
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTranscriptome analysis reveals GA induced apoptosis in HCT116 human colon cancer cells through calcium and p53 signal pathways
Chen Yang *abc,
Xiaoyu Xied,
Hu Tangabc,
Xuyan Dongabc,
Xiaodong Zhangd and
Fenghong Huang*abc
*abc,
Xiaoyu Xied,
Hu Tangabc,
Xuyan Dongabc,
Xiaodong Zhangd and
Fenghong Huang*abc
aOil Crops Research Institute, Chinese Academy of Agricultural Sciences, Wuhan 430062, P. R. China. E-mail: yangchen@caas.cn; huangfh@oilcrops.cn
bKey Laboratory of Oilseeds Processing, Ministry of Agriculture, Wuhan 430062, P. R. China
cHubei Key Laboratory of Lipid Chemistry and Nutrition, Wuhan 430062, P. R. China
dDepartment of Cell Biology, College of Life Sciences, Wuhan University, Wuhan 430072, P. R. China
First published on 3rd April 2018
Abstract
Gallic acid (GA) is a polyphenol widely found in numerous fruits and vegetables that has been reported to exert anticancer effects, including apoptosis, against cancer cell lines. However, little is known about the induction of apoptotic effects and the underlying mechanism. We used RNA-seq to examine differentially expressed genes in human colon cancer HCT116 cells after 12 h and 24 h exposure to GA. A total of 792 and 911 genes with known functions showed significantly different expression levels in 12 h and 24 h GA-treated HCT116 cells, respectively. KEGG enrichment analysis showed that the identified genes were involved in pathways such as cholinergic synapse, circadian entrainment, calcium signal processing and transport, arachidonic acid metabolism and the p53 signal pathway. Real-time quantitative PCR was used to validate the reliability of the results obtained by RNA-seq. The results of this study indicate that GA triggers apoptosis in HCT116 cells through obstructing the growth of cells in the early phase treatment by down-regulation of calcium channels and then up-regulation of the intrinsic p53 signal pathway through activation of apoptosis caspases, finally leading to the mitochondrial apoptosis pathway.
Introduction
Colorectal cancer (CRC) is the third most common type of cancer and the third leading cause of cancer-related death in the United States.1 CRC is common in both men and women and occurs when malignant tumors form on the inner wall of the large intestine.2 Epidemiological studies show that most cases (∼85%) of CRC are considered to be sporadic cases rather than due to hereditary factors.3 The sporadic development of colon cancer is tightly linked to environmental factors and individual lifestyle factors, including dietary habits, alcohol consumption and physical exercise.4 The conventional treatment of colon cancer includes surgery, chemotherapy and radiofrequency ablation; however, the effectiveness of chemotherapy is hampered by the disadvantages of toxicity and drug resistance.5 Thus, the identification of drugs with lower toxicity, such as plant-derived bioactive compounds, that might play a role in the remedy or prevention of CRC is vital.6,7Hydroxycinnamic and hydroxybenzoic acids are common dietary phenolic acids derived from many edible plants and micronutrients that exhibit significant biological activities in the human body.8 Gallic acid (GA) is a model hydroxybenzoic acid that is widely distributed in many edible plants such as vegetables, several legumes, fruits, hickory nuts.9,10 GA displays marked biological activities, including antioxidative,11 anti-inflammatory,12 antibacterial,13 and anti-virus activities.14 In recent years, GA has attracted much interest from scientists due to its capability of inducing apoptosis in several types of cancer cell lines, including HL-60RG,15 LL-2,16 MCF-7 (ref. 17) and SCC-4 (ref. 18) cells. In particular, a previous report showed that GA could induce cell death in CoLo205 and HCT-15 colon cancer lines.19 However, the apoptotic mechanism of GA in colon cancer lines has not been fully elucidated.
Over the past decade, gene expression profiles have been applied into drug development to understand and predict the activity of chemical compounds in pre-clinical test.20 High throughput data created by large scale “omic’’ approaches provide a comprehensive overview and easily reveal impacts on biological pathways and processes that had not previously been found before.21 RNA sequencing (RNA-seq) is an unbiased sequencing tool that uses whole-genome transcriptome profiling to detect gene expression changes in a cell or tissue sample.22,23
In this study, the apoptosis mechanism of GA was investigated in HCT116 colon cancer cells. We used RNA-seq technology to evaluate the changes in the gene expression profile induced by GA treatment of HCT116 colon cancer cells to identify the regulatory pathways and modulated genes that may represent potential therapeutic targets.
Experimental
Reagents and cell culture
The human cancer cell lines HCT116, RKO and SW48 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The HCT116 and RKO were cultured in McCoy's 5A (Invitrogen, Carlsbad, CA, USA), SW48 was grown in DMEM medium (HyClone, Logan, UT, USA). Both medium was supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA) and 1% penicillin–streptomycin (Invitrogen, Carlsbad, CA, USA). The cells were incubated in 5% CO2 at 37 °C. Cells in the logarithmic growth stage were used for the experiments. GA (Sigma Chemical, St. Louis, MO, USA) was dissolved in DMSO (Sigma Chemical, St. Louis, MO, USA) at a stock concentration of 10 mM and then diluted in water to a working concentration.Cell viability assay
Cells were seeded into 96-well flat-bottom plates at a density of 1 × 104 per well and cultured overnight, and then treated with increasing dosages of GA for 12 h, 24 h, or 48 h. Cell viability was assayed using the Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's instructions. Cells were incubated with 10 μL of CCK-8 solution for 1 h at 37 °C, and the absorbance was then determined by a spectrophotometer set ELx800 (BioTek, Winooski, VT, USA) at a wavelength of 450 nm.Flow cytometry assay
For apoptosis analysis, cells (1 × 105) were examined using Annexin V-FITC and PI (FITC Annexin V Apoptosis Detection Kit I, BD, CA, USA) according to the manufacturer's instructions. Briefly, HCT116 cells were collected, washed with ice-cold PBS and resuspended in 100 μL binding buffer. Then, 2 μL of Annexin V-FITC and 5 μL of PI were added to the cells. After 15 min at room temperature in the dark, an additional 400 μL of binding buffer was added and the cells were subjected to FACS cytometry (BD Biosciences, San Jose, CA, USA). The results were analyzed with Summit version 4.3 software.RNA extraction and cDNA synthesis
The cells were seeded on 100 × 20 mm dishes at 5 × 105 cells per dish and incubated overnight. Cells were harvested after treatment with 50 μM GA or with 0.5% DMSO (negative control) for 12 h or 24 h. Total RNA was extracted using TRIzol reagent (Life Technologies, Waltham, MA, USA) and each treatment had three duplicates. RNA purity was monitored on 1% agarose gels. RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA).Library preparation and transcriptome sequencing
Total RNA sample libraries were extracted using the Illumina TruSeq Stranded mRNA LT sample preparation kit (Illumina, San Diego, CA, USA), according to the standard manufacturer's instructions. rRNA was removed from the total RNA using the rRNA removal kit (Illumina, San Diego, CA, USA). cDNA was then synthesized from RNA using Super Script II Reverse Transcriptase (Invitrogen, CA, USA); the first strand of cDNA was synthesized with hexahedron random primers and reverse transcriptase, and the second strand cDNA was synthesized using the first strand of cDNA as template. After the library was constructed, the fragmented library was enriched by PCR amplification, and then fragments of 300–400 bp were selected. PCR was then performed with universal PCR primers and an Index (X) Primer. Finally, the PCR products were purified by AMPure XP system (Beckman coulter, Kraemer Boulevard, CA, USA) and the library quality was assessed using the Agilent Bioanalyzer 2100 system.Bioinformatic analysis
Transcriptome sequencing was performed by Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China) based on the Illumina HiSeq platform using a 150 cycle paired-end sequencing strategy. FastQC was used for quality control; a quality score Q > 20 of reads were used for downstream analysis. The reference genome sequence was downloaded from the Ensembl Genome Browser (http://www.ensembl.org/index.html) as the mapping reference. The reads were mapped against the reference using TopHat v2.0.1028,29 and Bowtie v2.1.030 after quality control.Bioinformatics analysis. HTSeq v0.6.1 was used to count the numbers of reads mapped to each gene and gene expression levels were presented as RPKM (reads per kilobase of exon model per million mapped reads). We used the DESeq (version 1.18.0) to detect DEGs with the gene symbol annotation (FDR < 0.05, fold change > 2). In addition, principle component analysis (PCA) was carried out on the significantly expressed genes among all different groups. The Gene Ontology (GO) annotations curated by the European Bioinformatics Institute were imported in the CLC Genomics Workbench v.7.0 and annotations of the DEGs were processed. Enrichment analysis of DEGs was conducted using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis.
Real-time quantitative PCR (qPCR)
Reverse transcription was performed using the TaKaRa PrimeScript™ RT-PCR Kit (TaKaRa, Tokyo, Japan) and qRT-PCR was performed with TransStart Top Green qPCR SuperMix (TRANSGEN, Beijing, China) on the CFX384 Touch™ (Bio-Rad, Hercules, CA, USA) following the manufacturer's protocol. The following thermal profile was used: an initial 30 second denaturation step at 95 °C, followed by 40 cycles at 95 °C for 15 second and at 55 °C for 15 second and finally at 72 °C for 20 second. The data were analyzed using 7500 software v.2.0.1 (Applied Biosystems, Foster City, CA, USA) for Ct determination. Gene expression levels were calculated using the comparative ΔΔCt method and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was used as an internal control (primer sequences 5′–3′ are CCCCACCACACTGAATCTCC and GTACATGACAAGGTGCGGCT). The results are provided as the mean and standard deviation of three technical replicates.Results
Effect of GA on cell proliferation
The effects of GA on colon carcinoma cell proliferation and growth were evaluated by CCK8 assay. As shown in Fig. 1a, GA treatment for 24 h remarkably decreased cell viability in HCT116 and SW48 cells in a dose-dependent manner. The susceptibility of HCT116 cells to GA was higher than that of SW48 and RKO cells. The IC50 value of GA treatment for HCT116 cells and SW48 cells were 54.8 μM and 41.4 μM, respectively, at 24 h. We also found that GA had an inhibitory effect on HCT116 cells in a time-dependent manner (Fig. 1b). The viability of HCT116 cells sharply dropped at 12 h to 60.5% and reached 34.8% at 24 h upon treatment with 50 μM GA. However, there were no significant differences between cell viability at 12 h and 24 h at lower GA concentrations (12.5 μM and 25 μM) examined.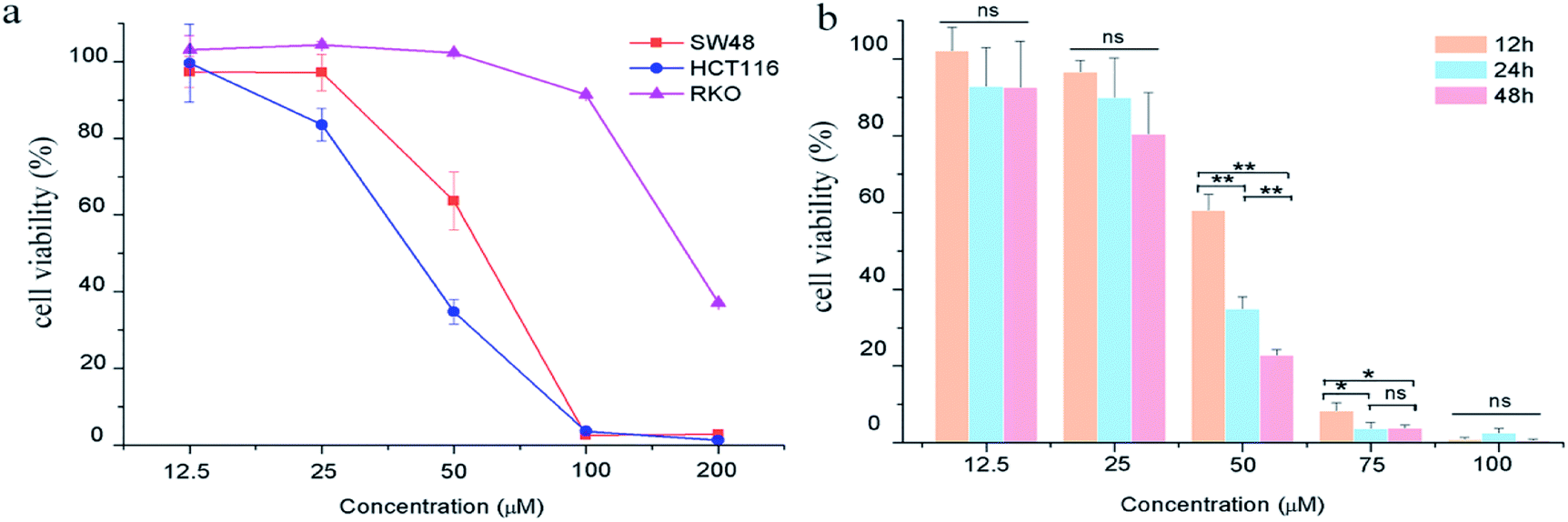 | ||
| Fig. 1 The effects of GA on colon carcinoma cell proliferation. (a) The cell viability of HCT116, RKO and SW48 cells treated with GA for 24 h. (b) The cell viability of HCT116 cells treated with GA for 12 h, 24 h and 48 h. Statistically significant differences p < 0.05 and p < 0.01 between different treated cells are denoted by * and ** respectively, “ns” means there was no significant difference between treated and untreated cell. | ||
Effect of GA on apoptosis in HCT116 cells
To further explore the mechanisms underlying the inhibitory effects of GA, cell apoptosis was assessed using Annexin V-PI dual staining and flow cytometric analysis (Fig. 2). The percentages of later apoptotic cells (in the upper right quadrant) were significantly increased by treatment with GA at 25 μM (12.3 ± 3.2%), 50 μM (62.2 ± 4.5%), and 100 μM (92.8% ± 7.7%) for 24 h in comparison with the control (7.2 ± 0.5%, P < 0.05). The early apoptotic cell proportions (in the lower right quadrant) in response to 25 μM, 50 μM and 100 μM GA were 6.3 ± 0.4%, 6.2 ± 0.6% and 1.6 ± 0.5%, respectively. These results suggested that GA could partially inhibit the proliferation of HCT116 cells by inducing apoptosis.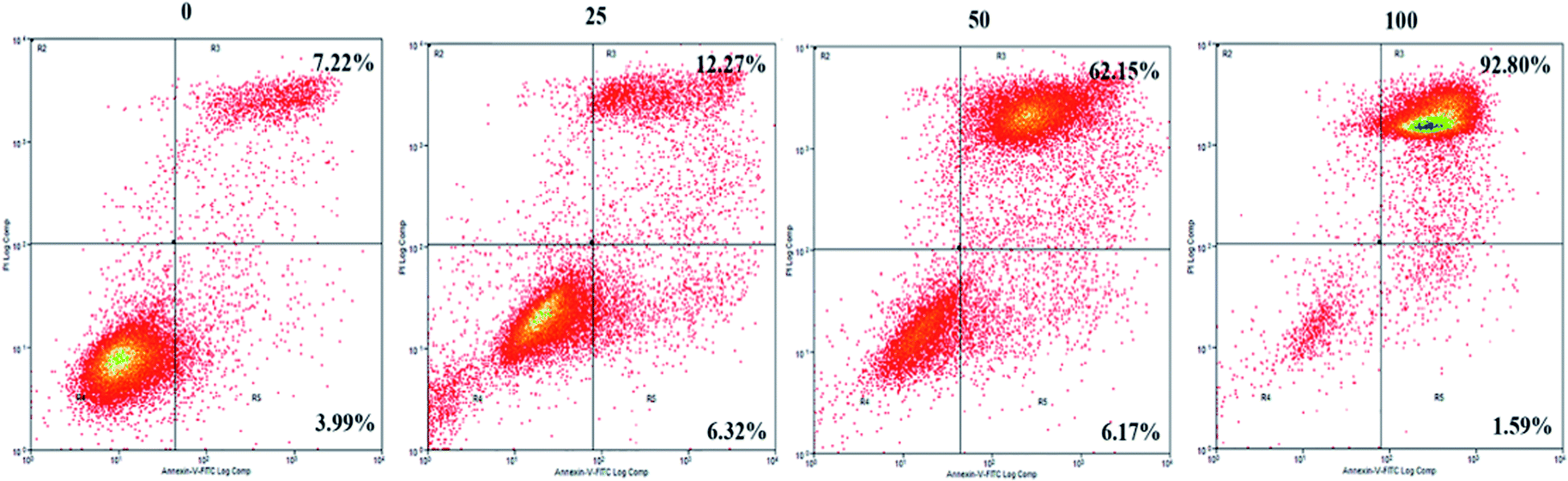 | ||
| Fig. 2 The effects of GA on apoptosis of HCT116 cells. Cell incubation for 24 h with 0, 25, 50 and 100 μM of GA was followed by analysis using flow cytometry. | ||
Effect of GA on gene expression patterns
To determine the expression patterns of mRNAs during GA treatment, heat maps were constructed to profile the overall transcriptome differences at different time points. More than 67% of genes were expressed (RPKM > 1) in each sequenced sample. Heat map representation of differently expressed transcripts revealed a similarity in the transcriptomes within the three triplicate runs of untreated group (C1, C2, C3), 12 h treatment group (GA12_1, GA12_2, GA12_3) and 24 h treatment group (GA24_1, GA24_2, GA24_3), respectively (Fig. 3a). Principal component analysis suggested that similar samples within a group clustered together, which was in agreement with the heat map (Fig. 3b). The first two principal coordinates explained total 93% of the variation observed among all the groups. The cells treated with GA for 12 h formed a distinct cluster from the untreated cells and cells treated for 24 h.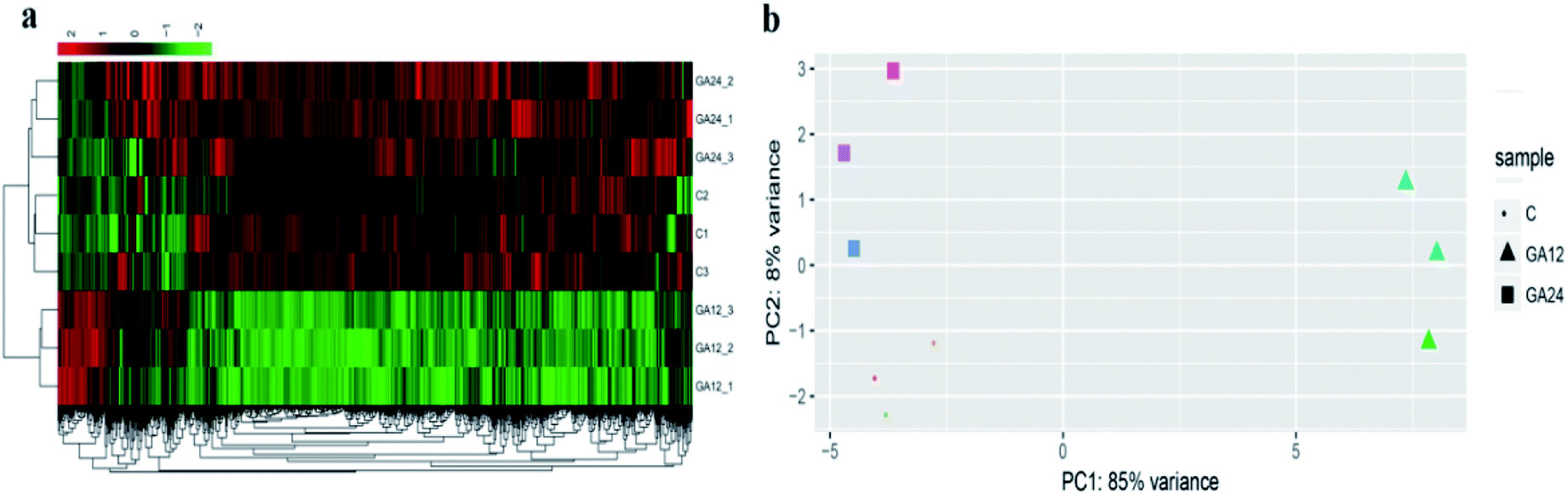 | ||
| Fig. 3 Gene expression profiles of HCT116 cells treated with GA. (a) Hierarchical clustering of differentially expressed genes in HCT116 cells based on log10-transformed expression values (RPKM). (b) Principle component analysis (PCA) on the complete transcriptome of HCT116 cells in the GA treatment group (GA group) and control group (C group). | ||
For differential gene expression analysis, pairwise comparisons of datasets from HCT116 cells treated with GA at different time points were performed. Only genes with a significant fold change (fold change ≥ 2; P < 0.05) were included in this analysis. As shown in Fig. 4a, the expressions of 61 genes (7.7%) were increased and 730 (92.3%) genes were decreased after 12 h GA treatment. In contrast, over 93.2% differentially expressed genes (DEGs) (849 genes) were up-regulated and only 6.8% DEGs (62 genes) were down-regulated during the treatment progress from 12 h to 24 h. There were 40 transcripts that fluctuated in the GA24 group compared with the untreated group C. We further identified the shared DEGs between each pairwise comparison groups and the numbers of DEGs among these samples are visualized in the Venn diagrams (Fig. 4b). Transcriptional profiling showed that 648 DEGs were modulated both in the first 12 h and later 12 h of the treatment process. Notably, the regulation pattern of these shared 648 genes was discrepant during the first 12 h and later 12 h treatment process. 611 down-regulated genes in the GA12-C group were turned into up-regulation in the GA24-GA12 group. 37 up-regulated genes in the GA12-C group then were down-regulated in the GA24-GA12 group. 14 genes DEGs were detected both in GA24-C group and GA24-GA12 group.
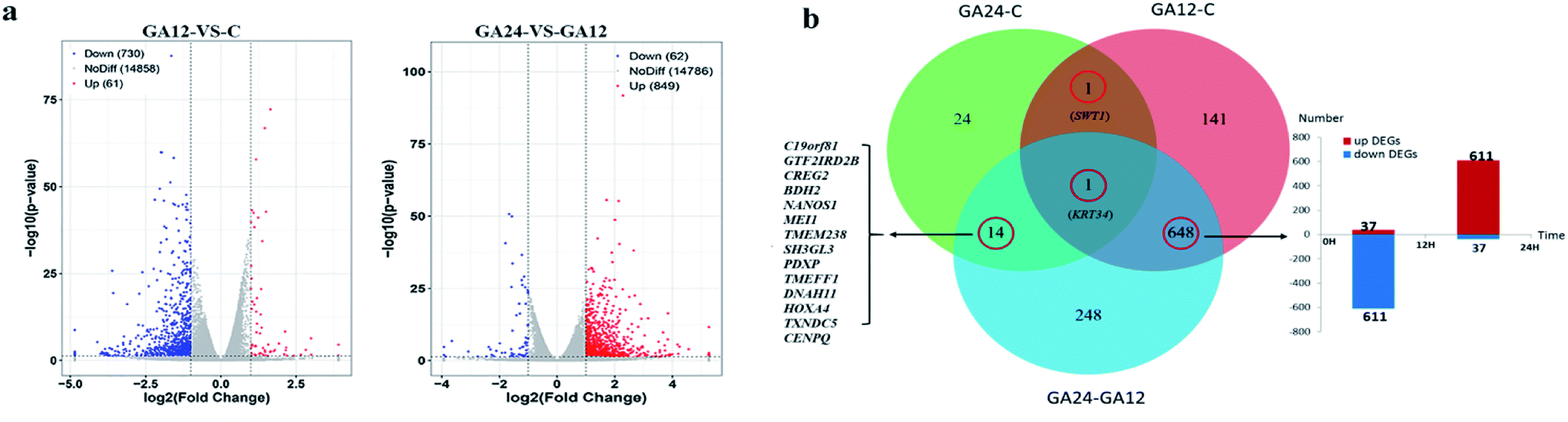 | ||
| Fig. 4 Overview of the differentially expressed genes for each comparison samples. (a) Volcano plots of differentially expressed genes between treated group GA12 with control group C, and treated group GA24 with treated group GA12 (b) Venn diagram depicting the differentially expressed genes common to different experimental exposure time. | ||
GO and KEGG enrichment analysis of DEGs
To understand the functions of the DEGs and the biological processes involved in GA treatment to HCT116 cells, we carried out GO enrichment analysis and classified them into three independent GO categories. Each DEG could be assigned to more than two GO terms and the top 30 enriched GO terms are shown in Fig. 5a. A total 738 out of 791 DEGs from the C-GA12 group could be assigned to a total of 4185 GO terms, including 3219 biological process terms, 558 molecular function terms and 408 cellular component terms. According to the three main categories of the GO classification, “calcium ion binding”, the “plasma membrane”, the “extracellular region” and “single-multicellular organism process” were most prevalent. Only a few DEGs classified under “Ras guanyl-nucleotide exchange factor activity” and “developmental maturation” were also observed.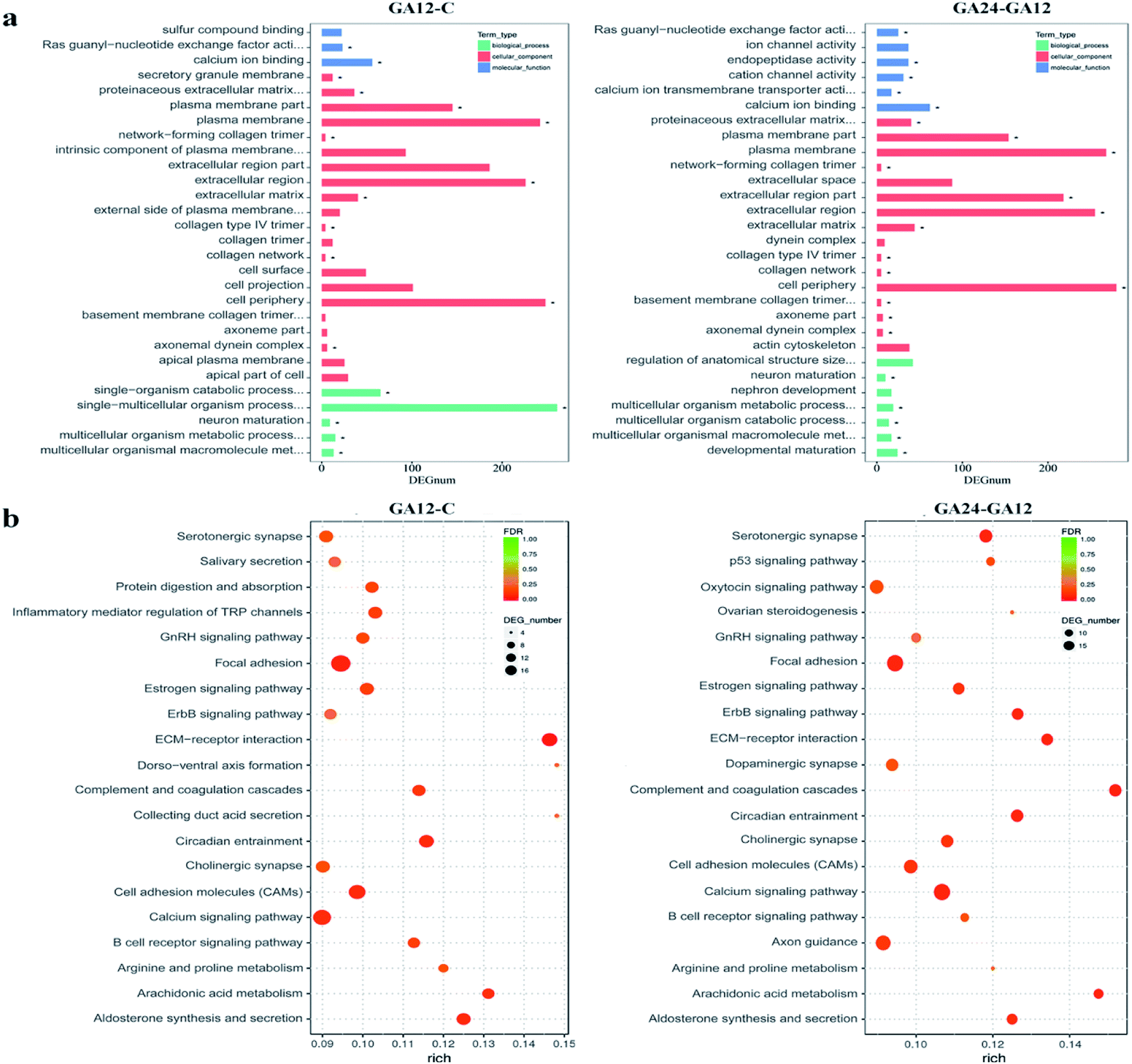 | ||
| Fig. 5 GO and KEGG enrichment analysis of differentially expressed genes. (a) Histograms of the top 30 enriched GO terms. Enriched items were selected with a corrected. p-Value 0.05. (b) Scatter plots of the top 20 enriched KEGG pathway terms. Enriched items were measured by the rich factor, q value (q < 0.05) and the number of genes. | ||
As treatment time extended, the number of DEGs increased. About 92.8% (845 out of 911) DEGs identified between GA12 and GA24 were categorized into 4577 GO terms. The “calcium ion binding” and related channel, “plasma membrane”, “extracellular region” and “cell periphery” showed the highest enrichment and the “network-forming collagen trimer” and “multicellular organism metabolic process” were much lower enrichment GO terms. Only the GO term “FMN binding” was significantly enriched when compared the untreated samples (C group) to cells treated with GA for 24 h (GA24 group).
All the DEGs were mapped to the reference pathway in the KEGG database to identify the response pathway operating during the treatment process. However, only a few genes could be annotated to the KEGG pathways and most were involved metabolism and signal transduction. Fig. 5b displays the top 20 significantly enriched KEGG pathways for the DEGs during the first 12 h and the later 12 h treatment with GA. Of the 791 DEGs detected in GA12 samples, only 178 had a KO ID and were associated with 119 pathways. The enriched pathways were mainly related to “Organismal Systems” and “Environmental Information Processing”. Among these pathways, the “calcium signaling pathway, “inflammatory mediator regulation of TRP channels”, “cholinergic synapse” and “ErbB signaling pathway” were highly enriched. The enriched “arginine and proline metabolism” and “circadian entrainment”, which were related to lipid metabolism and environmental adaptation were also detected. Further exposure to GA induced more DEGs, of which 182 genes have predicted involvement in 192 pathways. The enriched pathways were mainly related to “organismal systems” and “environmental information processing”. The highly enriched pathways were “Calcium signalling pathway”, “Arachidonic acid metabolism”, “Cell adhesion molecules (CAMs)” and “ErbB signalling pathway”. Two pathways “focal adhesion” and “p53 signalling pathway” associated with “cellular community” and “cell growth and death” were also observed.
Metabolic pathways related to proliferation and apoptosis
Genes from the top 10 significantly enriched pathways could be considered as the important gene expression regulators in response to GA treatment, especially the ones shared between the C-GA12 group and GA12-GA24 group (Table 1). Most gene expression patterns among other pathways also showed initial down-regulation and then up-regulation during the 24 h treatment. But the gene Per2 encoding period circadian clock 2 in the circadian entrainment pathway, gene NRG1 encoding neuregulin 1, and gene THBS1 encoding thrombospondin 1 were up-regulated in the C-GA12 group and down-regulated in the GA12-GA24 group. Genes involved in the arachidonic acid metabolism pathway were down-regulated in the first 12 h and then up-regulated in the last 12 h, such as ALOX5 and ALOX15 genes encoding members of the lipoxygenase gene family, which catalyze the synthesis of leukotrienes from arachidonic acid. The calcium signalling pathway involved the most number of DEGs, among them, CACNA1G, CACNA1A, CACNA1I encoded calcium voltage-gated channel (VGC) subunits. Purinergic receptor P2RX7 belonged to receptor-operated calcium channel (ROC), and calcium release-activated calcium modulator ORAI3 was considering as store-operated calcium channels (SOC). They were all found down-regulated in GA12-C group and up-regulated in GA24-GA12. Two transmembrane protein-coding genes PDGFRB, BDKRB2 encoding signal receptors were also found down-regulated first and then up-regulated. The left four DEGs in the calcium signal pathway: CAMK2B, PLCD4, PLCB4, ADCY4 and PRKCG genes encoding important enzymes also differentially expressed in other 7 pathways including cholinergic synapse, circadian entrainment, ErbB signalling pathway, estrogen signalling pathway, serotonergic synapse GnRH signalling pathway and Inflammatory mediator regulation of TRP channels. There were 7 DEGs found in the p53 signalling pathway, THBS1, SESN3 and ADGRB1 were down-regulated in the first 12 h and then up-regulated in the last 12 h as the same as most DEGs. However, TP53INP1, RASA4, IGFBP3 and BBC3 which are directly related to cell cycle and death, were up-regulated only in the GA12-GA24 group.| Gene name | Gene description | GA12-vs-C | GA24-vs-GA12 | ||
|---|---|---|---|---|---|
| log2 fold change | q value | log2 fold change | q value | ||
| a Significant differences p < 0.05, p < 0.01 and p < 0.001 of log2 fold change are denoted by “*”, “**” and “***” respectively. The genes without significantly different expression level were marked as “—”. Expression information of each DEG is listed when first mentioned or its name was boldly showed following the pathway name. | |||||
| Calcium signalling pathway (PDGFRB P2RX7 PLCD4 ORAI3 BDKRB2 PLCB4 CAMK2B SLC8A2 ADCY4 CACNA1G CACNA1A CACNA1I PRKCG) | |||||
| PDGFRB | Platelet derived growth factor receptor beta | −1.62 | * | 2.14 | *** |
| P2RX7 | Purinergic receptor P2X 7 | −1.40 | *** | 1.54 | *** |
| PLCD4 | Phospholipase C delta 4 | −1.42 | — | 1.40 | — |
| ORAI3 | ORAI calcium release-activated calcium modulator 3 | −1.56 | *** | 2.07 | *** |
| BDKRB2 | Bradykinin receptor B2 | −2.11 | ** | 1.70 | * |
| PLCB4 | Phospholipase C beta 4 | −1.47 | *** | 1.19 | * |
| CAMK2B | Calcium/calmodulin dependent protein kinase II beta | −1.53 | ** | 2.03 | ** |
| SLC8A2 | Solute carrier family 8 member A2 | −1.30 | ** | 1.27 | *** |
| ADCY4 | Adenylate cyclase 4 | −2.10 | * | — | — |
| CACNA1G | Calcium voltage-gated channel subunit alpha1 G | −1.63 | ** | 1.63 | ** |
| CACNA1A | Calcium voltage-gated channel subunit alpha1 A | −1.63 | ** | 1.82 | ** |
| CACNA1I | Calcium voltage-gated channel subunit alpha1 I | −1.29 | ** | 1.48 | *** |
| PRKCG | Protein kinase C gamma | −1.61 | *** | 1.52 | *** |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| p53 signalling pathway (THBS1 SESN3 ADGRB1 IGFBP3 BBC3 TP53INP1 RASA4) | |||||
| THBS1 | Thrombospondin 1 | 1.32 | ** | −1.41 | ** |
| SESN3 | Sestrin 3 | −2.18 | ** | 2.48 | ** |
| ADGRB1 | Adhesion G protein-coupled receptor B1 | −1.69 | ** | 1.67 | ** |
| IGFBP3 | Insulin like growth factor binding protein 3 | — | — | 1.49 | * |
| BBC3 | Bcl2 binding component 3 | — | — | 1.02 | *** |
| TP53INP1 | Tumor protein p53 inducible nuclear protein 1 | — | — | 1.97 | *** |
| RASA4 | RAS p21 protein activator 4 | — | — | 1.32 | *** |
|
|||||
| Arachidonic acid metabolism (PLA2G4D ALOX5 AKR1C3 ALOX15 PTGDS GGT5 CYP4F2) | |||||
| PLA2G4D | Phospholipase A2 group IVD | −4.11 | *** | 4.17 | *** |
| ALOX5 | Arachidonate 5-lipoxygenase | −1.90 | *** | 1.83 | *** |
| AKR1C3 | Aldo-keto reductase family 1 member C3 | −1.93 | ** | 2.45 | *** |
| ALOX15 | Arachidonate 15-lipoxygenase | −2.30 | ** | 2.44 | ** |
| PTGDS | Prostaglandin D2 synthase | −3.00 | *** | 2.79 | *** |
| GGT5 | Gamma-glutamyltransferase 5 | −1.49 | *** | 1.76 | *** |
| CYP4F2 | Cytochrome P450 family 4 subfamily F member 2 | −2.29 | ** | 2.14 | ** |
|
|||||
| Cholinergic synapse (CHRM4 PLCB4 FOS GNG13 CREB3L3 CAMK2B CACNA1A PRKCG ADCY4) | |||||
| CHRM4 | Cholinergic receptor muscarinic 4 | −1.20 | *** | 1.33 | *** |
| PLCB4 | Phospholipase C beta 4 | −1.47 | *** | 1.19 | * |
| FOS | Fos proto-oncogene, AP-1 transcription factor subunit | −1.99 | *** | 1.99 | *** |
| GNG13 | G protein subunit gamma 13 | −1.55 | *** | 1.78 | ** |
| CREB3L3 | cAMP responsive element binding protein 3 like 3 | −2.01 | *** | 2.13 | *** |
|
|||||
| Circadian entrainment (PER2 KCNJ5 CACNA1G CACNA1I PRKCG PLCB4 FOS GNG13 CAMK2B ADCY4) | |||||
| PER2 | Period circadian clock 2 | 1.36 | *** | −1.55 | *** |
| KCNJ5 | Potassium voltage-gated channel subfamily J member 5 | −1.74 | * | — | — |
|
|||||
| ErbB signalling pathway (NRG1 SHC3 CBLBJUN SHC2 SOS2 CAMK2B PRKCG) | |||||
| NRG1 | Neuregulin 1 | 1.67 | ** | −1.80 | ** |
| SHC3 | SHC adaptor protein 3 | −2.33 | *** | 2.35 | *** |
| CBLB | Cbl proto-oncogene B | −1.26 | *** | 1.06 | *** |
| JUN | Jun proto-oncogene, AP-1 transcription factor subunit | −1.13 | *** | 1.25 | *** |
| SHC2 | SHC adaptor protein 2 | −1.51 | *** | 1.73 | *** |
| SOS2 | SOS RAS/Rho guanine nucleotide exchange factor 2 | −1.14 | * | 1.35 | ** |
|
|||||
| Estrogen signalling pathway (MMP9 SHC3 KCNJ5 PLCB4 FOS JUN SOS2 SHC2 CREB3L3 ADCY4) | |||||
| MMP9 | Matrix metallopeptidase 9 | −1.76 | *** | 1.77 | *** |
|
|||||
| GnRH signalling pathway (LHB PLD1 PLA2G4D PLCB4 JUN SOS2 CAMK2B ADCY4) | |||||
| LHB | Luteinizing hormone beta polypeptide | −1.84 | *** | 1.62 | ** |
| PLD1 | Phospholipase D1 | −1.59 | *** | 1.82 | *** |
|
|||||
| Serotonergic synapse (CCNG2 CACNA1A ALOX15 PRKCG KCNJ5 PLA2G4D ALOX5 PLCB4) | |||||
| CCNG2 | Cyclin G2 | −1.80 | ** | 2.05 | ** |
|
|||||
| Inflammatory mediator regulation of TRP channels (ASIC4 TRPV4 TRPV2 BDKRB2 PRKCG PLA2G4D PLCB4 CAMK2B) | |||||
| ASIC4 | Acid sensing ion channel subunit family member 4 | −1.20 | ** | 1.15 | ** |
| TRPV4 | Transient receptor potential cation channel subfamily V member 4 | −1.24 | ** | 1.11 | * |
| TRPV2 | Transient receptor potential cation channel subfamily V member 2 | −1.52 | ** | 1.63 | ** |
Based on the GO and KEGG enrichment analysis, the primary fluctuation in HCT116 cells induced by GA treatment were sketched. As shown in Fig. 6, after exposure to GA for 12 h, the stress was generated and detected by the sensor-like proteins such as P2RX7, PDGFRB and BDKRB2 on the cell membrane. At the same time, the calcium release-activated calcium modulator (ORAI3) and the voltage-gated calcium channel proteins (CACNA1A, CACNA1I, and CACNA1G) were down-regulated and the concentration of calcium in the cytoplasm was restrained. Changed calcium signal was sensed through intermediate calcium sensors to decrease the activity of nitric oxide synthase (NOS1) and calcium/calmodulin (CALM)-dependent protein kinase (CAMK2B). The down-regulation of CAMK2B could lead to the arrest of the cell cycle and inhibit cell proliferation. NOS1 and protein kinase C (PRKCG), which are mediated by the intracellular calcium and second messenger diacylglycerol (DAG), also would affect other pathways involved in cell communication and the phosphatidylinositol signal pathway. This indicates that in order to respond to GA, HCT116 cells might reprogram the gene expression to optimize signaling transduction for more efficient and effective adaptation.
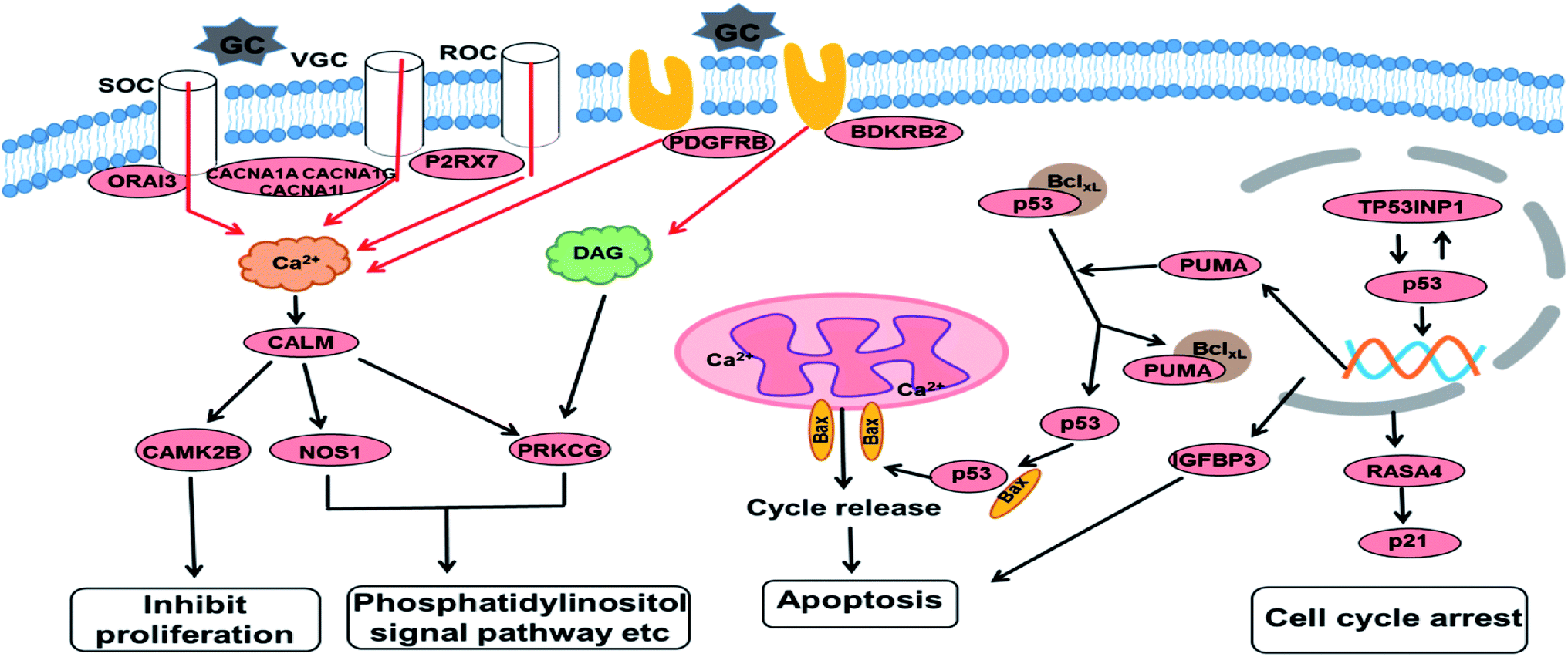 | ||
| Fig. 6 An overview of the molecular changes and their related functional outcomes induced by GA treatment in HCT116 cells. | ||
With the increase of exposure time, the down-regulated DEGs in the calcium signal pathway became up-regulated after 24 h of treatment. The expression level of four genes in the p53 signal pathway showed no significant differences in the GA12-C comparing group but were markedly enhanced in the GA12-GA24 group. TP53INP1 is a p53 target gene involved in a large panel of cellular processes, such as reactive oxygen species regulation, apoptosis, and cell cycle. The high expression level of TP53INP1 implicated in a positive feedback loop with transcriptional activity of p53. p53 directly activates the expression of many genes important for cell cycle regulation and apoptosis through binding to p53 DNA-binding sites. The up-regulation of Bcl2-binding component 3 (BBC3, also known as PUMA), a pro-apoptotic protein, would led to the release of cytochrome C and induction of cell apoptosis through the mitochondrion pathway. Increased IGFBP3 level would also induce cell apoptosis and suppress the IGF-1/mTOR pathway. Although there was no significant change in the p21 gene, the up-regulated RASA4 who associated with its activation was detected, which suggested that the G1 arrest of the cell cycle was also affected.
Validation RNA-seq by RT-qPCR
We randomly selected 12 DEGs involved in different metabolism pathways to verify their expression levels. The information of used primers and the calculated fold change in RT-qPCR experiments were shown in Table 2. The fold change values of each gene are the mean values of three parallel experiments. The RT-qPCR results were consistent with the RNA-seq findings, indicating that the RNA-seq results were quite credible.| Name | NCBI_GI | Forward primer | Reverse primer | log2 fold change (GA12/C) | log2 fold change (GA24/GA12) | ||
|---|---|---|---|---|---|---|---|
| qRT-PCR | RNA-seq | qRT-PCR | RNA-seq | ||||
| ALOX5 | NM_00698.4 | TGGCGCGGTGGATTCATAC | CGTCCATCCCTCAGGACAAC | −1.12 | −1.9 | 1.32 | 1.83 |
| CAMK2B | NM_172084.2 | CGGAATTTCTCAGCCCGGAA | TGAGCCGGATGTAAGCGATG | −0.97 | −1.53 | 1.78 | 2.03 |
| PRKCG | NM_002739.4 | GCCACTAGGTGTCCCCAAC | GAGAATATCGGGCTCCGCTC | −1.03 | −1.61 | 1.12 | 1.52 |
| GNAZ | NM_002073.3 | AGCGGCTACGACCTGAAACT | AAGCAGATGGTGAGCGGGAT | −0.81 | −1.37 | 0.94 | 1.41 |
| FOS | NM_005252.3 | AGGGCTGGCGTTGTGAAGA | TCAGCAGGTTGGCAATCTCG | −1.36 | −1.99 | 1.56 | 1.99 |
| PLCD4 | NM_032726.3 | CAGGGCTTCACCATTGTCTT | TAACCGCTGAACTTCTTGGA | −0.78 | −1.42 | 1.11 | 1.4 |
| CYP4F2 | NM_023944.3 | CCTTGGACAGTCTACAGAAATGC | CTGAGTGGGGAGGGTGCGAC | −1.84 | −2.29 | 1.56 | 2.14 |
| PLAC1 | NM_001316887.1 | TACACGAGGAGTCTGTCAAGG | CAGCCAATCAGATAATGAACCA | −1.03 | −1.66 | 1.09 | 1.66 |
| CCNG2 | NM_004354.2 | CTGAGTTTGATTGAGGCTAC | GTTTCACCTTCATAAGAGCC | −1.22 | −1.8 | 1.17 | 2.05 |
| SESN3 | NM_001271594.1 | TTGACAAGAGGACCAAGTGC | AACTGGCTCCGCAAGAAAGA | −1.34 | −2.18 | 1.36 | 2.48 |
| KRT6A | NM_005554.3 | CATCCAAGAGGTCACCGTCA | GCAGGGTCCACTTTGTTTCC | 2.57 | 3.92 | −4.15 | −3.93 |
| MASP2 | NM_006610.3 | GAGGACATTGACGAGTGCCA | TGGAGAGTTTGGGATACGGC | 1.58 | 2.13 | −2.11 | 1.29 |
Discussion
Gallic acid abundantly exist in several natural food and food products, including spices and drinks. More and more studies revealed an efficacy of GA administration to various health scenarios such as cardiovascular diseases,24 hypertension,25 hepatic ailments,26 or cancers27 in vivo experiments. For colorectal cancer, diet is thought to play a major role in its etiology. Gallic acid and gallates from dietary supplementation may exert beneficial effects and there is increasing evidence providing a basis for using them in the prevention and treatment of colorectal cancer.28In this study, we investigated whether GA inhibit proliferation in HCT116 cells and sought to identify the mechanisms of GA related to cell apoptosis. Our findings suggested that GA induced apoptosis of colon cancer cell lines through calcium and p53 signal pathways, which indicate GA as a novel promising therapy for the treatment of colon cancer.
Our results showed that GA could time- and dose-dependently inhibit cell proliferation and induce apoptosis as measured by CCK-8 assay and Annexin V-staining assay. We also used RNA-seq technology to determine the global regulation network for the response of HCT116 cells to GA treatment and identified some key pathways related to cell apoptosis.
GA was cytotoxic to HCT116 cells in a time- and dose-dependent manner, which was superior to the effect of GA on HCT-15 cell proliferation.29 As reported by Henning et al., one cup of green tea and black tea contains approximately 17 mg GA.30 The IC50 of GA on HCT116 cells after 24 h of treatment was approximately 54.8 μM and this concentration could be easily reached in the gastrointestinal tract, as 3 mg of GA is enough to yield about 88 μmol L−1 in the colonic volume of 200 mL. These results show that GA has insignificant anti-proliferative activity against colon cancer cells at a very low concentration within the range of biological availability.
Flow cytometric analysis revealed that the early and late apoptotic cell proportions increased along with increasing GA concentration. Furthermore, cell viability only exhibited a significant drop between 12 h and 24 h treatment by GA at concentration higher than 50 μM. These results suggested that the inhibition of the proliferation of HCT116 cells by GA relies on inducing the typical apoptotic pathway and massive death of cells probably occurred during 12 to 24 h exposure to GA.
Transcriptome data can offer accurate genome information quickly and economically, especially in revealing impacts on biological pathways and processes that had previously not been considered.31 We identified 792 and 911 genes with known functions that were differentially expressed in 12 h and 24 h GA-treated HCT116 cells, respectively. These tremendous changes reflect not only protein cascades directly related to apoptosis, but also many genes and transcription factors involved in calcium channels, circadian entrainment, serotonergic synapse and fatty acid metabolism. The fluctuant DEGs involving cell cycle detected in HCT116 cells indicate that like other anticancer agents, GA has the ability to disturb the cell cycle in cancer cells, which might contribute to the induction of apoptosis.32 Our results also revealed that GA modulated the expression of many cell signaling molecules in the p53 signal pathway, such as Bcl2 binding component BBC3, which is agreement with previous studies showing that GA triggered the intrinsic or mitochondrial pathway to lead to enhanced cancer cell death.33,34
The calcium ion (Ca2+) is one of the key intracellular signals that is implicated in the regulation of cell functions such as impregnation, cell proliferation, differentiation and death.35 Cells use this external source of Ca2+ by activating various entry channels with widely different properties.36 In the first 12 h of treatment with GA, considerable calcium channels subunit-coding genes involved in three calcium channels were down-regulated. These calcium channels mediate the entry of calcium ions into excitable cells, and thus their down-regulation in the early phase of GA exposure would decrease the intracellular calcium concentration. Moreover, the expression level of two membrane protein-coding genes PDGFR and BDKRB2 were also decreased. Although the functional pathway between these proteins and GA was unclear, suppressed expression of these factors could activate multiple downstream targets inside the cell to decrease intracellular free calcium. Protein CaMK2B which is involved in many signaling cascades is regulated by the Ca2+/CALM complex.37 We inferred that the down-regulation of this gene was caused by loss of intracellular calcium and finally lead to suppress of the cell proliferation process in the first 12 h exposure to GA.
The calcium signal also plays an important role in cell death and survival; there is a close relation among intracellular Ca2+ concentration, Bcl2 family proteins and cell apoptosis.35 Recent studies have shown that several pro-apoptotic factors including members of the Bcl2 family proteins and reactive oxygen species regulate Ca2+ sensitivity through both the Ca2+ release channels in the endoplasmic reticulum and mitochondria.38 In this study, with the prolongation of GA treatment time to 24 h, the expression pattern of factors in the calcium signaling pathway was changed to up-regulation. However, the cell proliferation continued to be suppressed and the proportion of cell apoptosis reached as high as 62.2%. A previous report showed that cadmium elevated the intracellular concentration of Ca2+ and CALM and finally induced apoptosis in gill cells of crabs.39 Lange et al.40 also reported an increased calcium signaling pathway that resulted in apoptosis in neuroblastoma cells. These researches showed that the high concentration intracellular Ca2+ and CALM were closely related to apoptosis in cells enduring chemical stress. Our results indicate that long time GA treatment induced an alteration of calcium signaling pathway genes and increased mitochondria calcium, which eventually led to activation of the apoptotic pathway.41
Induction of apoptosis is considered an effective approach to prevent and treat cancer. The tumor suppressor protein p53 plays a crucial role in the regulation of cell cycle arrest and induction of carcinoma cell apoptosis.42 p53 exhibits its anticancer role through triggering the expression of genes involved in cell cycle regulation and apoptosis, which ultimately results in the inhibition of cancer progression and malignant transformation.43 p53 inhibits cell cycle progression at the G1 or G2/M phases through induction of the cell cycle inhibitor p21 and promotes apoptosis through induction of pro-apoptotic Bax expression.44 Previous reports showed that the treatment of a polyphenol-curcumin could upregulate total and activated p53, as well as increase the expression of p53-regulated p21, PUMA and Bax in human colon cancer.45 It was reviewed that increased Bcl2 family members including PUMA, NOXA, and BIM, induced the mitochondrial apoptotic pathway by triggering Bak/Bax activation.46 In this study, p53 inducible nuclear protein TP53INP1, BBC3 (PUMA) and p21 activated kinase RASA4 were overexpressed after 24 h treatment of GA. Thus we inferred that the up-regulated p53 signal pathway triggered mitochondrial apoptosis in HCT116 cells.
In summary, here we investigated the effect and mechanism of the phenolic compound GA on the growth of HCT116 colon cancer cells by RNA-seq. KEGG analysis showed that the DEGs mainly encoded proteins involved in cholinergic synapse, circadian entrainment, calcium signal processing and transport, arachidonic acid metabolism and the p53 signal pathway. The regulation of the expression of calcium ion binding proteins and related channels between the first 12 h and later 12 h after treatment with GA suggests that the calcium signal pathway plays important roles in both cell proliferation and cell apoptosis. The results also suggest that GA-triggered apoptosis in human colon cancer HCT116 cells was associated with the up-regulation of the intrinsic p53 signal pathway through the activation of caspases and finally led to apoptosis through the mitochondrial pathway. Our findings reveal the mechanism of GA on inducing colon cancer cells and also provide insight for GA as a potential chemotherapeutic agent for colon cancer. However, further investigation in clinical settings are needed to promote GA as a likely candidate for anticancer agent.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Agricultural Science and Technology Innovation Project of Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2013-OCRI), the National Natural Science Foundation of China (31701560) and Director Fund of Oil Crops Research Institute (1610172017009).References
- A. W. Lowe and R. H. Moseley, Gastroenterology, 2012, 143, 1125–1126 CrossRef
.
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Ca-Cancer J. Clin., 2011, 61, 69–90 CrossRef PubMed
- H. F. A. Vasen, I. Tomlinson and A. Castells, Nat. Rev. Gastroenterol. Hepatol., 2015, 12, 88–97 CrossRef PubMed
- L. Anning, N. Koo, J. Neely, A. Wells, S. K. Clark, R. Miller and O. Will, Colorectal Dis., 2011, 13, E297–E302 CrossRef CAS PubMed
- E. P. Misiakos, N. P. Karidis and G. Kouraklis, World J. Gastroenterol., 2011, 17, 4067–4075 CrossRef PubMed
- G. D. Stoner and H. Mukhtar, J. Cell. Biochem., 1995, 169–180 CrossRef CAS
- J. R. Araujo, P. Goncalves and F. Martel, Nutr. Res., 2011, 31, 77–87 CrossRef CAS PubMed
- J. Liu, H. T. Wu, J. F. Lu, X. Y. Wen, J. Kan and C. H. Jin, Chem. Eng. J., 2015, 262, 803–812 CrossRef CAS
- J. Serrano, R. Puupponen-Pimia, A. Dauer, A. M. Aura and F. Saura-Calixto, Mol. Nutr. Food Res., 2009, 53, S310–S329 Search PubMed
- D. Li, Z. J. Liu, W. J. Zhao, Y. L. Xi and F. L. Niu, Process Biochem., 2011, 46, 2210–2214 CrossRef CAS
- A. Abdelwahed, I. Bouhlel, I. Skandrani, K. Valenti, M. Kadri, P. Guiraud, R. Steiman, A. M. Mariotte, K. Ghedira, F. Laporte, M. G. Dijoux-Franca and L. Chekir-Ghedira, Chem.-Biol. Interact., 2007, 165, 1–13 CrossRef CAS PubMed
- S. H. Kim, C. D. Jun, K. Suk, B. J. Choi, H. Lim, S. Park, S. H. Lee, H. Y. Shin, D. K. Kim and T. Y. Shin, Toxicol. Sci., 2006, 91, 123–131 CrossRef CAS PubMed
- M. S. Kang, J. S. Oh, I. C. Kang, S. J. Hong and C. H. Choi, J. Microbiol., 2008, 46, 744–750 CrossRef CAS PubMed
- J. M. Kratz, C. R. Andrighetti-Frohner, P. C. Leal, R. J. Nunes, R. A. Yunes, E. Trybala, T. Bergstrom, C. R. Barardi and C. M. Simoes, Biol. Pharm. Bull., 2008, 31, 903–907 CAS
- M. Inoue, R. Suzuki, T. Koide, N. Sakaguchi, Y. Ogihara and Y. Yabu, Biochem. Biophys. Res. Commun., 1994, 204, 898–904 CrossRef CAS PubMed
- M. Kawada, Y. Ohno, Y. Ri, T. Ikoma, H. Yuugetu, T. Asai, M. Watanabe, N. Yasuda, S. Akao, G. Takemura, S. Minatoguchi, K. Gotoh, H. Fujiwara and K. Fukuda, Anticancer Drugs, 2001, 12, 847–852 CrossRef CAS PubMed
- K. Wang, X. Zhu, K. Zhang, L. Zhu and F. F. Zhou, J. Biochem. Mol. Toxicol., 2014, 28, 387–393 CrossRef CAS PubMed
- S. W. Weng, S. C. Hsu, H. C. Liu, B. C. Ji, J. C. Lien, F. S. Yu, K. C. Liu, K. C. Lai, J. P. Lin and J. G. Chung, Anticancer Res., 2015, 35, 2077–2084 CAS
- M. Inoue, R. Suzuki, N. Sakaguchi, Z. Li, T. Takeda, Y. Ogihara, B. Y. Jiang and Y. Chen, Biol. Pharm. Bull., 1995, 18, 1526–1530 CAS
- A. Mortazavi, B. A. Williams, K. Mccue, L. Schaeffer and B. Wold, Nat. Methods, 2008, 5, 621–628 CrossRef CAS PubMed
- K. W. J. Derks, B. Misovic, M. C. G. N. van den Hout, C. E. M. Kockx, C. P. Gomez, R. W. W. Brouwer, H. Vrieling, J. H. J. Hoeijmakers, W. F. J. van IJcken and J. Pothof, RNA Biol., 2015, 12, 30–42 CrossRef PubMed
- U. Nagalakshmi, K. Waern and M. Snyder, Curr Protoc Mol Biol., 2010, ch. 4, Unit 4, vol. 11, pp. 11–13 Search PubMed
- K. O. Mutz, A. Heilkenbrinker, M. Lonne, J. G. Walter and F. Stahl, Curr. Opin. Biotechnol., 2013, 24, 22–30 CrossRef CAS PubMed
- J. R. Xie and L. N. Yu, Acta Anaesthesiol. Scand., 2007, 51, 909–913 CrossRef CAS PubMed
- L. Jin, Z. H. Piao, S. M. Sun, B. Liu, G. R. Kim, Y. M. Seok, M. Q. Lin, Y. Ryu, S. Y. Choi, H. J. Kee and M. H. Jeong, Sci. Rep., 2017, 7, 15607 CrossRef PubMed
- O. Karimi-Khouzani, E. Heidarian and S. A. Amini, Pharmacol. Rep., 2017, 69, 830–835 CrossRef CAS PubMed
- H. A. Aglan, H. H. Ahmed, S. A. El-Toumy and N. S. Mahmoud, Tumor Biol., 2017, 39, 6 Search PubMed
- R. Y. Kosuru, A. Roy, S. K. Das and S. Bera, Mol. Nutr. Food Res., 2018, 62, 1700699 Search PubMed
- A. P. Subramanian, S. K. Jaganathan, M. Mandal, E. Supriyanto and I. I. Muhamad, World J Gastroenterol., 2016, 22, 3952–3961 CrossRef CAS PubMed
- S. M. Henning, P. Wang, N. Abgaryan, R. Vicinanza, D. M. de Oliveira, Y. Zhang, R. P. Lee, C. L. Carpenter, W. J. Aronson and D. Heber, Mol. Nutr. Food Res., 2013, 57, 483–493 CAS
- S. R. Zhao, W. P. Fung-Leung, A. Bittner, K. Ngo and X. J. Liu, PLoS One, 2014, 9, e7864 Search PubMed
- M. D. Garrett and I. Collins, Trends Pharmacol. Sci., 2011, 32, 308–316 CrossRef CAS PubMed
- R. X. Wang, L. J. Ma, D. Weng, J. H. Yao, X. Y. Liu and F. G. Jin, Oncol. Rep., 2016, 35, 3075–3083 CrossRef CAS PubMed
- K. Wang, X. Zhu, K. Zhang, L. Zhu and F. Zhou, J. Biochem. Mol. Toxicol., 2014, 28, 387–393 CrossRef CAS PubMed
- A. Danese, S. Patergnani, M. Bonora, M. R. Wieckowski, M. Previati, C. Giorgi and P. Pinton, Biochim. Biophys. Acta, 2017, 1858, 615–627 CrossRef CAS PubMed
- S. W. Jones, J. Bioenerg. Biomembr., 2003, 35, 461–475 CrossRef CAS PubMed
- T. Yamauchi, Biol. Pharm. Bull., 2005, 28, 1342–1354 CAS
- M. Swami, Nat. Rev. Cancer, 2010, 10, 738 CrossRef CAS PubMed
- J. X. Wang, P. P. Zhang, N. Liu, Q. Wang, J. X. Luo and L. Wang, PLoS One, 2015, 10, e0144392 Search PubMed
- I. Lange, J. Moschny, K. Tamanyan, M. Khutsishvili, D. Atha, R. P. Borris and D. L. Koomoa, Int. J. Oncol., 2016, 48, 1608–1616 CrossRef CAS PubMed
- X. Ma, X. Tian, X. Huang, F. Yan and D. Qiao, Mol. Cell. Biochem., 2007, 302, 99–109 CrossRef CAS PubMed
- X. D. Ma, X. M. Tian, X. X. Huang, F. Yan and D. F. Qiao, Mol. Cell. Biochem., 2007, 302, 99–109 CrossRef CAS PubMed
- S. Kim and S. S. A. An, Medicine, 2016, 95, e3993 CrossRef CAS PubMed
- F. B. Charrier-Savournin, M. T. Chateau, V. Gire, J. Sedivy, J. Piette and V. Dulic, Mol. Biol. Cell, 2004, 15, 3965–3976 CrossRef CAS PubMed
- J. L. Watson, R. Hill, P. B. Yaffe, A. Greenshields, M. Walsh, P. W. Lee, C. A. Giacomantonio and D. W. Hoskin, Cancer Lett., 2010, 297, 1–8 CrossRef CAS PubMed
- R. J. Youle and A. Strasser, Nat. Rev. Mol. Cell Biol., 2008, 9, 47–59 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2018 |