
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron-doped graphene as a metal-free catalyst for gas-phase oxidation of benzyl alcohol to benzaldehyde
Wenjun
Cheng
a,
Xueting
Liu
a,
Na
Li
a,
Jiatong
Han
b,
Shuangming
Li
*ac and
Sansan
Yu
 *ac
*ac
aCollege of Chemical Engineering, Shenyang University of Chemical Technology, Shenyang 110142, China. E-mail: ssyu@syuct.edu.cn; lishuangming@syuct.edu.cn; Fax: +86 24 89383760; Tel: +86 24 89381190
bCollege of Environmental and Safety Engineering, Shenyang University of Chemical Technology, Shenyang 110142, China
cKey Laboratory of Chemical Separation Technology of Liaoning Province, Shenyang University of Chemical Technology, Shenyang 110142, China
First published on 20th March 2018
Abstract
Boron-doped graphene samples (BGs) with tunable boron content of 0–2.90 at% were synthesized and directly used in the gas-phase oxidation of benzyl alcohol to benzaldehyde, and showed excellent performance. XPS results indicated that the graphitic sp2 B species (BC3) is the mainly boron dopant species incorporated in the graphene lattice, which could significantly improve the content of ketone carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) on the graphene. For instance, the contents of CO jumped from 1.93 to 4.19 at% while BC3 doped into the graphene lattice was only 0.35 at%. The CO is the active site of catalytic reaction, so BG has significantly improved catalytic activity. Compared to the un-doped graphene (G), the conversion of benzyl alcohol over BGs increased 2.35 times and the selectivity of benzaldehyde increased from 77.3% to 99.2%. Aerobic–anaerobic exchange experiments revealed that the superior catalytic performance of BG was achieved only under aerobic conditions. The study of the boron-doped carbocatalyst may also provide guidance for the design of surface modified carbon-based catalysts for the selective oxidation dehydrogenation of alcohols by regulating doping elements and their types.
O) on the graphene. For instance, the contents of CO jumped from 1.93 to 4.19 at% while BC3 doped into the graphene lattice was only 0.35 at%. The CO is the active site of catalytic reaction, so BG has significantly improved catalytic activity. Compared to the un-doped graphene (G), the conversion of benzyl alcohol over BGs increased 2.35 times and the selectivity of benzaldehyde increased from 77.3% to 99.2%. Aerobic–anaerobic exchange experiments revealed that the superior catalytic performance of BG was achieved only under aerobic conditions. The study of the boron-doped carbocatalyst may also provide guidance for the design of surface modified carbon-based catalysts for the selective oxidation dehydrogenation of alcohols by regulating doping elements and their types.
1. Introduction
Selective oxidation of alcohols to the corresponding aldehydes, involving the cleavage of O–H bonds followed by α-C–H bond activation, is one of the crucially important reactions in the production of fine chemicals and intermediates.1,2 The selective oxidation dehydrogenation (ODH) of benzyl alcohol to benzaldehyde is a typical example. Among these, the type and structure of the catalyst have an important influence on the formation of the target product. Over the last several years, the most commonly used catalysts for selective oxidation of benzyl alcohol are organometallic compounds (TEMPO, Ru(PPh3)Cl3/TEMPO, VOPO4/TEMPO, etc),3–5 chromium(VI) reagents, noble metal coordination compounds (Pd, Ru, Au, Pt, etc)6–8 and stoichiometric quantities of inorganic oxidant (manganese dioxide, Sarrett reagent, Jones reagent, etc).9 However, these catalysts are precious and can produce large amounts of toxic pollution during the reaction process, contradicting the principles of green chemistry and sustainable development from both economic and environmental concepts. Consequently, sustaining efforts have been devoted to choosing the efficient and environmentally catalysts for the oxidation reaction to meet the increasingly stringent ecological standards.10,11Carbon-based catalysts, including graphene, carbon nanotubes, nanodiamond and other nanocarbon based materials, have attracted great interest in the past decades due to their sustainable development, eco-friendly, high specific surface area, feasible surface modification and continuous large-scalable preparation.12–16 Additionally, compared to metal based catalysts, carbon based materials provide additional advantages induced by the existence of giant π structures in the sp2 hybridization carbon atoms, which promotes strong interactions with various reactants especially for compounds with benzene ring.17 In the past several years, significant progress has been made in the study of catalytic performance of the carbon materials. For instance, Zhang et al. reported that carbon nanotubes with modified surface functionality could efficiently catalyze the ODH of n-butane to butenes with a high selectivity, which was achieved for periods as long as 100 hours.18 Diao et al. used reduced porous graphene oxide as a metal free catalyst for the selective ODH of ethylbenzene to styrene, which showed the best catalytic performance compared with other carbon materials (routinely reduced graphene oxide, graphite powder and oxidized carbon nanotubes) and commercial iron oxide.19 Zhang et al. described the application of nano-diamond as a novel catalyst for direct dehydrogenation of ethylbenzene under steam-free conditions. The unique sp2–sp3 hybrid structure resulted in different behavior to other carbons, with superior activity and stability and freedom from coke formation.20
Among these carbon-based materials, graphene, a single layer of sp2-bonded carbon atoms arranged in a hexagonal honeycomb structure, attracts extensive attention in recent years,21 due to its unique properties such as good conductivity,22 high specific surface area23 and high mobility of charge carriers,24etc. It is regarded as a promising material for potential application in catalysis. The microstructure and surface chemistry of nanocarbons have been thought to play important role in carbon-catalyzed reactions. For example, CO functional groups, locating at the prismatic edges of stacked graphene sheets or at the surface defects, have been regarded as the active centers for the ODH reactions.53–56 Furthermore, heteroatoms doping can offer an effective method to modulate electronic properties and surface physicochemical features of graphene, introduce a considerable amount of defects into the lattice, and promote catalytic performance by introducing electron acceptors or donors.25–30
For the chemical doping of graphene, Denis et al. demonstrated that boron and nitrogen atoms are considered to be the best doping elements because their sizes are closed to the carbon atoms,31 resulting in strong valence bonds between them. To date, nitrogen-doped graphene (NG) has been applied to organic synthesis reaction. Long et al. reported the NG as metal-free carbon-catalyst applied to selective oxidation of alcohols.21 Li et al. prepared NG with different doping contents, which could efficiently catalyze the dehydrogenation of ethanol with 100% selectivity.32 However, BG is mainly applied as electrode material in solar cells and lithium ion batteries.33–36 So far, there is no report used BG as catalyst directly for organic synthesis reaction, especially in gas-phase reaction. Theoretical calculations indicate that O2 molecular can be easily adsorbed by boron dopant owing to the large difference of electronegativity between boron and oxygen, which is the precondition for the subsequent formation of O2−, O2−, and O22−.30 In addition, boron can substitute carbon atom at the trigonal sites (BC3),37 whose electron accepting nature makes boron more affinitive to oxygen compared to carbon.38,39 Therefore, we speculate that BG will have certain influence on the oxidation dehydrogenation reaction, compared to G.
Herein, boron-doped graphene was used as catalysts for ODH of benzyl alcohols in order to demonstrate that incorporating boron is a viable method to design efficient carbon-based catalysts for the gas-phase oxidation of benzyl alcohol. Long et al. have been studied NG was used as catalyst for ODH of benzyl alcohol,21 what is worth mentioning that the reaction was a liquid-phase-batch reaction. While in this work, BG was used for the gas-phase-continuous reaction. In this study, boron-doped graphene samples with tunable boron content of 0–2.90 at% were synthesized by regulating the boric acid/graphene oxide (GO) mass ratio. It was found that the multilayer graphene doped with boron heteroatoms can catalyze oxidative dehydrogenation of benzyl alcohol to benzaldehyde efficiently with the high selectivity.
2. Experimental section
2.1. Preparation of catalysts
Graphite oxide (GO, purity of 96.0%) was supplied from Jining Leadernano Technology Co., LTD. (Shandong Province, China). GO was prepared from natural graphite by a modified Hummers' method.40,41 BGs synthesized at 1173 K were conducted in argon atmosphere for 2 h with the different mass ratio (1/1, 2/1, 3/1, 4/1 and 5/1) of boric acid/GO, and the corresponding products were denoted as BG-1, BG-2, BG-3, BG-4, BG-5. In the typical procedure, GO and boric acid were mixed uniformly in a nickel boat, which was then placed in the center of a corundum tube. The furnace was heated to 1173 K at a ramping rate of 5 K min−1 with a continuous flow of argon, then kept at 1173 K for 2 h before cooling down to room temperature naturally. The obtained products were then refluxed in 3 M KOH aqueous solution for 2 h to remove the unreacted boron oxide. After filtration and water washing, the samples were dried in a vacuum at 333 K for overnight. For comparison, G was prepared by using the same procedure, but neither without adding boric acid nor using 3 M KOH aqueous solution for reflux washing.2.2. Characterizations
The structure and purity of the products were identified by XRD, which was carried out on a D8 Advance X-ray generator (Bruker AXS Company, Germany) by using Cu Kα1 radiation. The X-ray intensity was measured over a 2θ diffraction angle from 5° to 60° with a step size of 0.02° min−1. The morphologies of catalysts were observed by scanning electron microscopy (SEM) (S-4800 FESEM, Hitachi Ltd., Japan) and transmission electron microscopy (TEM) (TECNAI G2 F20, FEI Company, USA). The SEM and TEM images were obtained at an accelerating voltage of 10 and 200 kV, respectively. Brunauer–Emmett–Teller (BET) (Autosorb-iQ, Quantachrome, America) surface area analysis was done by nitrogen adsorption–desorption isotherms at liquid nitrogen temperature of 77 K. X-ray photoelectron spectroscopy (XPS) (ESCALAB250, Thermo Electron Corporation, USA) was employed to analyze the surface composition of synthesized materials.2.3. Measurement of catalytic performance
The ODH experiments were carried out at atmospheric pressure using 100 mg of each catalyst in a fixed-bed stainless steel reactor. The reactor was heated at 10 K min−1 up to the reaction temperature under 40 mL min−1 nitrogen, and after that the feed comprising benzyl alcohol (BA), oxygen (nO2/nBA = 2) in nitrogen (nN2/nBA = 6.2) was dosed in the reactor at a total flow of 40 mL min−1. The ODH reactions were studied under relatively gentle conditions (473 and 523 K). The liquid-phase product collected after cooling by ice water were sampled periodically and analyzed by High Performance Liquid Chromatography (HPLC, Agilent 1260, USA) equipped with an Agilent Zorbax SB-C18 column. The reaction tail gas was monitored by an on-line Gas Chromatograph (Techcomp GC7980, China) equipped with a thermal conductivity detector (TCD) and a Porapak-Q column. Blank experiments without carbon catalyst showed that reaction conversion were negligible (<0.2%).3. Results and discussion
3.1. Characterization
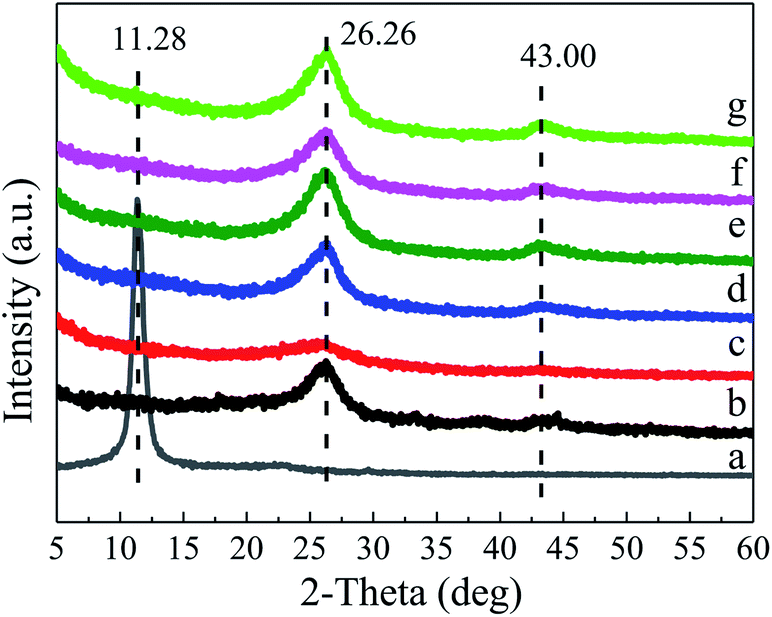 | ||
| Fig. 1 XRD patterns of (a) GO, (b) G, (c) BG-1, (d) BG-2, (e) BG-3, (f) BG-4 and (g) BG-5. | ||
 | ||
| Fig. 2 SEM, TEM and HRTEM images of the boron-doped graphene samples (BGs) and the un-doped graphene (G). | ||
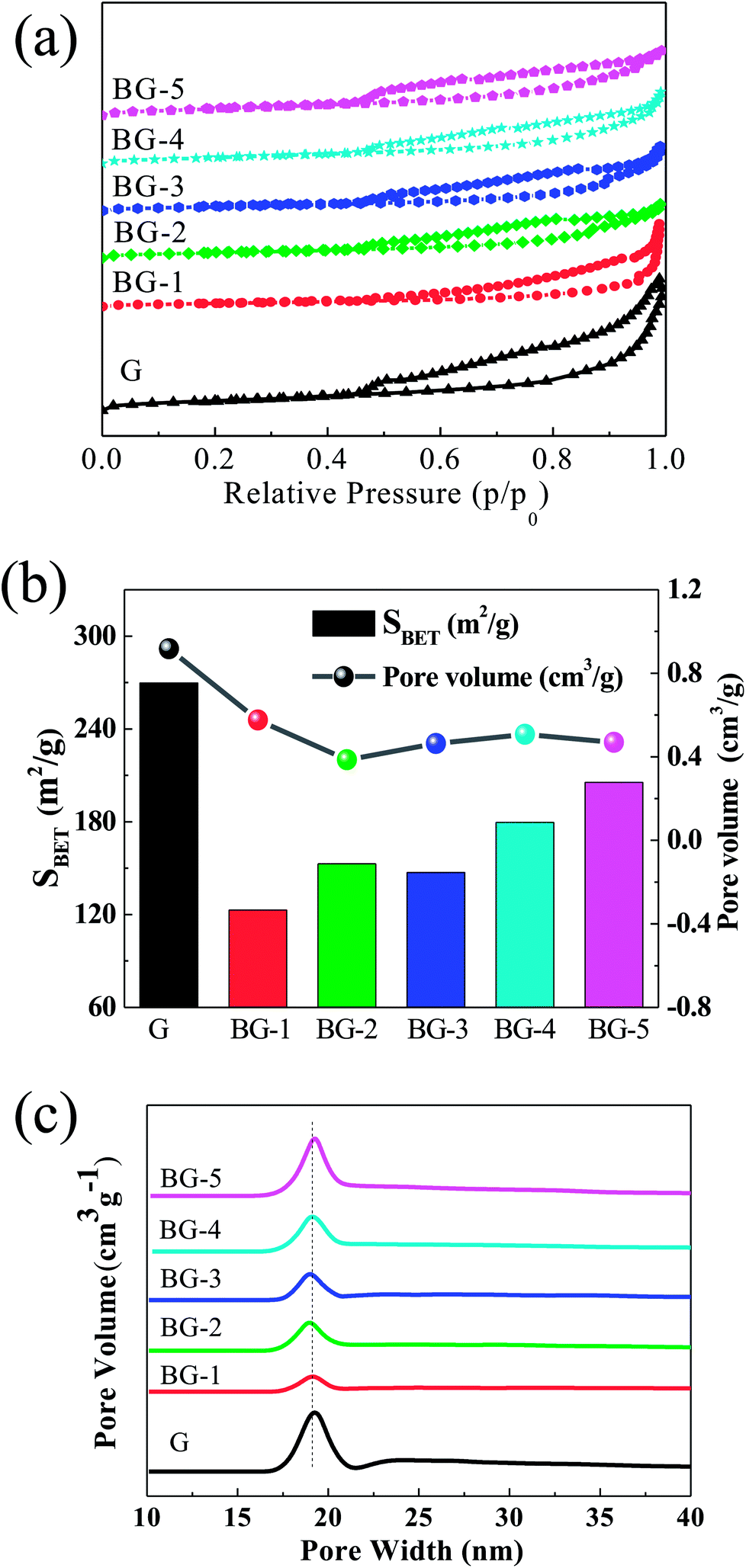 | ||
| Fig. 3 (a) Nitrogen adsorption–desorption isotherms, (b) specific surface area column (column graph) and pore volume (line graph) and (c) pore size distribution plot. | ||
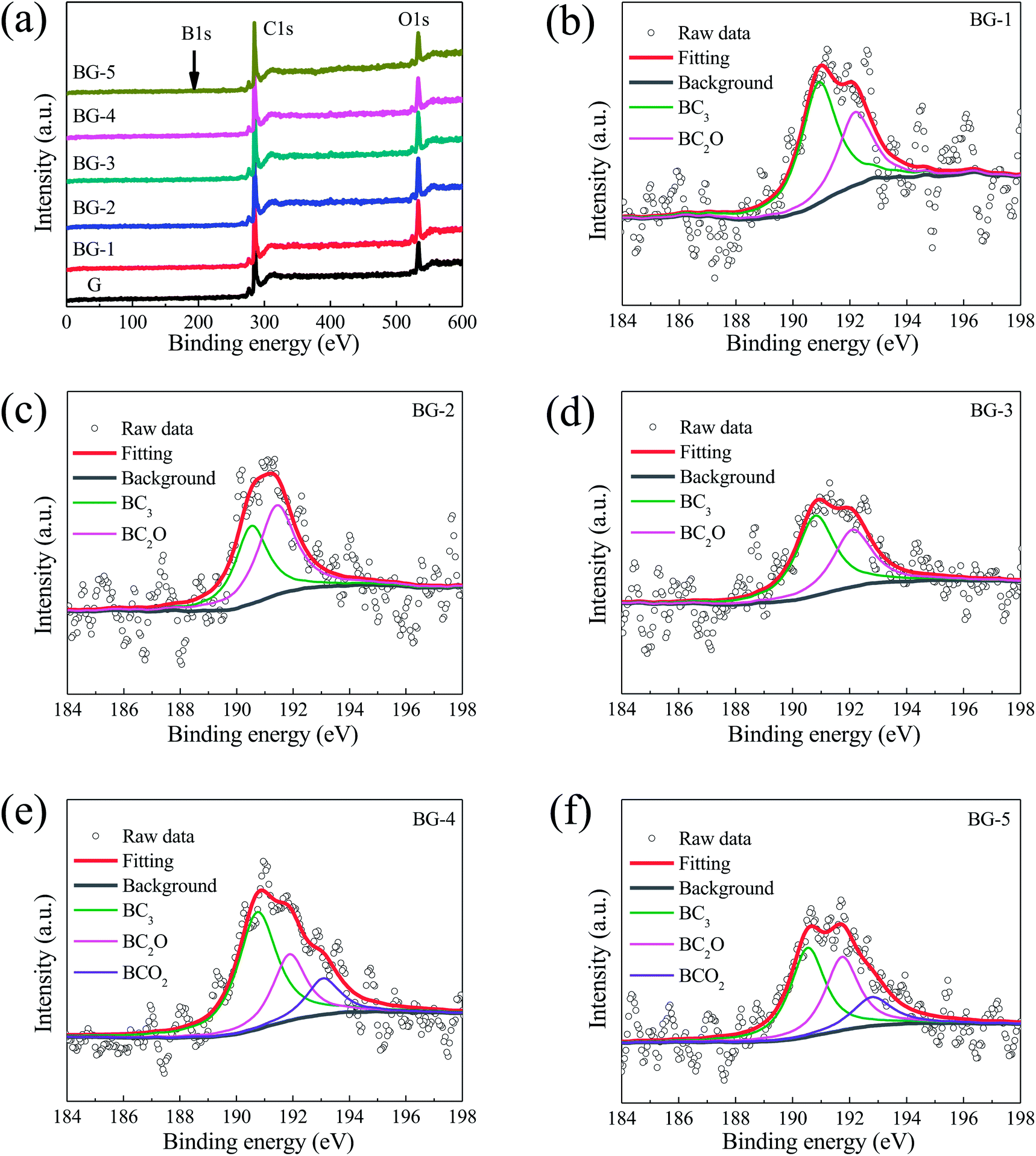 | ||
| Fig. 4 (a) XPS survey spectrum for the un-doped graphene (G) and BG samples with various boron dopant concentrations, (b)–(f) high resolution B1s spectra of the BG samples with various boron dopant concentrations, samples. | ||
In addition, the atomic contents of C, O, and B elements in these samples calculated from XPS spectra are summarized in Table 1. Interestingly, it is noted that the oxygen atomic percentage of the BG samples is higher than G and the oxygen content increases steadily with increasing boron content. Noticeably, the content of boron atoms in the BG samples increases with increasing of the mass ratio of boron acid and GO, and reaches the highest value of 2.90% for BG-4. Further increasing the mass ration to 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 does not improve the content of boron in BG, suggesting the boron-doping reaction is completed after increasing the mass ration of 4:1.46
:1 does not improve the content of boron in BG, suggesting the boron-doping reaction is completed after increasing the mass ration of 4:1.46
| Sample | O/Ca | C (at%)b | O (at%)b | B (at%)b | O | B | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| CO (at%)c |
O–CO (at%)c |
C–O–C/C–OH (at%)c | –BC3 (at%)c | BC2O (at%)c | BCO2 (at%)c | |||||
| a The ratio of surface O and C content. b Surface carbon, oxygen and boron contents were determined by XPS. c The content of various oxygen species and boron species obtained by deconvolution analysis. | ||||||||||
| G | 0.18 | 84.89 | 15.11 | — | 1.93 | 7.15 | 6.03 | — | — | — |
| BG-1 | 0.24 | 80.26 | 19.19 | 0.55 | 4.19 | 6.65 | 8.35 | 0.35 | 0.20 | — |
| BG-2 | 0.23 | 80.01 | 18.38 | 1.61 | 4.28 | 7.63 | 6.47 | 0.71 | 0.90 | — |
| BG-3 | 0.22 | 80.11 | 17.86 | 2.03 | 4.75 | 5.83 | 7.28 | 1.18 | 0.85 | — |
| BG-4 | 0.22 | 79.66 | 17.44 | 2.90 | 5.04 | 7.50 | 4.90 | 1.67 | 0.80 | 0.43 |
| BG-5 | 0.21 | 81.51 | 16.82 | 1.67 | 4.33 | 6.47 | 6.02 | 0.77 | 0.61 | 0.29 |
The effect of the precursors mass ratio on boron species was further analyzed by deconvoluting the B1s spectra. As shown in Fig. 4b–d, the high-resolution B1s peak can be fitted into two peaks centered at 191.28 and 192.60 eV, which indicates that there are two types of B species present in these BG nanosheets. However, Fig. 4e–f show that the B1s peak can be fitted into three peaks centered at 191.28 and 192.60 eV, and 193.50 eV, indicating that there are three types of B species present in BG-4 and BG-5 nanosheets. According to the literatures,21,47,48 the binding energies of 191.28, 192.60 and 193.50 eV are attributed to BC3, BC2O and BCO2, respectively. In addition, it can be seen from Table 1 that the BC3 are predominant in these B species, and a maximum content of 1.67 at% for BC3 species can be obtained upon the mass ratio at 4:1 (BG-4).
Fig. 5a displays the high-resolution O1s XPS spectra of the BGs and G. The broad O1s peak can be divided into three peaks centered at 531.20, 532.70, and 534.10 eV, belonging to CO, O–CO, and C–O–C/C–OH groups, respectively.49–52 As shown in Fig. 5b, the contents of CO increase with the boron incorporated in graphene enhancing, indicating that the boron dopant (BC3) in the lattice may contribute to the formation of the CO groups.
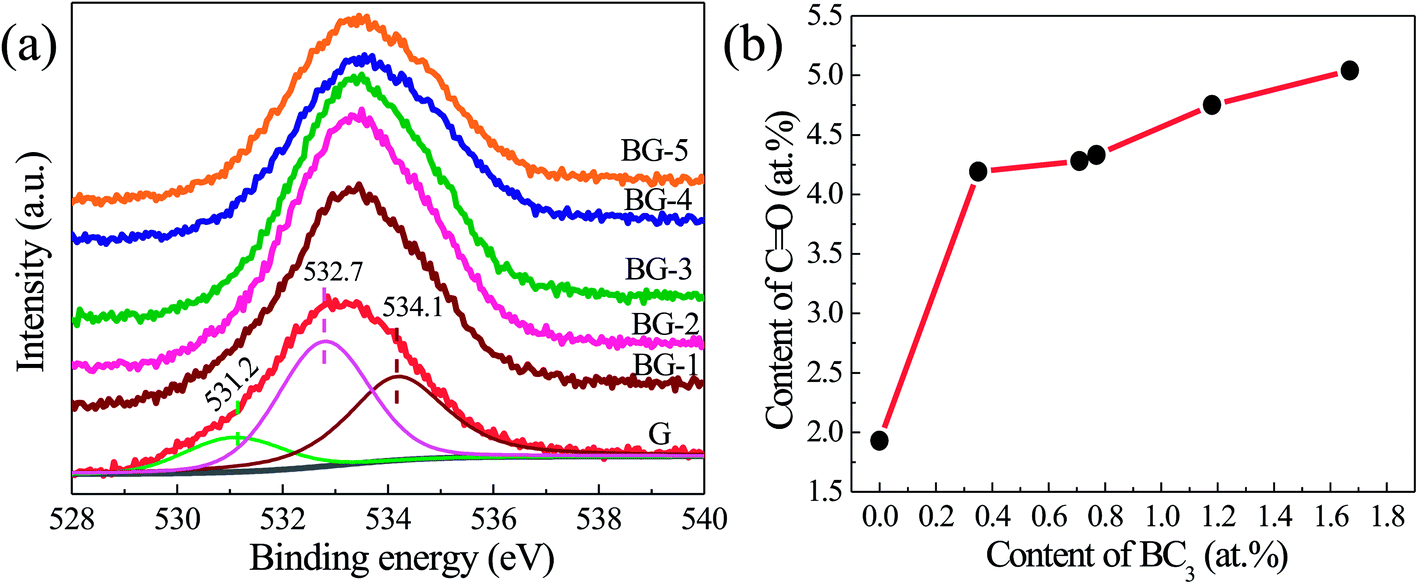 | ||
| Fig. 5 (a) High resolution O1s spectra of the un-doped graphene (G) and boron-doped graphene samples (BGs) and (b) the relationship between the content of the graphitic sp2 B species (BC3) and ketone carbonyl groups (CO) over different samples. | ||
3.2. Catalytic performance
Fig. 6a gives the conversion of benzyl alcohol over BGs with tunable boron content of 0–2.90 at% at 473 and 523 K. In the control experiment without catalyst, the conversion of benzyl alcohol detected by HPLC is no more than 0.20%. In the reference experiment with G as catalyst, the selectivity of benzaldehyde is 77.3% (473 K) and 85.1% (523 K), which is lower than 94.2–99.2% of BGs. It is indicated that the introduction of boron could produce benzaldehyde with the high selectivity. As shown in Table 2, results of the selective oxidation of alcohols to aldehydes over different metal-based catalysts and nano-carbon materials reported in recent years are summarized. Although the conversion efficiency of the metal-based catalyst is relatively high, the selectivity (no more than 82%) is generally lower than that of the BG and other nano-carbon catalyst (not less than 97%). Moreover, BGs are compared with other nano-carbon materials in the Table 2. The selectivity of the target products over the BGs and other nano-carbon catalyst are both high, and BGs have advantage in the conversion rate. In addition, the effect of the boron doping in the graphene is the enhancement of the conversion of raw materials. The highest catalytic activity of 11.7% (473 K) and 21.5% (523 K) for the conversion of benzyl alcohol can be obtained upon the boron doping content at 2.90 at% (BG-4). It can also be seen from the Fig. 6a that the amount of B doping is different, and the catalytic performance is different. The catalytic activities of the BGs for the aerobic oxidation increase with increasing boron content, for instance, the yield of benzaldehyde enhance from 5.6 to 11.0% at 473 K (9.9 to 20.9% at 523 K) with the content of boron from 0.55 to 2.90 at%. The similar changing trend between conversion of benzyl alcohol and the content of CO, as shown in Fig. 6b, indicates the CO groups are the active sites for the ODH reactions, which is consistent with the literature.20,53–56 More intuitively, from the Fig. 5b, the amount of CO increase with the content of BC3 enhancing. Based on these analyses, it was inferred that, doping of boron effectively enhanced the conversion of benzyl alcohol and the selectivity for benzaldehyde production. The CO alone cannot selectively oxidize benzyl alcohol, while only when BC3 and CO are both present, can a high yield of benzaldehyde be achieved.
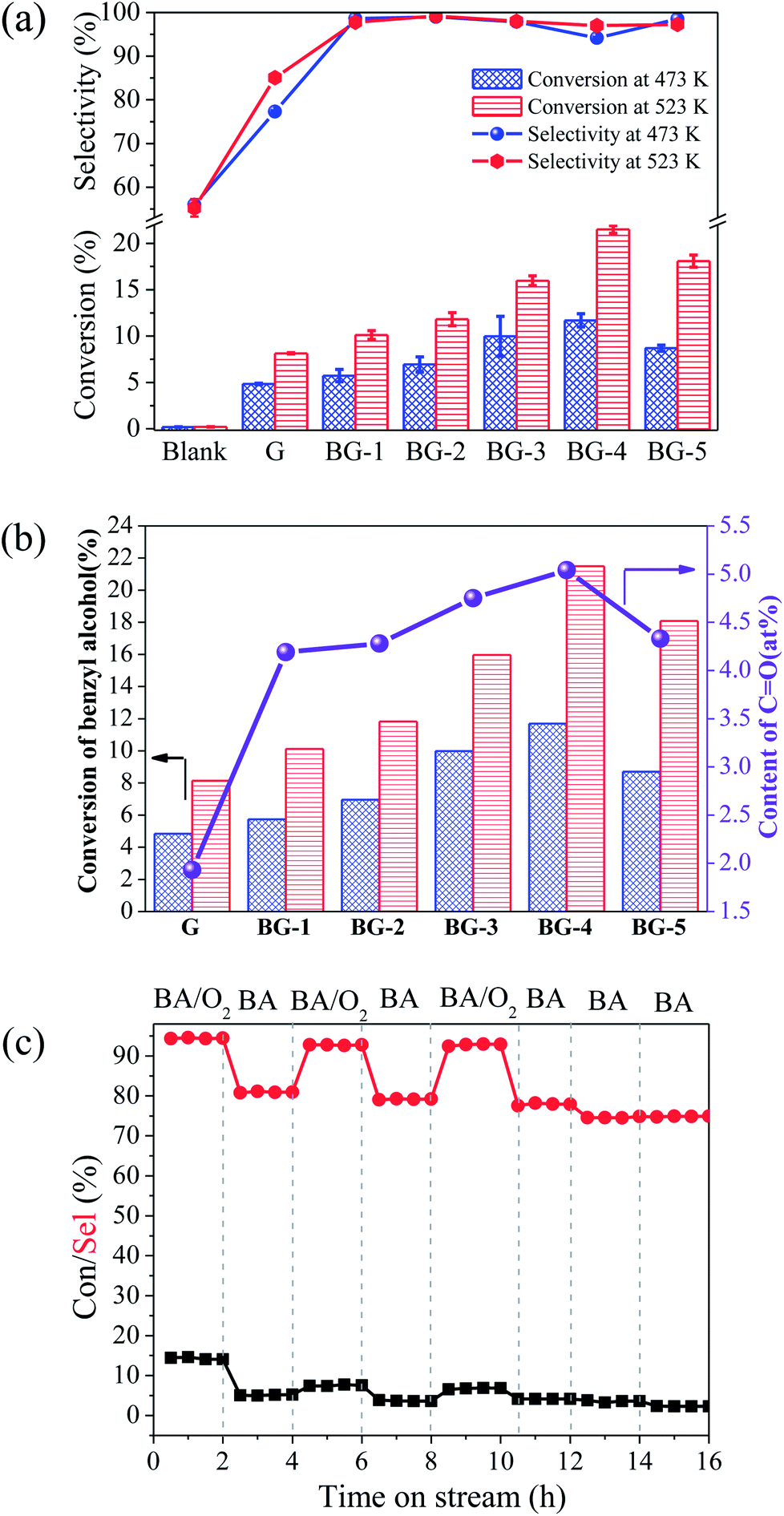 | ||
| Fig. 6 (a) The conversion of benzyl alcohol (column graph) and the selectivity of benzaldehyde (line graph) over different catalysts at 473 and 523 K, (b) the conversion of benzyl alcohol (column graph) and the content of CO (line graph) over different samples at 473 and523 K, (c) the conversion of benzyl alcohol (column graph) and the selectivity of benzaldehyde (line graph) on aerobic–anaerobic exchange experiments over BG-4 by switching BA/N2, and BA/O2/N2 at 473 K. BA = 9.0%, O2 = 18% with N2 as balance in aerobic conditions, and BA = 9.0% with N2 as balance in anaerobic conditions, total flow = 40 mL min−1. | ||
| Reaction systems | Catalyst | Doping amount (at%) | Phase/solvent | Reaction temperature (K) | Reaction type | Reaction time (h) | Conversion (%) | Selectivity (%) | Reference |
|---|---|---|---|---|---|---|---|---|---|
|
a The three N-doped graphene samples were denoted as NG-T, where T means nitridation temperature.
b The NG (x:1) means the nitrogen-doped graphene with different nitrogen doping contents, where x:1 means a mass ratio between urea and graphite oxide (GO).
c Boron-doped graphene samples were synthesized with the different mass ratio (1/1, 2/1, 3/1, 4/1 and 5/1) of boric acid/GO and the corresponding products were denoted as BG-1, BG-2, BG-3, BG-4 and BG-5.
|
|||||||||
| Benzyl alcohol to benzaldehyde | TiO2/Cu(II) | — | Liquid/H2O | 312 | Batch | 6 | 84 | 63 | 58 |
| Benzyl alcohol to benzaldehyde | Ru/g-C3N4 | — | Liquid/H2O | Room T | Batch | 4 | 73 | 72 | 59 |
| Benzyl alcohol to benzaldehyde | Au/MgO | — | Liquid/no solvent | 403 | Batch | 5 | 51 | 86 | 60 |
| Benzyl alcohol to benzaldehyde | Au/Al2O3 | — | Liquid/no solvent | 403 | Batch | 5 | 69 | 65 | 60 |
| Benzyl alcohol to benzaldehyde | Au/La2O3 | Liquid/no solvent | 403 | Batch | 5 | 52 | 69 | 60 | |
| Benzyl alcohol to benzaldehyde | Au/Sm2O3 | — | Liquid/no solvent | 403 | Batch | 5 | 44 | 75 | 60 |
| Benzyl alcohol to benzaldehyde | Au/CuO | — | Liquid/no solvent | 403 | Batch | 5 | 27 | 69 | 60 |
| Benzyl alcohol to benzaldehyde | NG-800a | 4.16 | Liquid/H2O | 343 | Batch | 3 | 6.5 | 100 | 21 |
| NG-900a | 3.48 | 343 | 3 | 12.8 | 100 | ||||
| NG-900a | 3.48 | 343 | 3 | 3.5 | 100 | ||||
| NG-1000a | 1.71 | 343 | 3 | 4.0 | 100 | ||||
| Ethanol to acetaldehyde | NG(30:1)b |
6.61 | Gas/— | 473–623 | Continuous | — | 8.0–10.7 | 100 | 32 |
| NG(200:1)b |
6.74 | — | 6.2–8.9 | 100 | |||||
| NG(300:1)b |
6.98 | — | 8.7–10.8 | 100 | |||||
| Benzyl alcohol to benzaldehyde | BG-1c | 0.55 | Gas/— | 523 | Continuous | — | 10.1 | 97.8 | This work |
| BG-2c | 1.61 | — | 11.8 | 99.2 | |||||
| BG-3c | 2.03 | — | 16.0 | 98 | |||||
| BG-4c | 2.90 | — | 21.5 | 97 | |||||
| BG-5c | 1.67 | — | 18.1 | 97.3 | |||||
In addition, aerobic-anaerobic exchanges by switching BA/N2 and BA/O2/N2 at 473 K experiments were carried out, and the results were as shown in Fig. 6c. About 14.0% of benzyl alcohol conversion and 94.0% of benzaldehyde selectivity are obtained under coexistence of benzyl alcohol and oxygen, whereas quick deactivation occurs once oxygen was removed. Fan et al. reported that the lower benzaldehyde yield in the absence of O2 implied oxygen is necessary to shift the equilibrium toward the carbonyl compound and accelerate the reaction.57 The reaction occurs without added oxygen supported the so-called classic dehydrogenation mechanism where the alcohol dehydrogenation and oxidant reduction steps are sequential, and not coupled. Therefore, the result convinced us that the superior catalytic performance of boron doped graphene was achieved only under aerobic conditions.
4. Conclusions
In summary, we study surface physical and chemical property of BGs and G and their catalytic performance in the gas-phase oxidation benzyl alcohol to benzaldehyde, which thoroughly revealing how the BGs enhanced the performance in the ODH reaction. The effect of the boron doping in the graphene is the enhancement of the conversion of benzyl alcohol and the selectivity of benzaldehyde. The superior catalytic performance of BGs is attributed to boron doping species (BC3) enhanced the O2 chemisorption on catalysts and further promoted forming the active sites (CO). It can be observed with XPS analysis, the BC3 could stimulate the increase in the amount of CO obviously, and thus promoted the C–H bond activation ability of BG. This study provides a direction to the heteroatom doped on the graphene for the oxidative dehydrogenation reaction of benzyl alcohol. We anticipate that the heteroatom doped on the surface microstructure of graphene can open a new and fascinating route for designing highly efficient nanocarbon catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Liaoning BaiQianWan Talents Program, China (No. 2013921046), and Natural Science Foundation of Liaoning Province, China (No. 2015020245 and 20170540714).References
- R. A. Sheldon, I. W. C. E. Arends, G. T. Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS.
- R. A. Sheldon, I. W. C. E. Arends and A. Dijksman, Catal. Today, 2000, 57, 157–166 CrossRef CAS.
- A. Dijksman, A. Marinogonzalez, A. M. I. Payeras, A. I. W. C. E. Arends and R. A. Sheldon, J. Am. Chem. Soc., 2001, 123, 6826–6833 CrossRef CAS.
- Z. Du, J. Ma, H. Ma, M. Wang, Y. Huang and J. Xu, Catal. Commun., 2010, 11, 732–735 CrossRef CAS.
- Z. Du, J. Ma, H. Ma, J. Gao and J. Xu, Green Chem., 2010, 12, 590–592 RSC.
- T. F. Blackburn and J. Schwartz, Chem. Commun., 1977, 157–158 RSC.
- M. J. Schultz, R. S. Adler, W. Zierkiewicz, T. Privalov and M. S. Sigman, J. Am. Chem. Soc., 2005, 127, 8499–8507 CrossRef CAS.
- M. Lee and S. Chang, Tetrahedron Lett., 2000, 41, 7507–7510 CrossRef CAS.
- J. Gross, K. Tauber, M. Fuchs, N. G. Schmidt, A. Rajagopalan, K. Faber, W. M. F. Fabian, J. Pfeffer, T. Haas and W. Kroutil, Green Chem., 2014, 16, 2117–2121 RSC.
- D. S. Mannel, S. S. Stahl and T. W. Root, Org. Process Res. Dev., 2014, 18, 1503–1508 CrossRef CAS.
- M. J. Schultz and M. S. Sigman, Tetrahedron, 2006, 62, 8227–8241 CrossRef CAS.
- C. Su and K. P. Loh, Acc. Chem. Res., 2013, 46, 2275–2285 CrossRef CAS.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS.
- X. Fan, G. Zhang and F. Zhang, Chem. Soc. Rev., 2015, 44, 3023–3035 RSC.
- H. Hu, J. H. Xin, H. Hu, X. Wang and Y. Kong, Appl. Catal., A, 2015, 492, 1–9 CrossRef CAS.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed.
- P. Serp and J. L. Figueiredo, Carbon materials for catalysis, John Wliey & Sons, Hoboken, New Jersey and Canada, 2009, pp. 20–23 Search PubMed.
- J. Zhang, X. Liu, R. Blume, A. Zhang, R. Schlogl and D. S. Su, Science, 2008, 322, 73–77 CrossRef CAS.
- J. Diao, H. Liu, J. Wang, Z. Feng, T. Chen, C. Miao, W. Yang and D. S. Su, Chem. Commun., 2015, 51, 3423–3425 RSC.
- J. Zhang, D. S. Su, R. Blume, R. Schlogl, R. Wang, X. Yang and A. Gajovic, Angew. Chem., Int. Ed. Engl., 2010, 49, 8640–8644 CrossRef CAS.
- J. Long, X. Xie, J. Xu, Q. Gu, L. Chen and X. Wang, ACS Catal., 2012, 2, 622–631 CrossRef CAS.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS.
- M. D. Stoller, S. J. Park, Y. W. Zhu, J. H. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem., 2010, 49, 9336–9344 CrossRef CAS.
- H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335–3345 RSC.
- M. Pumera and C. H. A. Wong, Chem. Soc. Rev., 2013, 42, 5987–5995 RSC.
- B. Guo, Q. Liu, E. Chen, H. Zhu, L. Fang and J. R. Gong, Nano Lett., 2010, 10, 4975–4980 CrossRef CAS.
- D. Geng, S. Yang, Y. Zhang, J. Yang, J. Liu, R. Li, T.-K. Sham, X. Sun, S. Ye and S. Knights, Appl. Surf. Sci., 2011, 257, 9193–9198 CrossRef CAS.
- R. Lv and M. Terrones, Mater. Lett., 2012, 78, 209–218 CrossRef CAS.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Electrocatalysis, 2011, 50, 7132–7135 CAS.
- P. A. Denis and C. Pereyra Huelmo, Carbon, 2015, 87, 106–115 CrossRef CAS.
- S. Li, W. Wang, X. Liu, X. Zeng, W. Li, N. Tsubaki and S. Yu, RSC Adv., 2016, 6, 13450–13455 RSC.
- Z. Sheng, H. Gao, W. Bao, F. Wang and X. Xia, J. Mater. Chem., 2012, 22, 390–395 RSC.
- T. Lin, F. Huang, J. Liang and Y. Wang, Energy Environ. Sci., 2011, 4, 862–865 RSC.
- X. Li, L. Fan, Z. Li, K. Wang, M. Zhong, J. Wei, D. Wu and H. Zhu, Adv. Energy Mater., 2012, 2, 425–429 CrossRef CAS.
- Z. Wu, W. Ren, L. Xu, F. Li and H. Cheng, ACS Nano, 2011, 5, 5463–5471 CrossRef CAS.
- D. W. Wang, F. Li, Z. G. Chen, G. Q. Lu and H. M. Cheng, Chem. Mater., 2008, 20, 7195–7200 CrossRef CAS.
- P. A. Denis, C. P. Huelmo and F. Iribarne, Comput. Theor. Chem., 2014, 1049, 13–19 CrossRef CAS.
- P. A. Denis and F. Iribarne, Chem. Phys. Lett., 2016, 658, 152–157 CrossRef CAS.
- H. Kim, M. Seo, M. H. Park and J. Cho, Angew. Chem., Int. Ed. Engl., 2010, 49, 2146–2149 CrossRef CAS.
- C. Zhu, S. Guo, Y. Fang and S. Dong, ACS Nano, 2010, 4, 2429–2437 CrossRef CAS PubMed.
- L. Tang, Y. Wang, Y. Li, H. Feng, J. Lu and J. Li, Adv. Funct. Mater., 2009, 19, 2782–2789 CrossRef CAS.
- H. Guo, M. Peng, Z. Zhua and L. Suna, Nanoscale, 2013, 5, 9040–9048 RSC.
- L. Niu, Z. Li, W. Hong, J. Sun, Z. Wang, L. Ma, J. Wang and S. Yang, Electrochim. Acta, 2013, 108, 666–673 CrossRef CAS.
- L. Wang, Y. Ye, X. Lu, Z. Wen, Z. Li, H. Hou and Y. Song, Sci. Rep., 2013, 3, 3568 CrossRef.
- T. Kwon, H. Nishihara, H. Itoi, Q. Yang and T. Kyotani, Langmuir, 2009, 25, 11961–11968 CrossRef CAS PubMed.
- C. H. Choi, S. H. Park and S. I. Woo, ACS Nano, 2012, 6, 7084–7091 CrossRef CAS.
- Z. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Mullen, Adv. Mater., 2012, 24, 5130–5135 CrossRef CAS.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. D. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. Field, C. Ventrice and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS.
- D. Long, W. Li, L. Ling, J. Miyawaki, I. Mochida and S. Yoon, Langmuir, 2010, 26, 16096–16102 CrossRef CAS.
- R. Arrigo, M. Hävecker, S. Wrabetz, R. Blume, M. Lerch, J. McGregor, E. P. J. Parrott, J. A. Zeitler, L. F. Gladde, A. Knop-Gericke, R. Schlögl and D. S. Su, J. Am. Chem. Soc., 2010, 132, 9616–9630 CrossRef CAS.
- L. Shi, W. Qi, W. Liu, P. Yan, F. Li, J. Sun and D. Su, Catal. Today, 2017, 301, 48–54 CrossRef.
- J. Zhang, X. Liu, R. Blume, A. Zhang, R. Schlogl and D. S. Su, Science, 2008, 322, 73–77 CrossRef.
- Y. Zhang, R. Huang, Z. Feng, H. Liu, C. Shi, J. Rong, B. Zong and D. Su, J. Energy Chem., 2016, 25, 349–353 CrossRef.
- W. Qi, W. Liu, X. Guo, R. Schlogl and D. Su, Angew. Chem., Int. Ed. Engl., 2015, 54, 13682–13685 CrossRef CAS.
- J. Wang, R. Huang, Z. B. Feng, H. Y. Liu and D. S. Su, Chemsuschem, 2016, 9, 1820–1826 CrossRef CAS.
- Y. Dai, X. Yan, Y. Tang, X. Liu, L. Xiao and J. Fan, ChemCatChem, 2012, 4, 1603–1610 CrossRef CAS.
- L. d. P. P. R. D. Spasiano, J. C. Olleros, S. Malato, R. Marotta and R. Andreozzi, Appl. Catal., B, 2013, 56–63 CrossRef.
- M. J. Lima, P. B. Tavares, A. M. T. Silva, C. G. Silva and J. L. Faria, Catal. Today, 2017, 287, 70–77 CrossRef CAS.
- V. R. Choudhary, A. Dhar, P. Jana, R. Jha and B. S. Uphade, Green Chem., 2005, 7, 768 RSC.
| This journal is © The Royal Society of Chemistry 2018 |