]amino}-6-fluoro-N-phenylbenzamide, 17
To a solution of 2-{[(2S)-2-amino(butoxycarbonyl)butanoyl]amino}-6-fluoro-N-phenylbenzamide (100 g; 241 mmol) in dichloromethane (200 mL), was added trifluoroacetic acid (200 mL) and stirred for 3 h at ambient temperature. After completion of the reaction which was monitored by TLC, added water (2000 mL) to the reaction mass and adjusted the pH to neutral with saturated sodium bicarbonate solution. The obtained precipitate was filtered, washed with water and dried to afford 2-{[(2S)-2-aminobutanoyl]amino}-6-fluoro-N-phenylbenzamide, 15. Mp by DSC: 195.46 °C; 1HNMR (300 MHz, DMSO-d6) δ 10.64 (s, 1H), 8.13 (brs, 1H), 7.14 (d, J = 8.4 Hz, 2H), 7.56 (m, 2H), 7.50 (m, 2H), 7.32 (m, 1H), 7.13 (m, 1H), 3.96 (t, J = 6 Hz, 1H), 1.74 (q, 2H), 0.89 (t, J = 7.5 Hz, 3H); 13CNMR (75 MHz, DMSO-d6) δ 168.43, 160.62, 158.42, 158.01, 157.28, 139.01, 135.70, 131.10, 128.70, 123.81, 120.59, 119.40, 115.29, 112.75, 53.66, 24.54, 8.82; MS: m/z 316(M+ + H). The obtained compound was dissolved in acetonitrile (500 mL) and added 6-chloro purine (39 g; 253 mmol), zinc chloride (98.4 g, 722 mmol) followed by diisopropylamine (36.5 g, 361 mmol) and maintained at 80–100 °C for 10–12 h. After completion of the reaction which was monitored by TLC, the reaction mass was quenched into water (2000 mL), stirred for 3 h and filtered. The obtained crude product was purified by leaching with methanol (200 mL) and THF (200 mL), filtered and dried to afford 2-{[(2S)-2-amino(7H-purin)butanoyl]amino}-6-fluoro-N-phenylbenzamide, 17 in 60% yield. Mp by DSC: 267.99 °C; 1HNMR (300 MHz, DMSO-d6) δ 12.96 (s, 1H), 10.47 (s, 1H), 10.10 (s, 1H), 8.14 (t, J = 10.2 Hz, 2H), 7.89 (d, J = 8.1 Hz, 2H), 7.82 (d, J = 5.4 Hz, 1H), 7.48 (m, 2H), 7.24 (m, 2H), 4.79 (br s, 1H), 1.96 (m, 2H), 0.94 (t, J = 7.2 Hz, 3H); 13CNMR (75 MHz, DMSO-d6) δ 171.41, 161.05, 160.46, 157.20, 154.02, 151.96, 150.00, 139.18, 138.35, 137.34, 131.50, 131.37, 128.57, 123.93, 119.71, 118.30, 116.50, 111.37, 111.06, 56.34, 24.39, 10.70; MS: m/z 434(M+ + H).Preparation of 5-fluoro-3-phenyl-2-[(1S)-1-(9H-purin-6-ylamino)propyl]-4(3H)-quinazolinone, 1(idelalisib)
To a solution of 2-{[(2S)-2-amino(7H-purin)butanoyl]amino}-6-fluoro-N-phenylbenzamide, 50 g, 115 mmol), in acetonitrile (250 mL) was added zinc chloride (51 g, 374 mmol), hexamethyldisilazane (55.9 g, 347 mmol) and maintained the reaction mass at reflux for 4 h. After completion of the reaction which was monitored by TLC, the reaction mass was quenched into water (1250 mL), stirred for 2–3 h, filtered and washed with water. The obtained crude product was dissolved in DCM (500 mL), adjusted pH to 2.0–3.0 with 10% aq. HCl. The DCM phase separated and fresh DCM (500 mL) was added to the aq. phase and neutralised with aq. sodium carbonate. The obtained product is filtered, washed with water, dried and finally recrystallized from acetonitrile (500 mL) to afford a white coloured 5-fluoro-3-phenyl-2-[(1S)-1-(9H-purin-6-ylamino)propyl]-4(3H)-quinazolinone, 1 in 60% yield and 99.9% purity. The spectral data of the compound were identical with those reported in the literature.5a,bConclusion
In summary, we successfully developed a facile, novel and scalable strategy to prepare idelalisib. Cyclisation of the diamide without affecting the neighboring chiral center which is a challenging step was achieved smoothly. We further optimized this process and demonstrated the preparation in larger scale in the laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Dr Satyanarayana Chava, CEO and Dr Srihari Raju Kalidindi, Executive Director, Laurus Laboratories Ltd for their continuous encouragement and support. We also thank Mr. Narasimha Rao DVL and other Senior Management for their helpful discussions in carrying out this work.Notes and references
- (a) D. J. Hart, ARKIVOC, 2010, 32 CAS; (b) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166 RSC; (c) J. P. Michael, Nat. Prod. Rep., 2007, 24, 223 RSC; (d) J. P. Michael, Nat. Prod. Rep., 2005, 22, 603 RSC; (e) J. P. Michael, Nat. Prod. Rep., 2004, 21, 650 RSC; (f) J. P. Michael, Nat. Prod. Rep., 2002, 19, 7197 Search PubMed; (g) J. P. Michael, Nat. Prod. Rep., 2003, 20, 47 Search PubMed; (h) A. L. D'yakonov and M. V. Telezhenetskaya, Chem. Nat. Compd., 1997, 33, 221 CrossRef; (i) I. Khan, A. Ibrar, N. Abbas and A. Saeed, Eur. J. Med. Chem., 2014, 76, 193 CrossRef CAS PubMed; (j) L. He, H. Li, J. Chen and X.-F. Wu, RSC Adv., 2014, 4, 12065 RSC; (k) M. Demeunynck and I. Baussanne, Curr. Med. Chem., 2013, 20, 794 CAS; (l) S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787 CrossRef CAS; (m) D. J. Connolly, D. Cusack, T. P. O'Sullivan and P. J. Guiry, Tetrahedron, 2005, 61, 10153 CrossRef CAS; (n) Z. Ma, Y. Hano and T. Nomura, Heterocycles, 2005, 65, 2203 CrossRef CAS; (o) A. Witt and J. Bergman, Curr. Org. Chem., 2003, 7, 659 CrossRef CAS; (p) S. R. Padala, P. R. Padi and V. Thipireddy, Heterocycles, 2003, 60, 183 CrossRef.
- U. A. Kshirsagar, Org. Biomol. Chem., 2015, 13, 9336 CAS.
- E-mail: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206545lBl.pdf.
- http://www.ema.europa.eu/ema/index.jsp?curl=/pages/medicines/human/medicines/00384%203/human_med_001803.jsp.
- (a) K. W. Fowler, D. Huang, E. A. Kesicki, H. C. Ooi, A. Oliver, F. Ruan, J. Treiberg and K. Deep Puri, US Pat., 8980901, 2015; (b) K. Edward and Z. Paul, PCT application, WO2005/113554, 2005; (c) Z. Paul, K. Edward, T. Jennifer, B. Lisa, R. Matthew, O. HuaChee, W. Stephen, J. Angela and F. David, Org. Lett., 2007, 9, 1415 CrossRef PubMed.
- L. Guangyong, L. Xiaojun, F. Mingwei and G. Fengluan, CN patent, 104130261, 2014.
- X. Xu, CN patent, 104262344, 2015.
- D. Channe Gowda, B. Mahesh and S. Gowda, Indian J. Chem., 2001, 40B, 75 Search PubMed.
- M. R. Yadav, S. T. Shirude, A. Parmar, R. Balaraman and R. Giridhar, Chem. Heterocycl. Compd., 2006, 42, 1038 CrossRef CAS.
- S. Fujita, P. Y. Reddy and T. Toru, US Pat., 5965746, 1999.
- X. Lanting, Y. Jiang and M. Dawei, Org. Lett., 2012, 14, 1150 CrossRef PubMed.
- U. A. Kshirsagar, B. Mhaske Santosh and P. Argade Narshinha, Tetrahedron Lett., 2007, 48, 3243 CrossRef CAS.
- S. K. Dehury, N. Mekala, B. Srinivasa Rao, I. V. Sunil Kumar and V. Umamaheswara Rao, PCT International Application, 191608A1, 2017.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00407b |
| This journal is © The Royal Society of Chemistry 2018 |