
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZn 1,3,5-benzenetricarboxylate as an efficient catalyst for the synthesis of cyclic carbonates from CO2†
Chao Feng,
Xianglei Cao,
Liugen Zhang,
Changyan Guo,
Naeem Akram and
Jide Wang*
and
Jide Wang*
Key Laboratory of Oil & Gas Fine Chemicals Ministry of Education, Xinjiang University, Urumqi 830046, Xinjiang, People's Republic of China. E-mail: awangjd@sina.cn
First published on 2nd March 2018
Abstract
Development of heterogeneous catalysts for the cycloaddition of CO2 with epoxides to produce cyclic carbonates is a hot issue in the field of chemical fixation of carbon dioxide. It is fairly promising as production of by-products is quite low. In this study, the [Zn3(BTC)2]/n-Bu4NBr catalytic system was investigated for the solventless cycloaddition of carbon dioxide with epoxides and had an excellent synergetic effect in promoting the reaction. The reaction parameters were moderate i.e. (130 °C and 13 bar CO2 pressure) and were selected by a study of the catalytic system. Under the optimal reaction conditions, the yield of cyclic carbonate reached 99%. A decrease in the yield of cyclic carbonate was not apparent after [Zn3(BTC)2] was reused three times, indicating that [Zn3(BTC)2] was stable. At the same time, the catalytic activity of the catalyst for other epoxides was also verified. The acidic and alkaline nature of the [Zn3(BTC)2] catalyst did not change obviously after recycling the catalyst three times. In this study it is also verified that the [Zn3(BTC)2] catalytic cycloaddition reaction was closely related to the Lewis acid/base distribution. In addition, a plausible mechanism for the synergistic effect of the catalyst (Lewis acid and base properties) and the co-catalyst was suggested.
Introduction
The emission of carbon dioxide in the environment followed by human activities and industrialization play a vital role in global warming. Designing scientific methods to convert carbon dioxide into some value-added products1–4 can be used as a promising strategy to reduce carbon dioxide emissions. As a rich resource, carbon dioxide has received widespread attention for developing various valuable strategies that can reduce CO2 emissions. CO2 conversion under mild conditions is still very attractive but remains a challenge. The use of carbon dioxide and epoxides to form cyclic carbonates is considered to be the most environmentally friendly way of chemically fixing carbon dioxide,5–8 as the utilization of the atoms is 100%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9,10 (Scheme 1). Cyclic carbonates are important compounds with a wide range of applications11,12 including polar aprotic solvents, lithium-ion battery electrolytes, synthesis of monomeric polymers, degreasing agents, chemical intermediates and cosmetics. But the major inadequacy is the chemical inertness of CO2, which require the assistance of a catalyst. So far a number of catalysts have been synthesized for this attractive reaction, including both homogeneous and heterogeneous catalysts. Homogeneous catalysts include metal halide13 and metallic oxide,14,15 transition metal complexes,16 organic base,17 ionic liquids18 and quaternary ammonium19 etc. Recovery and the separation of such catalyst is a crucial obstruction for its industrial application. So with these characteristics, heterogeneous catalyst has received researcher's attention. However, most heterogeneous catalysts have somewhat lower catalytic activity or involvement of harsh reaction conditions. Therefore, preparation of a cycloaddition reaction catalyst for efficiently catalyzing carbon dioxide under mild reaction conditions should be studied.
9,10 (Scheme 1). Cyclic carbonates are important compounds with a wide range of applications11,12 including polar aprotic solvents, lithium-ion battery electrolytes, synthesis of monomeric polymers, degreasing agents, chemical intermediates and cosmetics. But the major inadequacy is the chemical inertness of CO2, which require the assistance of a catalyst. So far a number of catalysts have been synthesized for this attractive reaction, including both homogeneous and heterogeneous catalysts. Homogeneous catalysts include metal halide13 and metallic oxide,14,15 transition metal complexes,16 organic base,17 ionic liquids18 and quaternary ammonium19 etc. Recovery and the separation of such catalyst is a crucial obstruction for its industrial application. So with these characteristics, heterogeneous catalyst has received researcher's attention. However, most heterogeneous catalysts have somewhat lower catalytic activity or involvement of harsh reaction conditions. Therefore, preparation of a cycloaddition reaction catalyst for efficiently catalyzing carbon dioxide under mild reaction conditions should be studied.
 | ||
| Scheme 1 Synthesis of cyclic carbonates from epoxides and CO2. | ||
In some of the past work,20–22 metal–organic frameworks (MOFs) have shown excellent heterogeneous catalytic activity due to their chemical functionalities and its easier recovery for recycling, from the reaction system making MOFs a potential candidate for heterogeneous catalysis.23 The affinity of MOFs for CO2 makes them admirable catalysts24 for the formation of cyclic carbonates using CO2 and epoxides. In 2009, first ever chemically fixation of carbon dioxide by generating cyclic carbonates with MOF catalysts (MOF-5)25 was studied. Since then several materials in this research field have been reported. Among MOFs materials, MOF-5, Cr-MIL-101,26 ZIF-68,27 and ZIF-9528 have been studied in the cycloaddition reaction of carbon dioxide for formation of cyclic carbonates with resulting good. According to earlier reported research works,29,30 the catalytic activity of a heterogeneous catalyst in the cycloaddition to form cyclic carbonates is related to the Lewis acid and Lewis base involved in the reaction of the catalyst. Lewis acid–base sites can harmonize and activate the substrate molecules and facilitate the reaction and products formed. However, many MOFs materials have the disadvantages of low catalytic activity, poor stability and harsh catalytic conditions, which limit their further usage as commercial applications. Therefore, the investigating, developing and screening of different MOFs materials with excellent catalytic performance is a trending research for cycloaddition of carbon dioxide.
The focus of our work was preparation of a MOF catalyst ([Zn3(BTC)2]) in cyclic carbonates synthesis with solvothermal method with good stability and sustainability. [Zn3(BTC)2] unit has Lewis acid and base catalytic sites with strong nucleophilic anionic promoter (TBAB) to play its good synergistic effect. Under solvent-free and relatively moderate reaction conditions, the [Zn3(BTC)2]/TBAB catalytic system was found to have high activity and selectivity. Under the optimum reaction conditions, the yield of cyclic carbonate was 99% and [Zn3(BTC)2] has relatively higher content of Lewis acids–bases, exhibiting satisfactory catalytic performance than that of its corresponding heterogeneous and homogeneous catalysts. The catalyst still has quite high activity after being recycled for several times. The high stability of [Zn3(BTC)2] indicates that it is an efficient heterogeneous catalyst for chemical fixation of carbon dioxide.
Experimental
Materials
Zn(acetate)2·2H2O and styrene oxide were purchased from Aladdin Bio-Chem Technology Co., Ltd (Shanghai, China). Tribenzoic acid (BTC) and tetrabutyl ammonium bromide were purchased from Adamas Reagent Co., Ltd (Shanghai, China). Ethanol and ethyl acetate were purchased from Shanghai Titan Scientific Co., Ltd (Shanghai, China). CO2 (gas, 99.9999%) was purchased from Beijing Oxygen Plant Specialty Gases Institute Co., Ltd (Beijing, China).Catalyst preparation
The procedure to prepare [Zn3(BTC)2] was similar to the work reported previously.31 7.497 mmol of Zn(acetate)2·2H2O was dissolved in 12.66 mL of distilled water and mixed with a solution of 9 mmol of BTC in 25.02 mL of ethanol. The mixture was stirred for 30 min at room temperature and then was transferred into a Teflon lined stainless autoclave of 100 mL. The autoclave was sealed, kept at 175 °C for 24 h and then cooled to room temperature. The white crystals were collected by centrifugation, thoroughly washed with ethanol and distilled water, and then was dried at 80 °C for 12 h under vacuum in order to remove the trapped water molecules from the pores.Catalyst characterization
Powder X-ray diffraction (PXRD) was performed on a Rigaku X-ray diffractometer (Rigaku Rint, Japan) at a scan rate of 6° min−1 in 2θ ranging from 5° to 30° with Cu Kα radiation (λ = 1.54178 Å). Scanning electron microscopy (SEM) was conducted on a Hitachi S-8010 with an accelerating voltage of 100 kV. The surface areas of [Zn3(BTC)2] were calculated using the Brunauer–Emmett–Teller (BET) method, Fourier transform infrared spectrometer (FTIR) was used for the determination of functional groups in samples (dispersed in potassium bromide and compressed into pellets), using EQUINOX-55 (Bruker Company, Germany) in transmittance (%) mode in the range of 4000–400 cm−1. Thermogravimetric analysis (TGA) was performed on a Shimadzu DTG-50 thermal analyzer from room temperature to 700 °C at a heating rate of 10 °C min−1 under open air atmosphere. Temperature programmed decomposition (TPD) was determined using a TP-5080 adsorption instrument with ammonia over a temperature ramp of 50–400 °C, rate ramp of 10 °C min−1 and flow rate of 60 mL min−1. Gas chromatographic (GC) analysis were performed using a Shimadzu GC 2010-Plus equipped with a flame ionization detector (FID) and an SPB-5 column (length = 30 m, inner diameter = 0.25 mm, and film thickness = 0.25 μm). GC-MS analyses were performed using a Shimadzu GC-MS-QP2010 Ultra with a ZB-5MS column (length = 30 m, inner diameter = 0.25 mm, and film thickness = 0.25 μm). MS spectra were compared with the spectra gathered in the NIST library.Catalyst performance testing
The cycloaddition of epoxide with CO2 was carried out in a 25 mL stainless steel autoclave equipped with a magnetic stirrer. In a typical reaction, the mixtures of [Zn3(BTC)2] (10 mg), styrene oxide (20 mmol), 0.31 × 10−4 mmol co-catalyst (tetrabutyl ammonium bromide) were added into the autoclave. Where after, CO2 (99.999%) was introduced into the autoclave, before this, air was replaced with CO2 until reaching the appropriated pressure. The autoclave was then heated to an appropriate temperature for several hours with continuous magnetic stirring (400 rpm). After the reaction completion, the autoclave was cooled in an ice-water bath, and venting the remaining CO2. The product was extracted with ethyl acetate and then the solid catalyst was filtered before GC analysis (Agilent 7890A) using a designed method file. The structure of the product was determined by GC-MS. The recycling catalyst was washed with anhydrous ethanol and separated by centrifugation, then dried under vacuum at 60 °C.Results and discussion
The XRD patterns of [Zn3(BTC)2] as a catalyst and the simulated patterns of the coordination polymer calculated from single crystal data available in the literature32 were compared in Fig. 1. Fig. 1a displays that the well-resolved patterns of fresh [Zn3(BTC)2] match well with the corresponding simulated patterns of [Zn6(OH)3(BTC)3(H2O)3]·7H2O, which proves the phase purity of the as-synthesized samples. After the recycled experiment processes, it is clear that the diffraction pattern of catalyst after recycling (Fig. 1b–d) were kept the same as for the fresh sample. However, a change in the intensity of some peaks is observed, this may be due to the strong adsorption of organic compounds during the reuse experiments. These results indicate that [Zn3(BTC)2] showed good stability in the cyclic addition reactions even after recycling the catalyst, so there is no apparent change in terms of catalytic performance.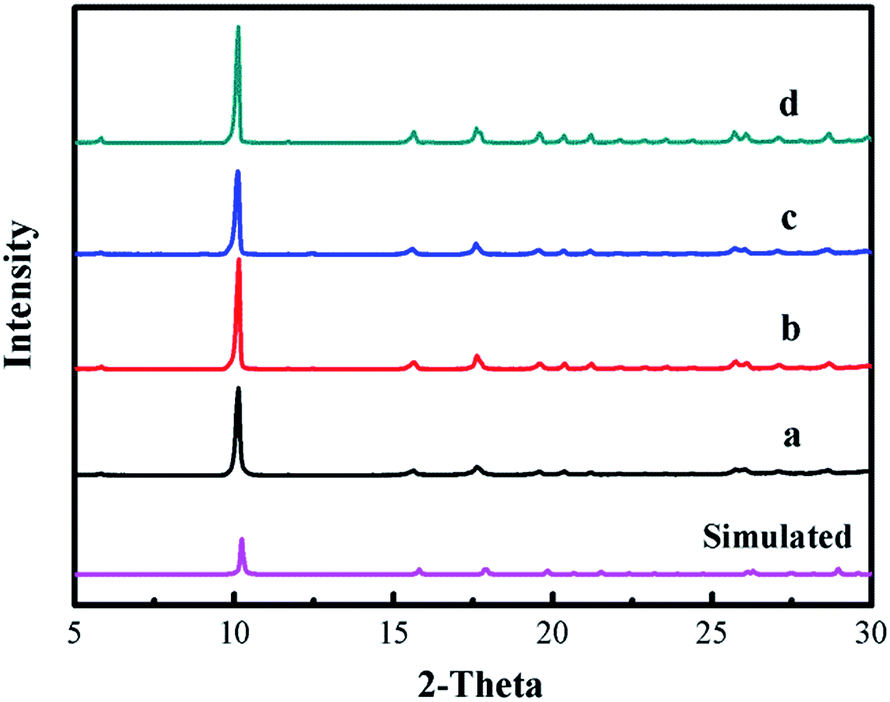 | ||
| Fig. 1 XRD patterns of [Zn3(BTC)2] catalyst: (a) fresh; (b) first recycle; (c) second recycle and (d) third recycle. | ||
In Fig. 2, the comparison between Fourier transform infrared spectroscopy (FT-IR) of [Zn3(BTC)2] and BTC showed that the bond formation between zinc(II) metal ions and BTC were observed from the noteworthy shift of carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) peak from carboxylate at 1715 cm−1 into 1613 cm−1. This transformation corresponded to the formation of coordination bonding in [Zn3(BTC)2]. A broad peak present at 3300–2500 cm−1 due to the stretching vibration of O–H bonds on H3BTC which also clearly transferred into 3450 cm−1, after coordination with the metal ion, the intensity conspicuously decreased. The absence of –COOH absorption bands (1730–1690 cm−1) shows that BTC is fitted in a single mode to the metal center. In addition, the absorption band at 760 cm−1 was assigned as a stretching vibration of Zn–O. These analyses indicated that Zn2+ had combined with organic ligands successfully.
O) peak from carboxylate at 1715 cm−1 into 1613 cm−1. This transformation corresponded to the formation of coordination bonding in [Zn3(BTC)2]. A broad peak present at 3300–2500 cm−1 due to the stretching vibration of O–H bonds on H3BTC which also clearly transferred into 3450 cm−1, after coordination with the metal ion, the intensity conspicuously decreased. The absence of –COOH absorption bands (1730–1690 cm−1) shows that BTC is fitted in a single mode to the metal center. In addition, the absorption band at 760 cm−1 was assigned as a stretching vibration of Zn–O. These analyses indicated that Zn2+ had combined with organic ligands successfully.
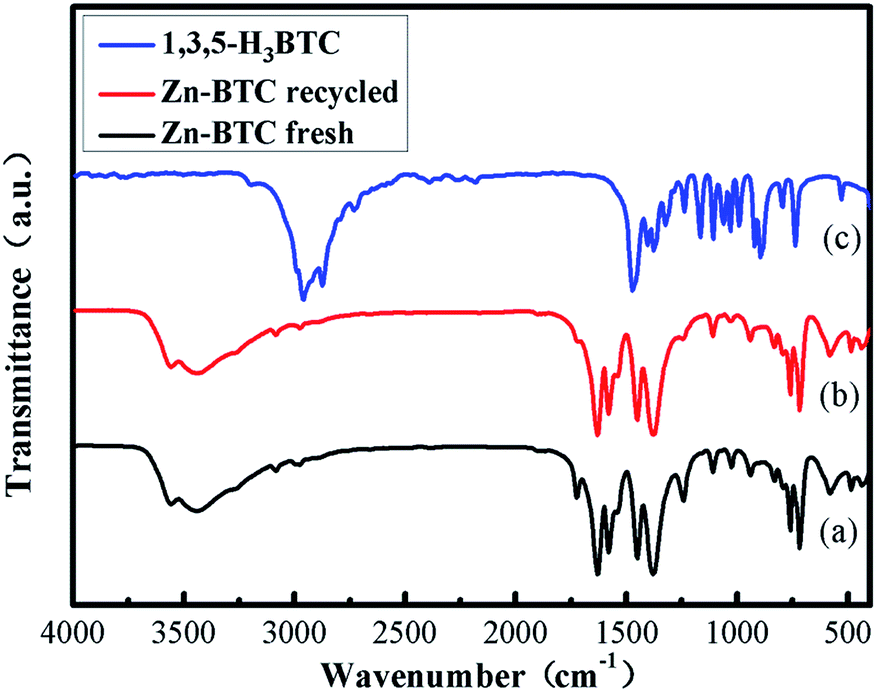 | ||
| Fig. 2 FT-IR spectra of [Zn3(BTC)2] (a) fresh; (b) recycled and (c) 1,3,5-H3BTC. | ||
The thermal stability of the catalyst is crucial in understanding the property of the framework construction. The thermal stability of fresh and recycled (3rd recycle) [Zn3(BTC)2] was studied by thermal gravimetric analysis (TGA) and is shown in Fig. 3. The weight loss steps were observed from 30 °C to 700 °C. The first weight loss up to 100 °C can be attributed to the solvent used for synthesis (H2O and ethanol) whereas the second weight loss around 250 °C is due to removal of guest molecules from open metal sites on [Zn3(BTC)2]. The TGA pattern comprising two-step weight reduction is an unparalleled characteristic of some MOF materials having open metal sites. The final major weight loss was observed within 400–480 °C for [Zn3(BTC)2], indicating that the catalyst is thermally stable up to 400 °C. This is a relatively high thermal stability among MOFs. The complete decomposition of [Zn3(BTC)2] was around 475 °C and thus [Zn3(BTC)2] is expected to exhibit good stabilities for catalytic reactions performed even at moderately high temperatures. The last loss corresponded to the decomposition of the organic ligand part occurred from 400 to 480 °C (found 47.50 wt%), after 480 °C leading to ZnO as the residue (found 40.00 wt%). According to the TGA profiles, the metal/ligand ratio could be estimated about 2:1, this is almost the same as the metal/ligand ratio calculated by Fu et al.32 In order to confirm the presence of Lewis acid sites and basic sites in the [Zn3(BTC)2] catalyst, CO2-TPD and NH3-TPD analysis were carried out between 50 °C and 400 °C. The TPD test temperature range is based on the thermal stability of the catalyst at 400 °C.
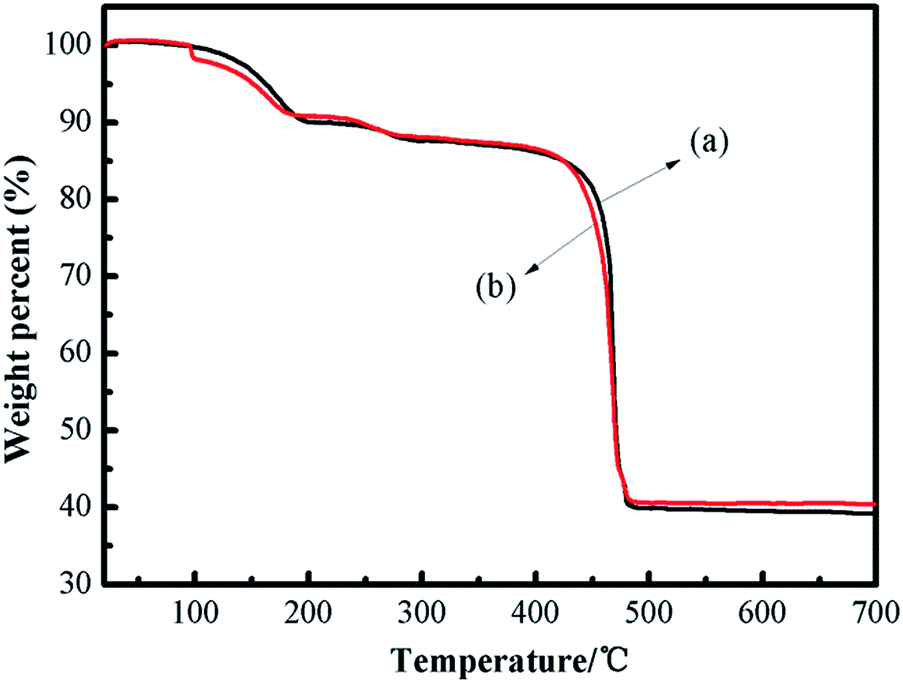 | ||
| Fig. 3 TGA profiles of fresh (a) and recycled (b) [Zn3(BTC)2] samples. | ||
Fig. 4 displays the scanning electron microscope (SEM) morphology of the fresh [Zn3(BTC)2] and the used one. The synthesized [Zn3(BTC)2] particles exhibited claviform shape with wide range of 0.5–2 μm and length range of 2–6 μm. After the catalyst is reused, the surface of the rod-shaped structure becomes no longer smooth. SEM pictures (Fig. 4b and d) showed that the rod like structure was slightly damaged. After each reuse, the catalyst surface defects will be more obvious, but its rod like structure is still clear. The surface morphologies of recycled catalyst had been slightly changed. This could be due to some damage of the skeletal structure of the recycled runs under vigorous stirring. Such structural change is not too significant, so the catalyst could indicate good recyclability. However, after recycling repeatedly, [Zn3(BTC)2] catalyst still shows a high catalytic, this also shows that it has better stability.
 | ||
| Fig. 4 SEM images of [Zn3(BTC)2] catalyst: (a) fresh; (b) first recycle; (c) second recycle and (d) third recycle. | ||
According to previous research reports,30 some of the Lewis acidic sites in MOFs were derived from metal species and the basic moiety was derived from non-metal atoms in the ligands. Both sites (acidic and basic) play key role in the catalytic activities of MOFs. The presence of Lewis acid sites and basic sites on heterogeneous catalysts can noteworthy enhance the interaction of CO2 with epoxides to form cyclic carbonates. The combination of Lewis acidic and basic sites may help the catalyst to improve its catalytic performance. Using NH3 and CO2 as probe gases, temperature-programmed desorption methods (TPD) were used to evaluate the acidity and alkalinity of fresh and recycled [Zn3(BTC)2] catalyst. The acid sites on the catalyst surface may serve as the adsorption of CO2 and styrene oxide site. Fig. 5 shows CO2-TPD and NH3-TPD spectra of [Zn3(BTC)2] fresh and recycled (3 times) catalysts. Fig. 5a shows two obvious CO2 desorption peaks around 145 °C and 280 °C, indicating that the presence of different surface basic sites. Based on the peak area, the catalyst surface has many basic sites manifests that [Zn3(BTC)2] has a larger CO2 adsorption capacity, providing more places for the activation of CO2. Fig. 5b shows that the desorption temperature decreases slightly after the catalyst has been reused three times, and there is a slight decrease in the peak intensity of CO2 desorption near 260 °C. This may be due to CO2 neutralizing some of the basic sites during the reaction, leading to desorption temperature and catalytic activity decreased, but still maintained strong alkaline sites, the cyclic carbonate still maintained a high yield. Fig. 5c shows the NH3-TPD results used to assess the acidity of the catalyst surface. A desorption peak centered at 270 °C was found in the figure, probably due to the fact that NH3 is adsorbed on Lewis acidic sites in one chemical state. Fig. 5d shows a desorption peak centered at 250 °C, it was formed by desorption of NH3 after three cycles of catalyst recycling. Focus partial shift in desorption peak compared to Fig. 5c may be due to partial neutralization of the acidity of the catalyst surface, resulting in a decrease in desorption temperature. The intensity of desorption peak was also slightly reduced, which may be the reason for the decrease in the catalytic activity of the catalyst. In summary, catalyst after recycled still maintained stronger Lewis acid and alkaline sites, this may be the reason why it still retains high catalytic activity.
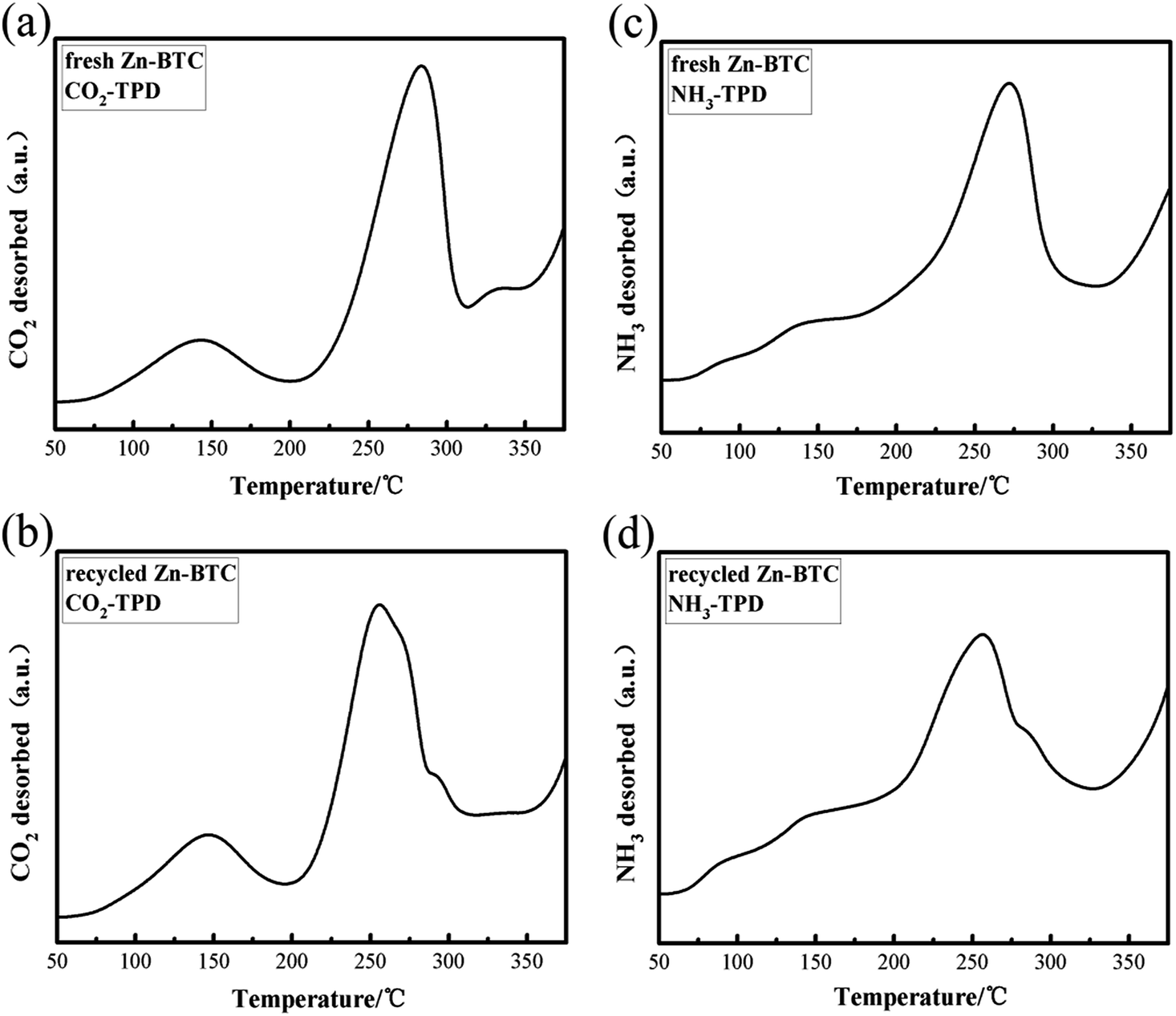 | ||
| Fig. 5 (a), (b) CO2-TPD and (c), (d) NH3-TPD profiles of fresh and recycled (3rd recycle) [Zn3(BTC)2] catalysts. | ||
The catalytic performance of [Zn3(BTC)2]/TBAB for the cycloaddition of CO2 with epoxides was studied to produce cyclic carbonates using styrene oxide as model substrate under relatively moderate reaction conditions (130 °C and 13 bar CO2 pressure). The control tests were analyzed with [Zn3(BTC)2], TBAB and the [Zn3(BTC)2] precursors as shown in Table S1.† Product formation was absolutely denied in the absence of the catalyst, even operating under the optimal conditions. A binary catalyst system of [Zn3(BTC)2]/TBAB procured styrene carbonate yields as higher as 99% under relatively moderate and solvent-free conditions. The result observed for just TBAB as a catalyst, disclosed lower yield of product (41.66%) than [Zn3(BTC)2]/TBAB system. Under the same reaction conditions, the mechanical mixture of [Zn3(BTC)2] precursors with TBAB, the styrene carbonate (SC) formation was lower in yield (47.92%) (33.07%) (38.91%). Even, the [Zn3(BTC)2] alone also showed lower activity (the product yield was 20.93%) at relatively moderate reaction conditions. The styrene carbonate yield of [Zn3(BTC)2] was increased to 84.32% at 160 °C in 24 h (13 bar CO2 pressure). However, the ultimate purpose of this research work was aimed lower energy conservation, so, [Zn3(BTC)2]/TBAB catalytic system was studies at relatively moderate reaction conditions (130 °C and 13 bar CO2 pressure).
Under normal circumstances, the reaction conditions for the synthesis of cyclic carbonate: carbon dioxide pressure is between 1 atm to 100 atm, the reaction temperature is between 70 °C and 200 °C. Excessive reaction temperature and pressure not only just wastage of energy, but also put higher requirements on the experimental equipment, so moderate experimental conditions are an important factor to examine the catalyst. Our prime focus in this study was to achieve high conversion of raw materials and high product selectivity under relatively moderate reaction conditions. The effects of different reaction conditions, such as temperature, pressure, amount of catalyst and reaction time, on the cycloaddition of carbon dioxide with styrene oxide were investigated in [Zn3(BTC)2]/TBAB binary system, screening out the optimal reaction conditions. As shown in Fig. 6, among the reaction parameters, temperature and pressure have the most significant influence on the reaction. We also ran a blank experiment without addition of catalyst to check either the reaction is possible or not, but there wasn't any reaction without catalyst.
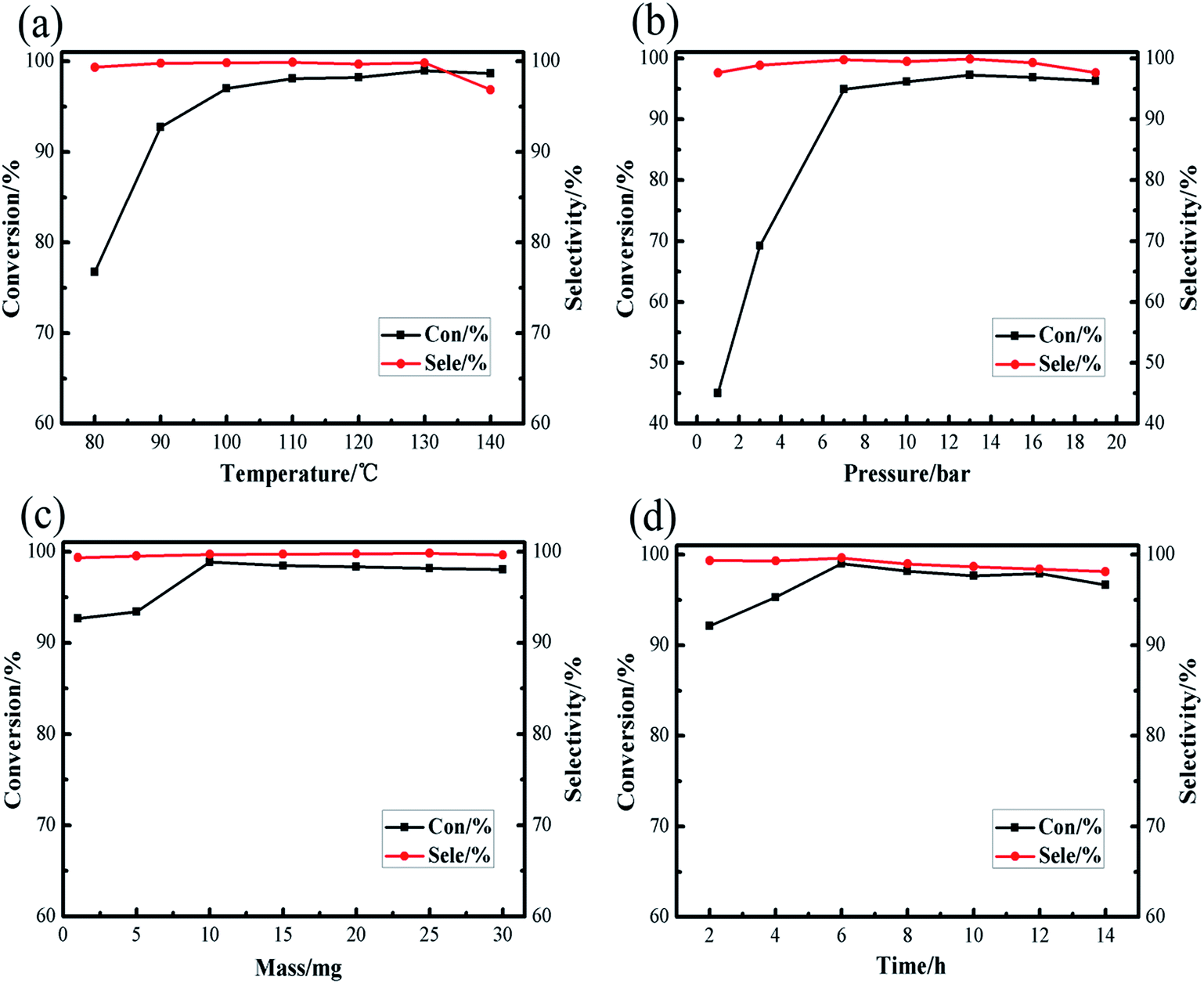 | ||
| Fig. 6 Effect of different reaction conditions on the conversions and selectivities of cyclic carbonate: (a) reaction temperature; (b) initial CO2 pressure; (c) catalyst amount; (d) reaction time. | ||
The reaction temperature is considered as an important factor in controlling the overall efficiency of the reaction. As shown in Fig. 6a, the reaction temperature screening interval is 80–140 °C, when the reaction temperature was 80 °C, the conversion of styrene oxide was only 76.76%, but as the temperature increased to 130 °C, the conversion of raw materials increased sharply to 98.96%. It is remarkable that at the appropriate reaction temperature (130 °C), the cyclic carbonate selectivity has been maintained above 99%. When the temperature is slightly raised to 140 °C, the conversion of styrene oxide and the selectivity of cyclic carbonate slightly decrease. May be at high temperature conditions, the product was decomposed or poly, formed by-products of cyclic carbonate, such as 2-phenylacetaldehyde and 1-phenylethyl glycol and 2,5-diphenyl-1,4-dioxane analyzed by gas chromatography-mass spectrometry (GC-MS), this was consistent with previous reports,33 thus the optimal reaction temperature selected in this study as 130 °C.
The pressure of CO2 is generally a key point affecting the cycloaddition reaction between CO2 and styrene oxide. As Fig. 6b revealed, the reaction pressure increases from 1 bar to 7 bar, the styrene oxide conversion rapidly increases from 45.04% to 94.9%. Conversion rate of the raw materials slightly increased within the pressure range of 7–13 bar, continue this increase in pressure to 19 bar, here the conversion of styrene oxide decreased. In the entire stress test range, the cyclic carbonate selectivity is still satisfactory (maintained at about 99%). The reaction system is composed of the gas phase and the liquid phase. As the CO2 pressure increases, the CO2 concentration in the liquid phase also increases. From the reaction kinetics point of view, this is a positive factor for the reaction, but as the CO2 pressure gradually increases, resulting in a dilution effect, an excessively high CO2 pressure no longer significantly promotes the reaction. The conversion of the feedstock and the selectivity of the product gradually decline, may be due to the decomposition of raw materials due to high pressure. Therefore, a relatively moderate CO2 pressure of 13 bar was enough for the satisfactory synthesis of cyclic carbonate.
Fig. 6c and d show the effect of amount of catalyst and reaction time on the catalytic performance of [Zn3(BTC)2]/TBAB binary system. The results showed that the conversion rate of styrene oxide showed an upward trend when the addition amount of catalyst was between 1–10 mg. When the amount of catalyst added was 10 mg, the conversion rate of raw material reached the highest 98.85%. Continue this increase in amount of catalyst, the conversion rate change is not obvious, and the cyclic carbonate selectivity still continues to be above 99%. The effect of the reaction time on the cycloaddition has also been gradually investigated. It was found that when the reaction reached 6 h, the conversion of styrene oxide reached the maximum of 99.02%. As time goes on, the conversion of styrene oxide and the selectivity of cyclic carbonate decreased slightly, may be longer reaction time promotes the formation of by-products. Overall, the amount of catalyst and the reaction time have little effect on the cycloaddition reaction relative to the reaction temperature and reaction pressure.
The recycling of the catalyst was surveyed to further testify the persistence of the catalytic activity and excellent stability in cycloaddition reaction. The catalytic activities of the reused [Zn3(BTC)2] catalyst are summarized in Fig. 7. For each cycle, the used catalyst was separated through centrifugation and thoroughly washed with ethanol and distilled water to remove the products adhered to the surface of the catalyst, and then was dried at 80 °C for 12 h under vacuum in order to remove the trapped ethanol and water molecules in the pores, finally, reused catalyst directly for the next run. In each run, both styrene oxide conversion and styrene carbonate yield showed a minimal reduction. However, after the catalyst was recycled three times, both the conversion of the feedstock and the yield of the product started to decrease slightly. Although the satisfied selectivity to styrene carbonate did not change obviously but after recycling the catalyst 5 times the conversion rate lowered to 95%, while recycling it in 6th run, the conversion rate observed was close to 90%. Partial loss of catalyst occurs. The [Zn3(BTC)2]/TBAB catalyst system was found to be applicable to a variety of epoxides under the optimized conditions determined as explained above. The results are summarized in Table 1. All epoxides showed high conversion and satisfactory product yield. We compared the catalytic performance of the [Zn3(BTC)2] with other MOFs heterogeneous catalysts reported by many researchers (Table 2). A comparison of the catalytic performance of the [Zn3(BTC)2]/TBAB system with other MOFs catalysts under the same reaction system was observed in this work. All the other MOFs reported in the literature, always need the assistance of a co-catalyst. All these MOFs catalytic reaction were performed at relatively mild conditions, that is, temperature and CO2 pressure varies between 60–140 °C and 10–20 bar respectively. In Table 2, the yield of the product and the TOF value exhibited by [Zn3(BTC)2]/TBAB in the cycloaddition reaction were compared with the different MOFs. The amount of co-catalyst used in this study is also very small, only (0.31 × 10−4 mmol). These results demonstrate the effectiveness of [Zn3(BTC)2]/TBAB in the field of carbon dioxide conversion to cyclic carbonates. Higher yields are important for the separation of the product/catalyst from the reaction mixture.
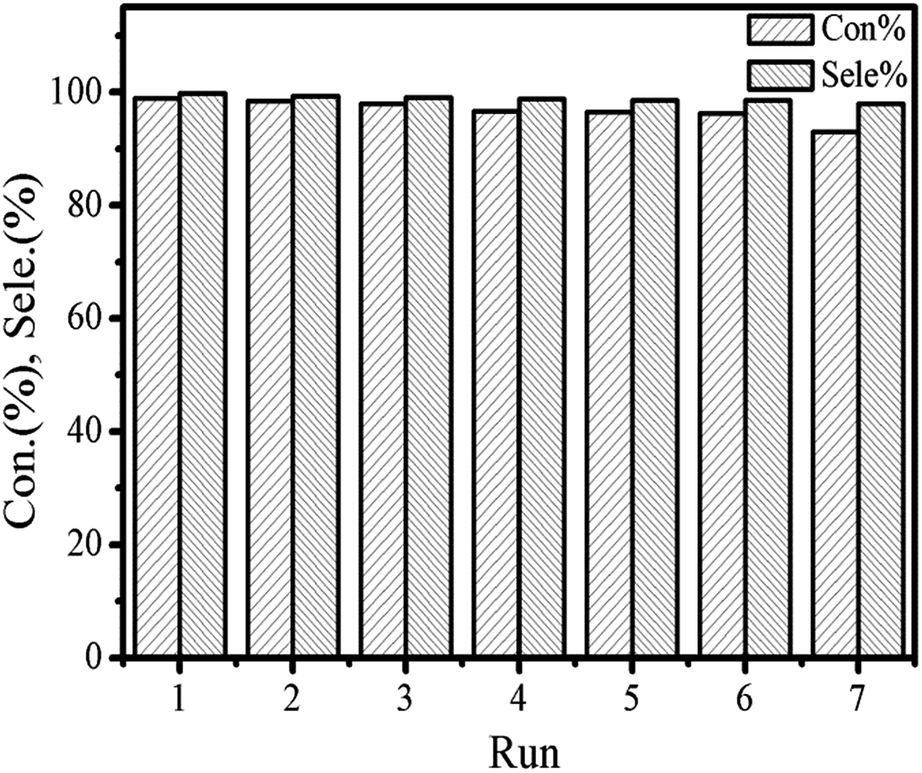 | ||
| Fig. 7 Catalytic performance of fresh and recycled [Zn3(BTC)2]/TBABr in the synthesis of styrene carbonate from carbon dioxide and styrene oxide. | ||
| Entry | Epoxide | Product | T (°C) | P (MPa) | t (h) | Reaction results | |
|---|---|---|---|---|---|---|---|
| C (%) | S (%) | ||||||
| a Reaction condition: epoxide (20 mmol); catalyst (10 mg); TBABr (0.31 × 10−4 mmol). C: conversion rate; S: product selectivity; all based on GC analyses. | |||||||
| 1 | 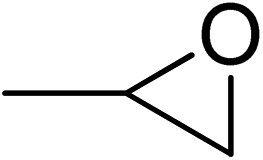 |
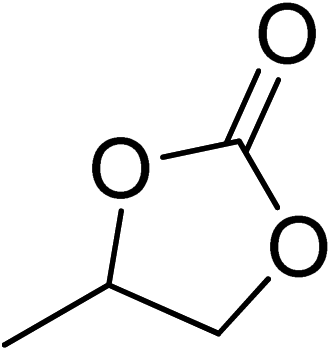 |
130 | 1.3 | 6 | 99.05 | >99 |
| 2 |  |
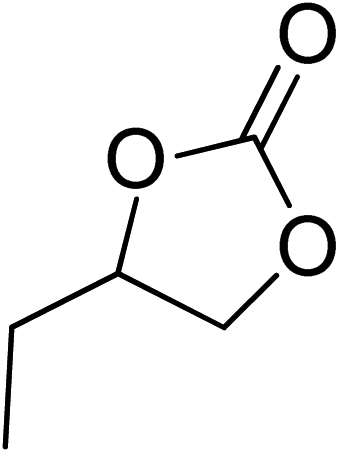 |
130 | 1.3 | 6 | 98.90 | >99 |
| 3 | 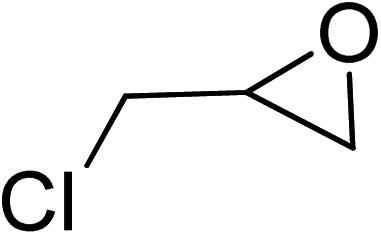 |
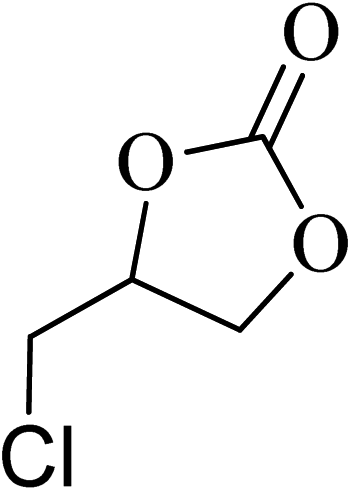 |
130 | 1.3 | 6 | 99.10 | >99 |
| 4 |  |
 |
130 | 1.3 | 6 | 98.96 | >99 |
| 5 |  |
 |
130 | 1.3 | 6 | 98.99 | >99 |
| 6 |  |
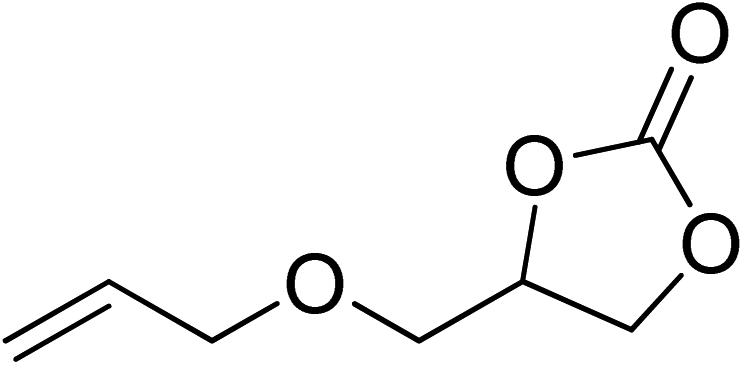 |
130 | 1.3 | 6 | 98.88 | >99 |
| 7 | 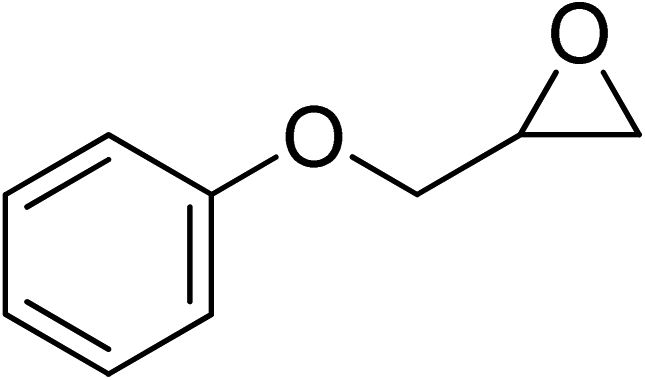 |
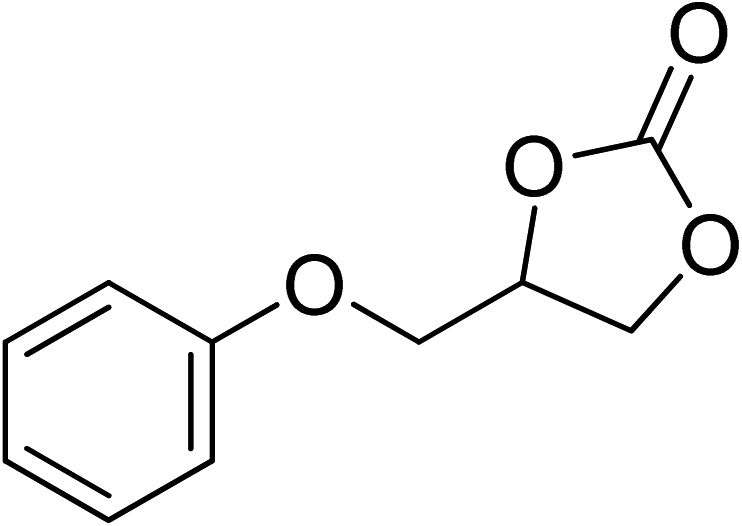 |
130 | 1.3 | 6 | 96.96 | >99 |
| 8 | 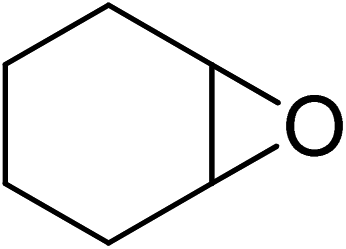 |
 |
160 | 3 | 24 | 55.35 | 90.5 |
| Entry | Catalyst | Co-catalyst/mmol | T (°C) | P (MPa) | t (h) | Yield (%) | TOF | Ref. |
|---|---|---|---|---|---|---|---|---|
| TOF = moles of cyclic carbonate formed/moles of metal in MOF catalyst/t. | ||||||||
| 1 | Al-MON | n-Bu4NCl/0.043 | 60 | 1 | 12 | 51 | 85 | 34 |
| 2 | Cr-MON | n-Bu4NCl/0.043 | 60 | 1 | 12 | 66 | 110 | 34 |
| 3 | Co-MON | n-Bu4NCl/0.043 | 60 | 1 | 12 | 75 | 125 | 34 |
| 4 | Ni-TCPE1 | n-Bu4NBr/0.3 | 100 | 1 | 12 | 99 | 167 | 35 |
| 5 | Ni-TCPE2 | n-Bu4NBr/0.3 | 100 | 1 | 12 | 86.2 | 143 | 35 |
| 6 | gea-MOF-1 | n-Bu4NBr/0.15 | 120 | 2 | 6 | 85 | 94 | 36 |
| 7 | In2(OH) (BTC) | n-Bu4NBr/0.5 | 80 | 2 | 4 | 91.5 | 169 | 37 |
| 8 | PCN-224 (Co) | n-Bu4NCl/0.0716 | 100 | 2 | 4 | 42 | 115 | 38 |
| 9 | Cd2[Ni(salen)] | n-Bu4NBr/0.05 | 80 | 1 | 12 | 91 | 15 | 39 |
| 10 | ZIF-95 | n-Bu4NBr/0.074 | 80 | 1.2 | 2 | 93.2 | 104 | 28 |
| 11 | [Zn3(BTC)2] | n-Bu4NBr/0.31 × 10−4 | 130 | 1.3 | 6 | 99 | 138 | This work |
| 12 | ZnGlu | n-Bu4NBr/0.4 | 80 | 1.2 | 6 | 99 | 35 | 40 |
| 13 | MIXMOF-5 | n-Bu4NBr/0.1 | 140 | — | 3 | 63 | 117 | 41 |
Potential mechanism of epoxide cycloaddition catalyzed by [Zn3(BTC)2]/TBAB catalytic system to produce cyclic carbonates is shown in Scheme 2. Previous reports based on experimental and density functional theory (DFT) studies,15,28,29,42 the presence of Lewis acidic sites and strong nucleophilic anions can effectively activate the epoxides and thus promote the ring-opening process. While carbon dioxide can be activated by a nucleophilic attack initiated by Lewis basic sites. Br− isolated from n-Bu4NBr is used to attack and activate fewer hindered carbon atoms in the epoxide, resulting in epoxides open-loop formation of reactive oxygen anion. Subsequently, the oxygen anion of the opened epoxide interacts with CO2 to form an intermediate. Finally, Br− is eliminated by a ring-closing step to produce the corresponding cyclic carbonate from the intermediate while regenerating the catalyst. The next cycle reaction immediately starts. Therefore, we verified that the catalyst contained both acidic sites and alkaline sites at the same time benefit for the reaction of carbon dioxide and epoxide. The synergistic effect of [Zn3(BTC)2] and n-BuN4Br is the major cause for the high catalytic activity of the catalyst system under relative moderate conditions.
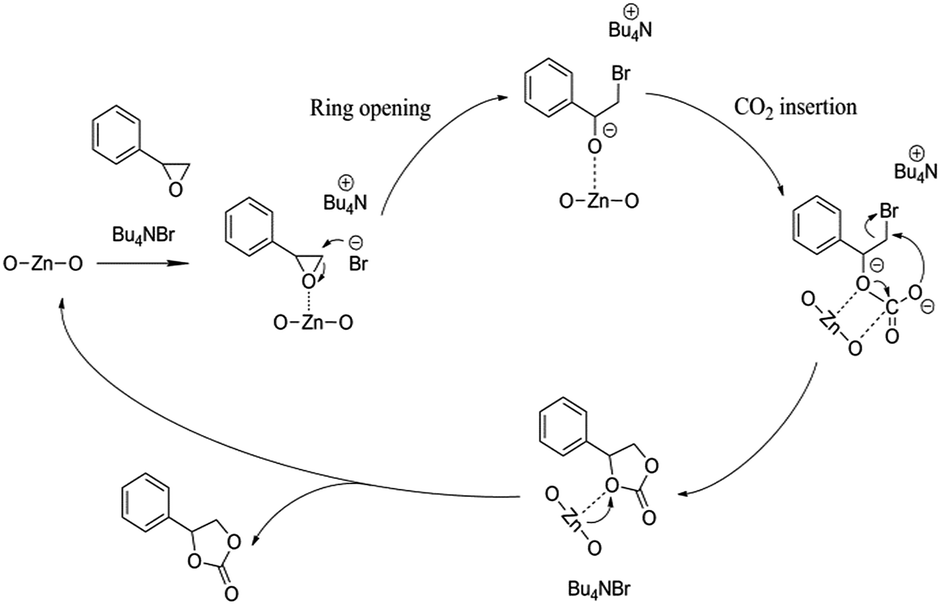 | ||
| Scheme 2 Proposed mechanism for the cyclic addition reaction of CO2 and styrene oxide in the presence of [Zn3(BTC)2]/TBABr. | ||
Conclusions
In summary, we report a virgulate 3D MOF has been synthetized by hydrothermal method using benzene-1,3,5-tri-carboxylic acid as ligand. Compared to hitherto known catalysts, [Zn3(BTC)2], a heterogeneous catalyst combined with n-Bu4NBr displayed efficient catalytic activity. Synergistic effects of [Zn3(BTC)2]/n-Bu4NBr catalysis systems have been investigated. Styrene carbonate yields as high as ∼99% under relatively moderate and solvent-free conditions (130 °C and 13 bar CO2 pressure) were surveyed for the [Zn3(BTC)2] catalysts. The catalytic activity of [Zn3(BTC)2] is better than the corresponding homogeneous species and having both high populations of Lewis acid and base sites, demonstrating the advantages of MOF catalysts for CO2 conversion reactions. Moreover, this MOF can be easily separated and reused (for three successive runs) without distinct loss of catalytic activity and structural deterioration. [Zn3(BTC)2] displayed particular chemical and thermal stability, which is crucial for its usage as industrial application. We conjecture [Zn3(BTC)2] will related reactions of epoxides with inhibition formation of byproducts while abide by the principles of green chemistry and atom economy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge technical assistance and material support from the Key Laboratory of Oil and Gas Fine Chemicals, Ministry of Education & Xinjiang Uygur Autonomous Region and acknowledge funding support from National Natural Science Foundation of China (No. 21162027 and 21261022).References
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2013, 114, 1709–1742 CrossRef PubMed.
- P. Lanzafame, G. Centi and S. Perathoner, Chem. Soc. Rev., 2014, 43, 7562–7580 RSC.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703 RSC.
- J.-C. C. Toshiyasu Sakakura and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef PubMed.
- M. A. Kazutoshi Ukai, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2006, 128, 8706–8707 CrossRef PubMed.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC.
- M. H. Beyzavi, C. J. Stephenson, Y. Liu, O. Karagiaridi, J. T. Hupp and O. K. Farha, Frontiers in Energy Research, 2015, 2, 63 CrossRef.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS.
- I. S. Metcalfe, M. North, R. Pasquale and A. Thursfield, Energy Environ. Sci., 2010, 3, 212–215 CAS.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169 CAS.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- J. Ma, J. Liu, Z. Zhang and B. Han, Green Chem., 2012, 14, 2410 RSC.
- R. Srivastava, D. Srinivas and P. Ratnasamy, Appl. Catal., A, 2005, 289, 128–134 CrossRef CAS.
- K. E. Kazuya Yamaguchi, T. Yoshida and A. K. K. H. Yoshida, J. Am. Chem. Soc., 1999, 121, 4526–4527 CrossRef.
- F. Li, L. Xiao, C. Xia and B. Hu, Tetrahedron Lett., 2004, 45, 8307–8310 CrossRef CAS.
- L. Wang, K. Kodama and T. Hirose, Catal. Sci. Technol., 2016, 6, 3872–3877 CAS.
- J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS.
- T. Ema, K. Fukuhara, T. Sakai, M. Ohbo, F.-Q. Bai and J.-y. Hasegawa, Catal. Sci. Technol., 2015, 5, 2314–2321 CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H.-L. Jiang and Q. Xu, Chem. Commun., 2011, 47, 3351 RSC.
- H. G. a. A. Corma and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef PubMed.
- R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS.
- H.-Y. Cho, D.-A. Yang, J. Kim, S.-Y. Jeong and W.-S. Ahn, Catal. Today, 2012, 185, 35–40 CrossRef CAS.
- J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031 RSC.
- O. V. Zalomaeva, A. M. Chibiryaev, K. A. Kovalenko, O. A. Kholdeeva, B. S. Balzhinimaev and V. P. Fedin, J. Catal., 2013, 298, 179–185 CrossRef CAS.
- L. Yang, L. Yu, G. Diao, M. Sun, G. Cheng and S. Chen, J. Mol. Catal. A: Chem., 2014, 392, 278–283 CrossRef CAS.
- K. M. Bhin, J. Tharun, K. R. Roshan, D.-W. Kim, Y. Chung and D.-W. Park, J. CO2 Util., 2017, 17, 112–118 CrossRef CAS.
- J. Zhu, T. Diao, W. Wang, X. Xu, X. Sun, S. A. C. Carabineiro and Z. Zhao, Appl. Catal., B, 2017, 219, 92–100 CrossRef CAS.
- J. Kim, S.-N. Kim, H.-G. Jang, G. Seo and W.-S. Ahn, Appl. Catal., A, 2013, 453, 175–180 CrossRef CAS.
- F. Farzaneh and S. S. Mortazavi, React. Kinet., Mech. Catal., 2016, 120, 333–344 CrossRef.
- Y. Fu, G. Li, F. Liao, M. Xiong and J. Lin, J. Mol. Struct., 2011, 1004, 252–256 CrossRef CAS.
- C. M. Miralda, E. E. Macias, M. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2012, 2, 180–183 CrossRef CAS.
- J. Chun, S. Kang, N. Kang, S. M. Lee, H. J. Kim and S. U. Son, J. Mater. Chem. A, 2013, 1, 5517 CAS.
- Z. Zhou, C. He, J. Xiu, L. Yang and C. Duan, J. Am. Chem. Soc., 2015, 137, 15066–15069 CrossRef CAS PubMed.
- V. Guillerm, Ł. J. Weseliński, Y. Belmabkhout, A. J. Cairns, V. D'Elia, Ł. Wojtas, K. Adil and M. Eddaoudi, Nat. Chem., 2014, 6, 673–680 CrossRef CAS PubMed.
- L. Liu, S.-M. Wang, Z.-B. Han, M. Ding, D.-Q. Yuan and H.-L. Jiang, Inorg. Chem., 2016, 55, 3558–3565 CrossRef CAS PubMed.
- D. Feng, W.-C. Chung, Z. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS PubMed.
- Y. Fan, J. Li, Y. Ren and H. Jiang, Eur. J. Inorg. Chem., 2017, 43, 4982–4989 CrossRef.
- A. C. Kathalikkattil, R. Babu, R. K. Roshan, H. Lee, H. Kim, J. Tharun, E. Suresh and D.-W. Park, J. Mater. Chem. A, 2015, 3, 22636–22647 CAS.
- W. Kleist, F. Jutz, M. Maciejewski and A. Baiker, Eur. J. Inorg. Chem., 2009, 24, 3552–3561 CrossRef.
- M. W. H. Donald and J. Darensbourg, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00152a |
| This journal is © The Royal Society of Chemistry 2018 |