
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of nanoparticle surface modification on the mechanical properties of polystyrene-based nanocomposites†
A. Kockmanna,
J. C. Porsiela,
R. Saadata and
G. Garnweitner *ab
*ab
aTechnische Universität Braunschweig, Institute for Particle Technology, Volkmaroder Straße 5, 38104 Braunschweig, Germany. E-mail: g.garnweitner@tu-braunschweig.de
bTechnische Universität Braunschweig, Laboratory for Emerging Nanometrology, 38106 Braunschweig, Germany
First published on 20th March 2018
Abstract
Nanocomposites consisting of metal oxide nanoparticles in a polymeric matrix enable the improvement of material properties and have become highly relevant for numerous applications, such as in lightweight structures with an enhanced Young's modulus for automotive and aircraft applications. The mechanical properties can be adjusted by controlling the amount of particles, their degree of agglomeration and their direct interaction with the matrix. Whilst the latter aspect is particularly promising to achieve high reinforcement at low filler contents, the mechanisms behind this effect are still not fully understood, preventing the rational design of a particle–polymer system with customized properties. In this work, a two-step modification strategy is used to tailor the particle–matrix interface via chemical groups bound to the surface of zirconia nanoparticles. Two modifications featuring terminal vinyl functions as potentially polymerizable groups are compared. Moreover, an inert reference modification is used to determine the influence of the terminal vinylic groups. In contrast to previous studies, all groups are covalently linked to the particle surface, thereby excluding effects such as detachment or weak coordination and ensuring that changes in the mechanical properties can be correlated to chemical groups on the particle surface. After embedding modified particles in polystyrene, the mechanical properties as well as the cross-linkage between the particles and the matrix are characterized, clearly showing the significant impact of a covalent particle–matrix linkage, with an increase of the Young's modulus by up to 28% with only 3 wt% filler content.
Introduction
The enhancement of the properties of polymers via the incorporation of inorganic filler particles is a widely-used approach in modern materials science and engineering.1–7 Typically, inorganic nanoparticles are dispersed in a polymeric resin which is then moulded and cured to form a nanocomposite (NC). There are many possible advantages over the neat polymer, from an enhancement of electrical8–11 and thermal12–14 conductivity, control of hydrophobicity,15–19 tailored optical properties20–22 to a great improvement of mechanical properties23–26 like scratch resistance27,28 or Young's modulus.26,29 In general, to achieve cost and weight reduction, smaller quantities and a homogenous distribution of the nanofiller are the key factors for an effective mechanical reinforcement.30 The main mechanism for the enhancement of the mechanical properties lies in the interplay between the relatively hard inorganic filler and the softer matrix,2 with the interface between filler and matrix playing a decisive role.30,31 The exact mechanisms leading to the reported benefits however are often hard to determine, and not in all cases the expected improvement in the mechanical properties can indeed be achieved. This still substantially impedes the commercial application of nanocomposites in various fields. In some cases, the addition of nanoparticles even results in a degradation of desired properties.32Although the particle distribution in the matrix can be adjusted to some degree by using various dispersing techniques, both of the above key factors that determine the mechanical properties are mainly dependent on the chemical nature of the nanoparticle surface. In many cases, the formation of strong bonds between the nanoparticles and the surrounding matrix is preferred to achieve high enhancement of the mechanical properties.4,25 As an example, Guo et al. showed a 58% increase of the Young's modulus by embedding 3 wt% of alumina particles in a vinyl-ester resin.4 An adjustment of the particle–matrix interphase, mostly influenced by the nanoparticle surface chemistry, is an important approach for the deeper understanding of the mechanisms of reinforcement of NCs and therefore, the development of materials with more specific and enhanced properties. In this way, similar or even better results can be obtained with potentially lower filler contents.
In general, for an optimization of the mechanical properties like the Young's modulus, the scratch resistance or higher breaking strength, the surface of the particles needs to be chemically altered to strengthen interface bonding. Theoretical approaches to explain and predict such an effect describe mechanisms like crack-pinning or crack-blunting, where a progressing crack strikes a hard filler particle which is too hard to break and therefore the crack has to evade it.33–38 Additionally, for nano-sized filler particles, another very important factor is the distribution of the particles within the matrix, because big agglomerates greatly limit the positive effect of a high specific surface area.39 One of the biggest advantages of particles in the nanometer range is the large surface-to-volume ratio. This on the one hand can improve the mechanical properties; on the other hand, nanoparticles tend to agglomerate due to their extensive surface and its high specific surface energies. If large agglomerates are incorporated in a matrix, a strong decrease of the mechanical strength can be expected. The mechanical weakening is often caused by the weak particle–particle interfaces in agglomerates, which possibly act as a weak spot, and less to no crack pinning can be observed.34,35,37,38
Metal oxide nanoparticles represent a preferred class of filler particles, due to their high chemical stability and moderate cost. In many cases, however, the precise control and adjustment of the surface chemistry of metal oxides is difficult and the nature of the particle–ligand interaction is often unknown. For best mechanical and chemical stability, a covalent attachment is preferred.32,37,39 A number of strategies have been presented to covalently modify the surface of nanoparticles for the linkage to a polymer.5,7,40 These strategies can generally be categorized into two approaches, the so-called grafting-to and grafting-from methods. In the grafting-from technique, an initiator moiety is bound to the nanoparticle, which results in the polymerization being started at the particle surface. The grafting-to technique on the other hand utilizes an already polymerized polymer which then is bound to the particle surface. In this work, a “grafting-through” strategy is used, where the particles feature polymerizable groups bound to their surface. Particles and styrene as monomer are mixed together and afterwards the polymerization is started by external initiators. During this polymerization, the reaction progresses from the matrix to the particle surface and on into the matrix, and thus a firm bond between the particles and the matrix is established.
We have recently developed a two-step modification strategy that enables the covalent binding of a wide range of carboxylic acids to the surface of different metal oxide nanoparticles.41 In this work, we utilized this strategy to tailor the surface chemistry of zirconia nanoparticles with long aliphatic as well as aromatic organic moieties as illustrated in Fig. 1. By linkage of ligands featuring polymerizable terminal double bonds a grafting-through strategy was realized, whilst saturated ligands were used as unreactive modifications for comparison. Subsequently, the modified particles were incorporated into monomeric styrene which was then polymerized with a peroxide initiator.
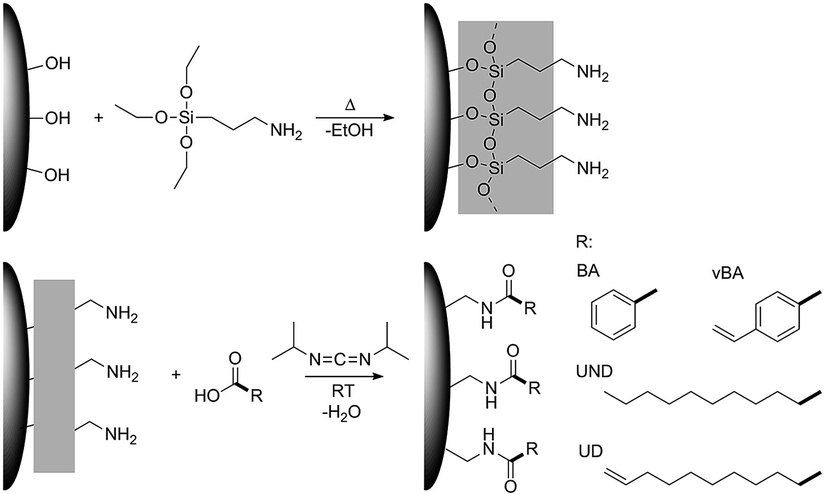 | ||
| Fig. 1 Schematic illustration of the modification strategy used in this study. | ||
This strategy was used to realize highly comparable particle systems with a uniform covalent linkage between different ligands and the particle surface, whereas different interactions between the ligands and the polymer matrix could be expected and compared. Undecanoic acid and undecylenic acid were used as aliphatic ligands and benzoic acid and vinylbenzoic acid as aromatic ligands. Undecylenic acid as well as vinylbenzoic acid feature terminal double bonds that in principle can react with styrene during the polymerization process. Undecanoic and benzoic acid on the other hand are highly unlikely to react with the styrene matrix in the polymerization. Due to these very similar functional groups, a resilient connection of these with the particles as well as a highly comparable density of functional groups on the particle surface, the effects of the terminal double bonds on the mechanical properties can directly be investigated. Furthermore, the usage of small angle X-ray scattering and transmission electron microscopy also suggests very comparable size distributions and structures of the particles in the final composite, thereby excluding further possible effects on the mechanical properties. To the best of our knowledge, such a high degree of a direct correlation between the specific functional group forming the particle–matrix interface and the resulting mechanical properties has not been yet achieved. By the application of rheological techniques, the strength of the interaction between the particles and the matrix is monitored to verify the resulting bond formation and cross-linkage between the particles and the polymer. The corresponding mechanical properties were characterized by determination of the Young's modulus from macroscopic indents (DIN EN ISO 2439:2009-05) as well as performing scratch tests. In this way, a strong influence of reactive chemical groups on the particle surface could be evidenced, achieving strong improvements of the Young's modulus and the scratch resistance for the optimum particle–matrix interface configuration.
Methods and materials
Materials
3-Aminopropyltriethoxysilane (APTES, 99%), N,N′-diisopropylcarbodiimide (DIC, 98%), 10-undecenoic acid (UD, 98%), undecanoic acid (UND, 98%), benzoic acid (BA, 99.5%) and 4-vinylbenzoic acid (vBA, 97%) were purchased from Sigma Aldrich and used without further purification. The solvents ethanol, ethyl acetate and acetone (technical grade) were obtained from VWR International. For the formation of the nanocomposites, monostyrene (with inhibitor) from BÜFA and CUROX M-312 as hardener from United Initiators were used as received.Synthesis of ZrO2 nanoparticles
Zirconia (ZrO2) nanoparticles were synthesized via the non-aqueous sol–gel synthesis as described earlier.42,43 Briefly, 12.80 ml of Zr(IV) n-propoxide in 1-propanol (70 wt%, Sigma Aldrich) was added to 920 ml of benzyl alcohol and the mixture heated in a closed vessel to react for 24 h at 250 °C. After cooling, a suspension of agglomerated 3.5 nm-sized ZrO2 nanoparticles was obtained.First step: APTES modification of ZrO2 nanoparticles
For the first modification step, the as-synthesized zirconia nanoparticle suspension was taken from the reactor and APTES with a mass ratio of 45 wt% ATPES per theoretical ZrO2 content (assuming a 100% ZrO2 yield) was added to the white suspension. The mixture was heated under reflux conditions at about 110 °C and kept at this temperature under stirring for 12 h. After the reaction, the APTES-modified ZrO2 nanoparticles (ZrO2-APTES) were precipitated from the formed clear yellow liquid via the addition of ethyl acetate and separated by centrifugation at 6280 rcf for 10 minutes. The obtained precipitate was dispersed in ethanol, reprecipitated with ethyl acetate and centrifuged, which was repeated for three times to remove remaining APTES and possible side products. Then, the ZrO2-APTES nanoparticles were stored in ethanol at room temperature prior to further modification.Second step: coupling of carboxylic acids on the ZrO2 nanoparticles
The particles were dispersed in ethanol with a concentration of 0.01 g ml−1 and sonicated in an ultrasonic bath for 20 minutes. A mixture of the respective carboxylic acid and DIC with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 in ethanol was added to the ZrO2-APTES dispersion and stirred at room temperature. After 12 h, the suspension was centrifuged, and the precipitated nanoparticles dispersed in ethanol and again precipitated via centrifugation for three times. This purification and stripping of unbound excess ligand was additionally repeated twice by using acetone instead of ethanol, and finally the particles were dried in a rotary evaporator at 2 mbar and 60 °C for 1 h. In this way, particles functionalized with 1-undecylenic acid (ZrO2-APTES-UD), undecanoic acid (ZrO2-APTES-UND), vinylbenzoic acid (ZrO2-APTES-vBA) and benzoic acid (ZrO2-APTES-BA) as carboxylic acids were obtained that could be used in the subsequent processing.
:1.5 in ethanol was added to the ZrO2-APTES dispersion and stirred at room temperature. After 12 h, the suspension was centrifuged, and the precipitated nanoparticles dispersed in ethanol and again precipitated via centrifugation for three times. This purification and stripping of unbound excess ligand was additionally repeated twice by using acetone instead of ethanol, and finally the particles were dried in a rotary evaporator at 2 mbar and 60 °C for 1 h. In this way, particles functionalized with 1-undecylenic acid (ZrO2-APTES-UD), undecanoic acid (ZrO2-APTES-UND), vinylbenzoic acid (ZrO2-APTES-vBA) and benzoic acid (ZrO2-APTES-BA) as carboxylic acids were obtained that could be used in the subsequent processing.
Preparation of the nanocomposites
For the preparation of the nanocomposite, the modified ZrO2 nanoparticles were dispersed via ultrasonication in monostyrene in an ultrasonic bath for 30 minutes. Subsequently, the peroxide hardener was added to the suspension with a content of 5 wt%. The obtained mixture was stirred and heated to 150 °C for 15 minutes, which resulted in a strong increase in viscosity due to polymerization of the styrene. Afterwards, the mixture was mold-casted at 120 °C and cured at 80 °C for another 24 h in an oven.Characterization
The FT-IR measurements were taken on a BRUKER Vertex V70 with an ATR crystal measurement setup, recording 32 scans from 4500 to 100 cm−1 with a resolution of 1 cm−1. Small angle X-ray scattering (SAXS) measurements were carried out on a SAXSess mc2 system from Anton Paar using a Cu Kα (λ = 0.15418 nm) radiation source and a CCD detector. The cured composites were measured as a fixed thin film with a sample to detector distance of 309 mm and the scattering patterns were measured 5 times, each with an exposure time of 10 seconds. The obtained 1D scattering curves were evaluated and corrected regarding the sample background and slit smearing effects of the setup by using SAXSquant (Anton Paar, version 3.91.2866.48). Furthermore, to obtain the real space information from the scattering data, indirect Fourier transformations were completed using GIFT (Generalized Indirect Fourier Transformation) from the PCG software package.44 Thermogravimetric and differential thermoanalytic analyses (TGA/DTA) were performed using a Mettler Toledo TG/SDTA 851e device, applying a heating rate of 10 K min−1 from 25 to 950 °C in air. The obtained thermograms were normalized to 100% at 200 °C to eliminate the influence of moisture and solvent residues present in the initial samples.Rheological measurements were carried out using a Malvern Rotonetic drive 2 rheometer. The tests were performed at 20 °C, with a plate–plate geometry (radius 20 mm, distance 150 μm) in the viscoelastic region at frequencies between 0.1 and 10 Hz. For the sample preparation, the composite as well as pure polystyrene as reference were grounded in a mortar and mixed with toluene in a weight ratio of 1:1 and kept under stirring for 24 h before testing.
The Young's modulus was determined via an indentation test based on DIN EN ISO 2439:2009-05. The cylindrical samples with a diameter of 15 mm and a height of 10 mm were placed between round stamps of hard metal of 10 mm diameter which were then pressed into the nanocomposite. Force–displacement curves were recorded with the material testing device Retroline by Zwick Roell. The slope of the linear region in the force–strain curves was evaluated and fitted to calculate the Young's modulus. For statistical reliability, 8 samples of each composite were measured, melted, and measured two further times.
Scratches for the scratch test were applied with a hard metal scriber, which was pressed at an angle of 45° with a force of 2 N against a flat 3 mm casting of the nanocomposite on glass and pulled over a distance of 15 mm, whereas five scratches were measured from two casts for each composition. The profile of the resulting scratches was subsequently measured with a BRUKER DektakXT profilometer with a 5 μm tip, a weight of 0.05 μN and a scanning speed of 10 nm s−1. The maximum indicated depths of the scratches are evaluated as the scratch depths.
The ninhydrin test was carried out using a 2.5 g l−1 ninhydrin solution in ethanol, which was added to a sample of the particle dispersion, followed by ultrasonication for 15 min in an ultrasonic bath and then heating up to 60 °C for 30 min.
For the transmission electron microscopy analysis, a FEI-Tecnai G2 Spirit instrument was used with a voltage of 200 kV. Prior to the measurement, the nanocomposite with 1 wt% filler content for each surface modification was ground to a fine powder that was subsequently placed on a carbon-coated TEM grid (3.05 mm mesh 300 from PLANO) and melted at 190° for 5 min to obtain a thin uniform sample.
Results and discussions
In this part, the modification of the nanoparticles (both steps), the distribution of the particles in the resulting matrix, and the properties of the final nanocomposites are discussed. Since the aim of our study is to directly and comparably monitor the influence of the terminal functional group in the modification (which may enable the formation of a firm particle–matrix connection) on the mechanical properties, the interaction of the polymer and the particles is characterized via rheological measurements.Nanoparticle surface modification
The used modification strategy was already introduced in our earlier publication.41 Nevertheless, careful characterization of the modified nanoparticles was performed to ensure stable linkage of the ligands to the particle surface. Fig. 2 shows the results of TGA/DTA measurements of the ZrO2 nanoparticles before (ZrO2) and after (ZrO2-APTES) the modification with APTES. An increase of the weight loss from 6.1 to 11.3 wt% can be observed; the main weight loss thereby was detected between 390 and 420 °C. This can be explained by the coupling of the silane to the particle surface.45,46 The propylamine moiety decouples from the silicon atom in a wide temperature range above 350 °C,47,48 whilst synthesis residues and side products (mostly benzoate) desorb at temperatures of around 400 °C.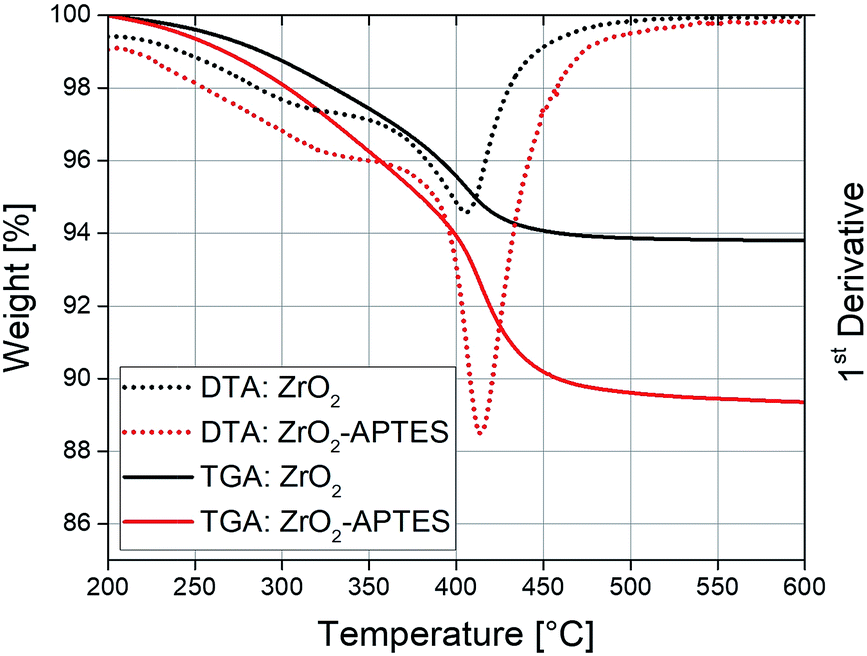 | ||
| Fig. 2 TGA/DTA curves of ZrO2 nanoparticles before and after the reaction with APTES. | ||
Ninhydrin tests of thoroughly washed particles were conducted to ensure the presence of reactive amino moieties (Fig. 3).49 The positive ninhydrin test even after many washing steps proves a strong binding between the amino group-bearing ligands and the nanoparticles.
 | ||
| Fig. 3 Ninhydrin test of ZrO2 nanoparticles before (left) and after (right) the modification with APTES. | ||
A comparison of the FT-IR spectra before and after the modification step with APTES shows a strong increase in the organic content (Fig. 4). The Zr–O stretching vibration peak at 583 cm−1 can be observed in both spectra. In the spectrum of the unmodified particles, typical benzoate peaks of the asymmetric and symmetric CH vibrations at 1536 and 1419 cm−1 can be found.2 Benzoate is formed as a side product in the utilized synthesis of the ZrO2 nanoparticles, as described in our previous work.42 Furthermore, at 1595 cm−1 the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations can be identified.
C stretching vibrations can be identified.
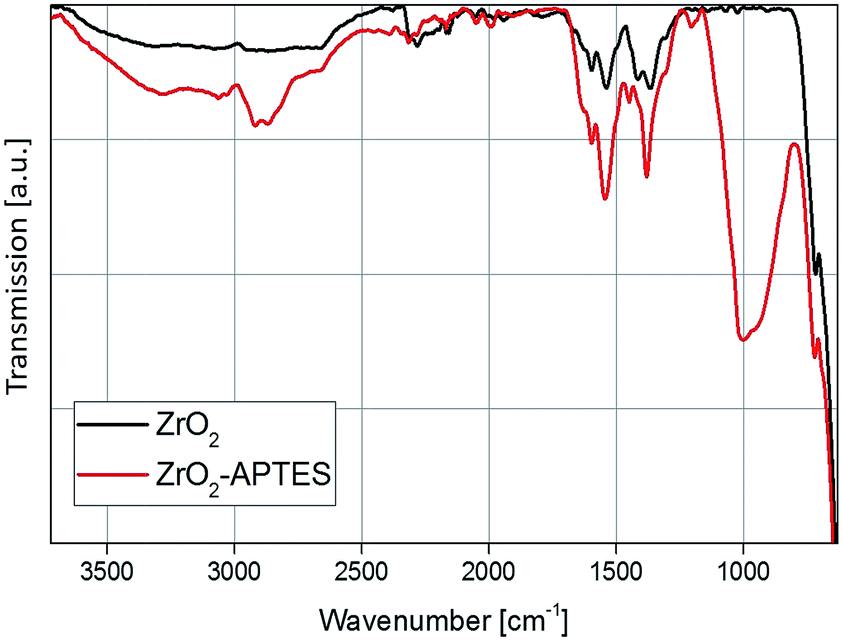 | ||
| Fig. 4 FT-IR spectra of the ZrO2 nanoparticles before and after the modification with APTES. | ||
The biggest difference between the spectra is the appearance of the asymmetric and symmetric stretching vibrations of the CH2 and CH3 bending vibrations at 2950, 2856 and 1450 cm−1, as well as the broad band of the Si–O vibration between 1250 and 900 cm−1 for the modified ZrO2 nanoparticles. This indicates a change in the surface chemistry due to the addition of the APTES to the particles via the Si–O coupling, which confirms the results from the TGA measurement and the ninhydrin test.
To investigate the subsequent reaction of the amino group with the carboxylic acid to form an amide bond, the FT-IR spectra of all modified particles are shown in Fig. 5 and 6. In Fig. 5, the spectra for ZrO2-APTES-UND and ZrO2-APTES-UD are compared. The asymmetric and symmetric C–H vibrations at 2950 and 2856 cm−1 are clearly visible, as can be expected for a C10 linear alkane chain. In the magnification inset, the characteristic region from 1800 to 1480 cm−1 is shown and the double peak for the amide bond at 1545 and 1640 cm−1 becomes clearly visible. Furthermore, for the ZrO2-APTES-UD particles the CC stretching vibration of the terminal double bond can be found at 1645 cm−1, in contrast to the ZrO2-APTES-UND particles, which shows the successful binding of the carboxylic acid via the postulated amide bond and the presence of the double bond at the particle surface.
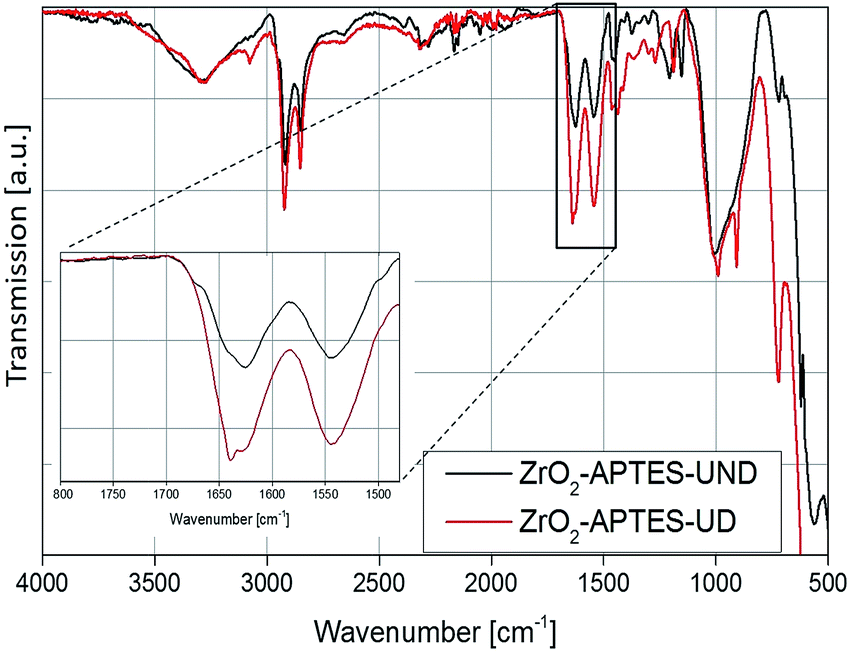 | ||
| Fig. 5 FT-IR spectra of ZrO2-APTES-UND and ZrO2-APTES-UD. | ||
 | ||
| Fig. 6 FT-IR spectra of ZrO2-APTES-BA and ZrO2-APTES-vBA. | ||
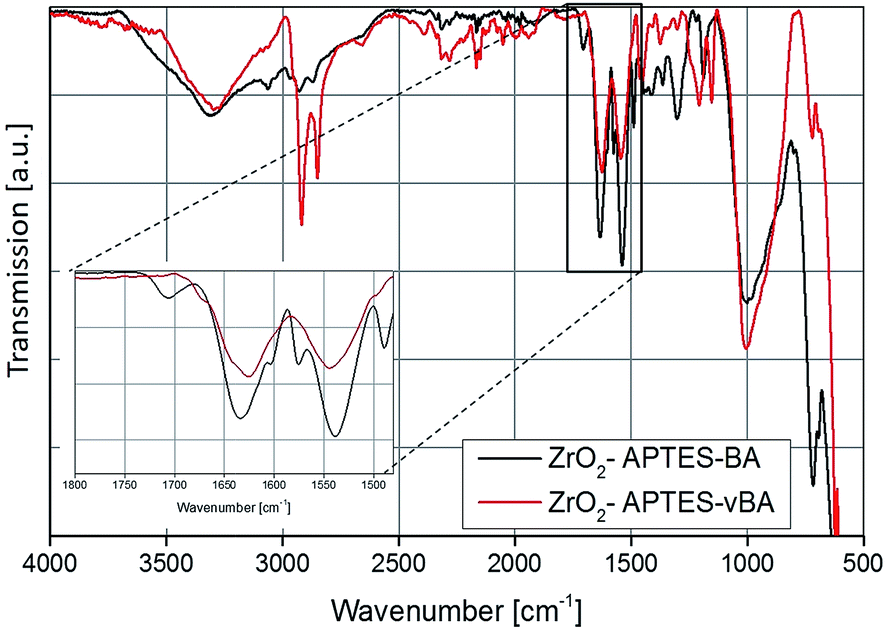
The FT-IR spectra of the ZrO2-APTES-BA and ZrO2-APTES-vBA samples are shown in Fig. 6. The most obvious differences between the two spectra is the presence of sharp peaks for the C–H vibrations at 2950 and 2856 cm−1 for the ZrO2-APTES-vBA in contrast to broader signals at much lower intensities for the ZrO2-APTES-BA particles. This can be attributed to the terminal vinyl group of the ZrO2-APTES-vBA particles, which is not present in the ZrO2-APTES-BA particles. Both particle systems furthermore possess propyl chains from the APTES moieties, explaining the small C–H peaks remaining for ZrO2-APTES-BA. Analogously to the aliphatic modifications, for ZrO2-APTES-vBA at 1645 cm−1 a side band for the vinyl group can be detected. Additional peaks in the fingerprint region show the existence of aromatic moieties.
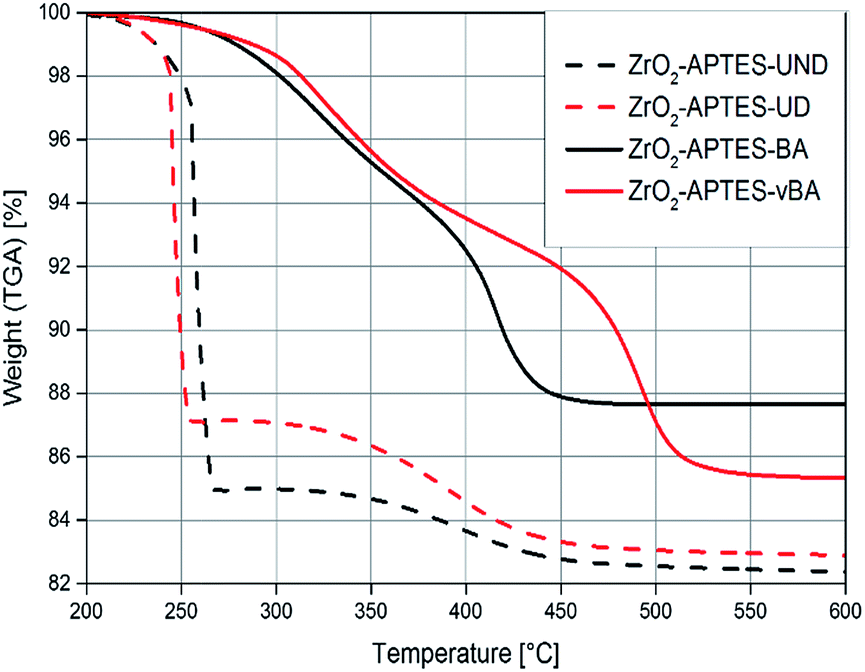
To assess the successful coupling of the used carboxylic acids after the second modification step, TGA/DTA was used (Fig. 7). The modifications with an aromatic group (–BA/–vBA) and the modification with aliphatic groups (–UD/–UND) each are similar to one another and only differ in the terminal vinyl group. Therefore, the decomposition temperatures and the amount of organic groups bound to the particle surface are comparable in each pair of ligands used. The weight loss of the particles modified with the relatively larger –UND and –vBA ligands are higher than for the –UD and –BA-modified particles, as can be expected.
 | ||
| Fig. 7 TGA of ZrO2 nanoparticles modified with APTES and different carboxylic acids as terminal groups. | ||
Structural characterization of nanocomposites
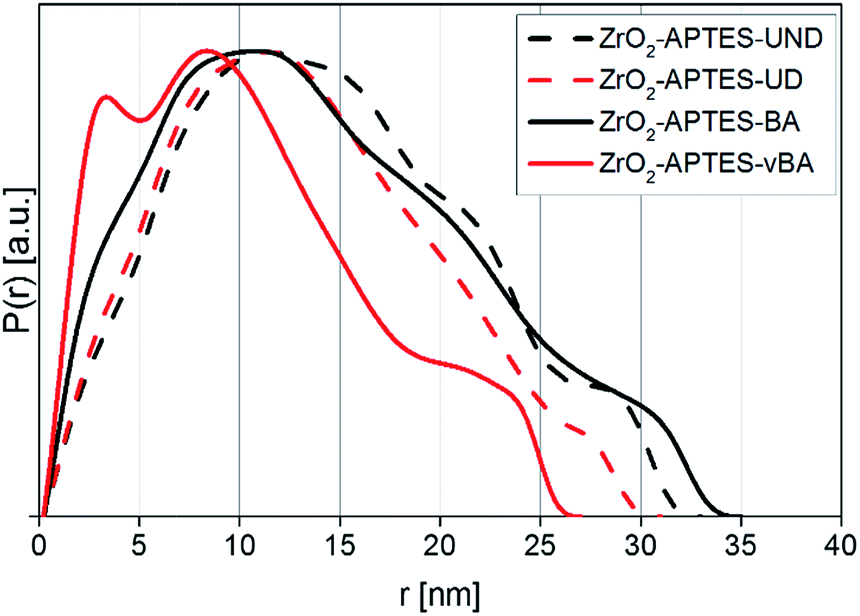
After the modification, the differently modified nanoparticles were incorporated in polystyrene in a grafting-through process and the distribution in the matrix was characterized to ensure that all nanocomposites show a homogeneous distribution of the NPs and the resulting properties are solely an effect of the particle–matrix interaction.Since the electron density of the nanofillers used in this work provided a high enough contrast to the chosen matrix, SAXS measurements could be performed to characterize the primary and secondary particle size, as well as detailed morphological features of the system in its actual final state in the nanocomposite. In Fig. 8, the pair distance distribution functions (PDDFs) as determined from the desmeared scattering curves using GIFT are plotted, showing the relative occurrence of all paired-sets of distances between all electrons in our samples.
 | ||
| Fig. 8 PDDF of nanocomposites formed from 3 wt% of differently modified particles measured by SAXS. | ||
The PDDFs clearly visualize that all samples contain larger structures rather than only single nanoparticles, with a number of maxima or shoulders being visible. Whilst the first maximum typically is correlated to primary particles, the following maxima can be attributed to small agglomerates that develop from the same primary particles. Thereby, the PDDFs do not present a true particle size distribution; however the maxima – the most numerous distances within a system – can be correlated to an agglomerate radius, whilst the right tail resembles the diameter. All utilized modifications in the final nanocomposites indicate a primary particle radius of around 3 nm, most clearly visible as a distinguished signal for ZrO2-APTES-vBA, but still visible for all other modifications as shoulders at the same position. A primary particle diameter of 6 nm is as expected for the applied synthesis.28,29
Since SAXS measures the sample as an ensemble and individual features of certain particle types are additive in the collected scattering curve, the values within the PDDF can be seen as the relative occurrence of each distance. Therefore, in ZrO2-APTES-vBA there is a significant portion of individual primary particles present, indicating the lowest tendency to form clusters compared to the other systems. For ZrO2-APTES-BA also a small fraction of non-agglomerated primary particles is clearly visible, whilst the aliphatic modifications result in a stronger tendency to form larger structures. This can be explained by the high chemical similarity of vBA and BA to styrene, resulting in better compatibility compared to the aliphatic ligands UD and UND. The PDDFs of ZrO2-APTES-BA, ZrO2-APTES-UD and ZrO2-APTES-UND show the presence of agglomerates with secondary particle diameters between 10 and 20 nm, with a small fraction of largest structures of about 35 nm in size. In contrast, ZrO2-APTES-vBA does not contain structures greater than 27 nm, with most agglomerate structures being less than 15 nm in size and a significant amount of primary particles present. ZrO2-APTES-BA tends to agglomerate to a similar degree as ZrO2-APTES-UD and ZrO2-APTES-UND, but it has very distinct shoulders and therefore contains both a significant amount of primary particles and of larger agglomerates.
It is obvious that distances based on the primary particle size frequently appear, but slightly differ between the samples in their exact position. This can be explained by the different lengths of the ligands. Especially for small particles, the agglomerate size is strongly influenced by the ligand shell of the primary particles. Moreover, a certain degree of polydispersity of the nanoparticles needs to be taken into account.
TEM pictures of thin films of the nanocomposites directly show the comparable distribution of the nanoparticles in the surrounding matrix for all used modifications. In Fig. 9, small agglomerates of around 6 nm-sized primary particles are visible possessing a secondary particle size of around 25 nm in all samples.
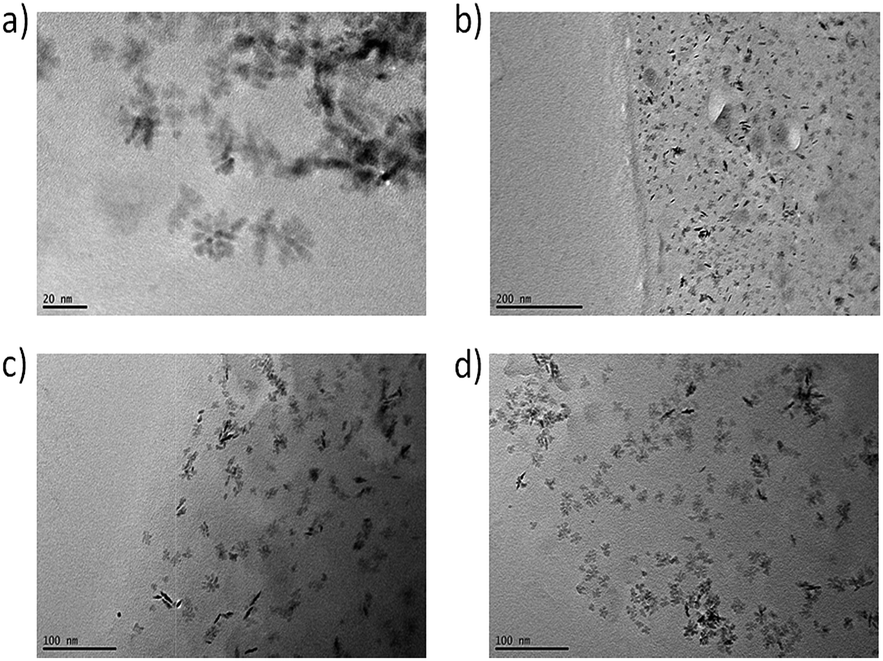 | ||
| Fig. 9 TEM pictures of NCs with 3 wt% of modified particles of (a) ZrO2-APTES-BA, (b) ZrO2-APTES-vBA, (c) ZrO2-APTES-UD, (d) ZrO2-APTES-UND. | ||
The formed structures possess comparable sizes for all modifications and are in good agreement with the SAXS measurements. In some TEM pictures, the phase boundary of the composite can be observed; as thinner sample spots showed better contrast of single structures, the edges of the obtained thin films were utilized for the measurements. Whilst in the composite with ZrO2-APTES-vBA, most agglomerates indeed contain only a few primary particles and are less than 10 nm in size, in all other composites mostly larger clusters are visible that are around 10–34 nm in size. In the TEM pictures, only particles close to the surface of the composite are clearly visible, whereas for samples with larger thickness, the contrast decreases and the state of agglomeration of the nanoparticles cannot be accurately assessed (images not shown).
Optically, there is little to no difference in the transparency of the different cast nanocomposites. All nanocomposite samples (prepared with a uniform thickness of 3 mm) look clear and (semi)transparent (Fig. 10), confirming the absence of larger agglomerates than observed in the SAXS and TEM measurements.
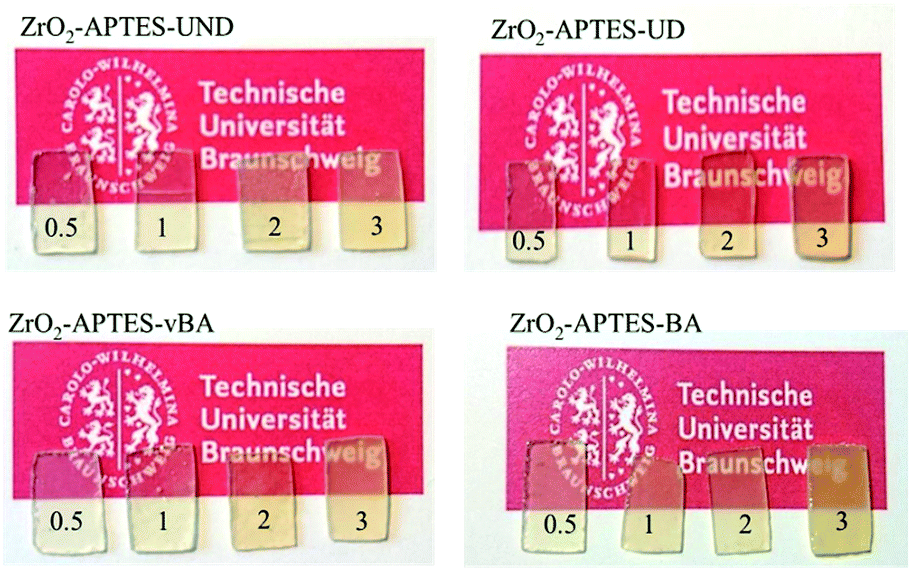 | ||
| Fig. 10 Photos of cured thin film polystyrene–zirconia NCs filled with different amounts of modified nanoparticles as indicated. | ||
Only the sample with ZrO2-APTES-BA at a filler content of 3 wt% shows a visible turbidity, which may result from the slightly larger agglomerate sizes (and broader size distribution) due to which the agglomerates would reach a size leading to scattering of the visible light in the composite. Another possibility that we cannot exclude could be the formation of a very small concentration of a few large agglomerates (>100 nm). A minute amount of greater agglomerates may not provide sufficient scattering intensity for detection in SAXS and may be easily be overseen due to the local focus of the studied TEM pictures although we have tried our best to obtain representative images, but still may have a significant impact on the mechanical and optical properties.
Mechanical properties
The controlled scratching and the successive measurement of the scratch profile is a macroscopic and application-oriented characterization method to test the resistance of a coating against scratches, which we applied to test the suitability of the different nanocomposites for coatings. In Fig. 11, typical profiles of scratches are shown for NCs each filled with 3 wt% of the differently modified nanoparticles. Comparing the modifications with saturated terminal groups (–UND; –BA) with the ones featuring terminal double bonds (–UD; –vBA), a significantly lower scratch depth and hence a greater scratch resistance is observed for the reactive modifications.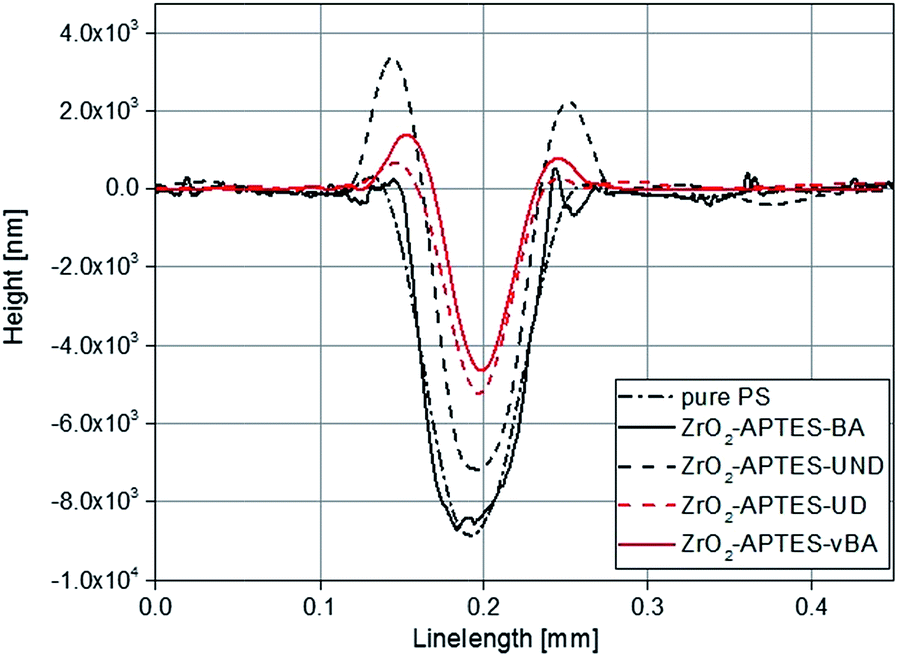 | ||
| Fig. 11 Overlay of typical scratch profiles of polystyrene NCs filled with 3 wt% of differently modified nanoparticles in comparison to pure polystyrene (PS). | ||
In Fig. 12, the average scratch depths of the nanocomposites with embedded ZrO2-APTES-BA, ZrO2-APTES-vBA, ZrO2-APTES-UND and ZrO2-APTES-UD particles are shown as a function of the content of embedded nanoparticles. All nanocomposites show a decrease in the scratch depth with rising particle content. However, with the embedment of ZrO2-APTES-UND, only a slight decrease in scratch depth (13%) can be observed, whilst the embedding of ZrO2-APTES-UD resulted in a significant decrease (44%). For the ZrO2-APTES-vBA, the decrease in the scratch depth is even greater with about 50%. Notably, the modification with similar chemical compatibility but without terminal double bonds (ZrO2-APTES-BA) leads to a decrease in the scratch depth of only 4% for 3 wt% of embedded particles. For the particles modified with aromatic as well as for the aliphatic ligands, the difference between the expected reactive (–vBA, –UD, red curves in Fig. 12) and non-reactive (–BA, –UND, black curves in Fig. 12) surface is highly pronounced.
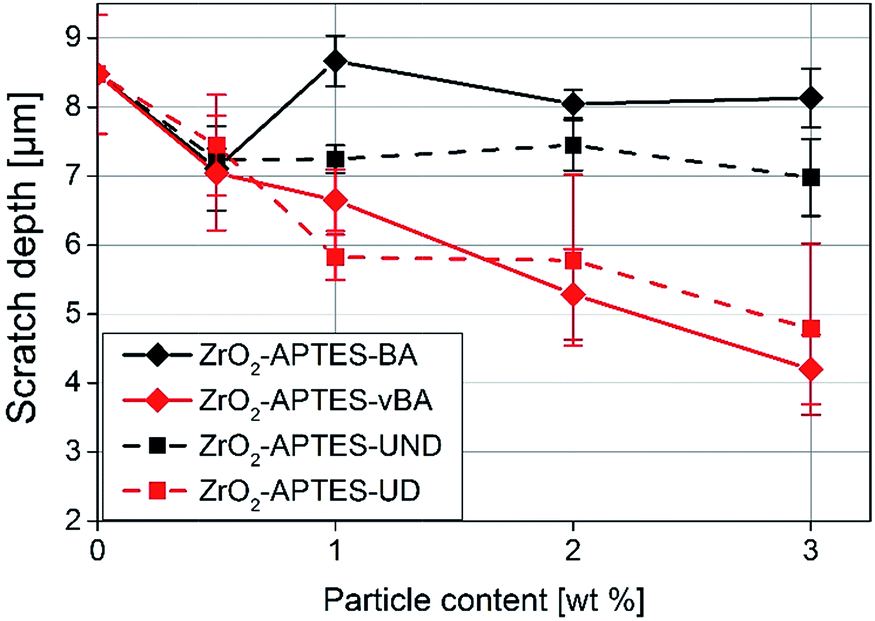 | ||
| Fig. 12 Scratch depth of NCs filled with different amounts of modified nanoparticles as indicated. | ||
As a general trend for the scratch development, it can be recognized that the scratch depth decreases almost in a linear fashion with the addition of increasing amounts of the nanoparticles for modifications with terminal double bonds whilst only slight effects are obtained for the other modifications.
Since the scratch depth only characterizes mechanical properties of the surface to a depth of only a few μm, indention tests were performed to characterize the macroscopic mechanical properties. In Fig. 13, typical force–strain measurements for NCs filled with 3 wt% of the differently modified particles in macro-indentation experiments are shown. From these experiments, the Young's modulus can be determined as the slope of the linear region (200–400 μm strain) of the curves. The calculated Young's moduli for nanocomposites with different contents of ZrO2-APTES-UND and ZrO2-APTES-UD as well as ZrO2-APTES-BA and ZrO2-APTES-vBA are shown in Fig. 14.
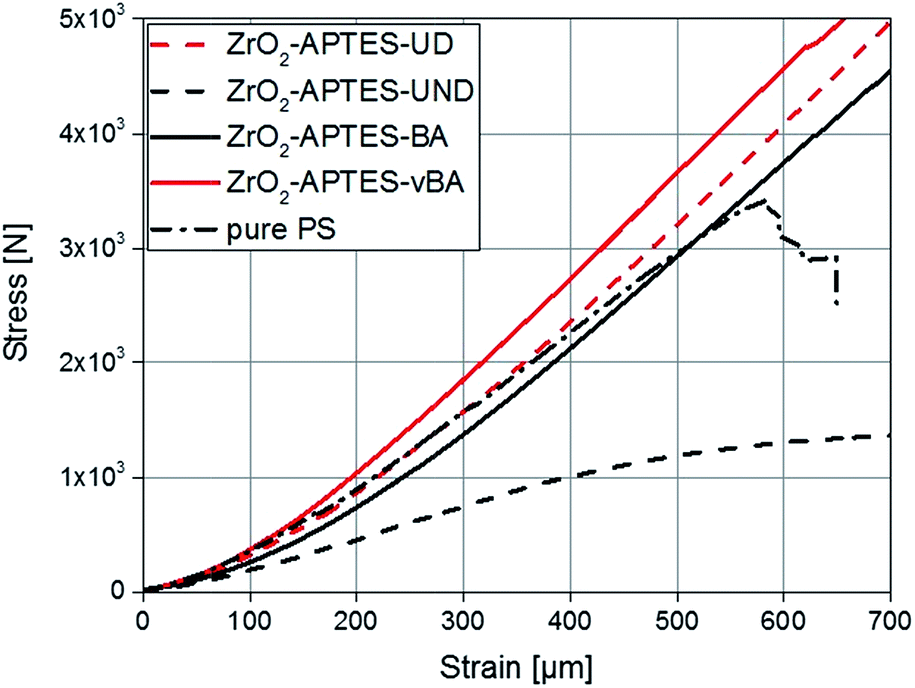 | ||
| Fig. 13 Stress–strain curves for composites with a filler content of 3 wt% in comparison with pure PS. | ||
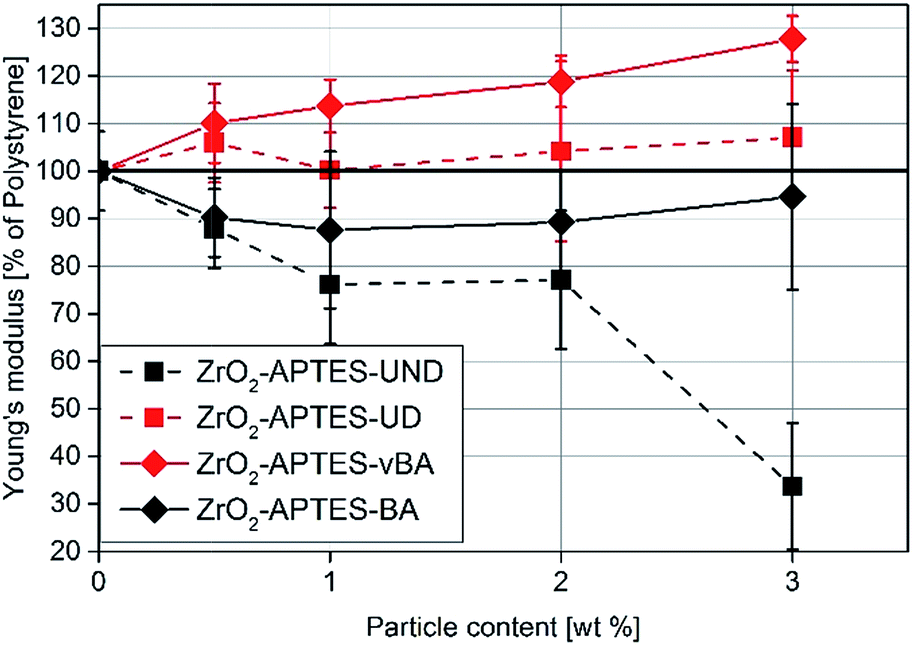 | ||
| Fig. 14 Young's modulus of NCs featuring the differently modified particles related to the detected modulus of pure PS. | ||
First, the Young's moduli of the NCs containing ZrO2-APTES-UND and ZrO2-APTES-UD are compared. Whilst the latter increases by about 8% for a filler content of 3 wt%, which lies within the standard deviation, for the ZrO2-APTES-UND composite, a steep decrease down to 35% of the modulus of pure polystyrene is observed. This is also directly visible after the curing step as these nanocomposites appeared more plastic and soft, in contrast to the solid composites obtained for all other modifications. The sudden change at 550–650 μm indicates the point of breakage of the pure polystyrene specimen. The results of the ZrO2-APTES-BA nanocomposite compared with the ZrO2-APTES-vBA system also show that the modification potentially resulting in particle–matrix coupling leads to an increase, the particles with inert modifications on the other hand rather to a decrease of the Young's modulus. The embedding of the ZrO2-APTES-vBA modified particles enabled an increase of 28% for a nanofiller content of only 3 wt%. For the ZrO2-APTES-BA system, no significant influence of the particle content on the calculated Young's moduli was observed. These trends reflect the results of the scratch test. The higher scratch resistance can be attributed to the higher Young's modulus, because the tip will penetrate the material more easily for composites with a small modulus.
Rheology
Rheological measurements can help to shed further light on the microstructure of nanocomposites, for the ratio of the elastic to the viscous modulus of the complex viscosity (tanδ) can be taken as a reference to describe the degree of crosslinking throughout the polymer.50–52 In Fig. 15, the determined values for tanδ are shown for composites featuring the differently modified particles as a function of the particle content. With a particle content above 2 wt%, for the non-reactive particles (–BA, –UND) a highly similar value is observed as for the pure polymer.
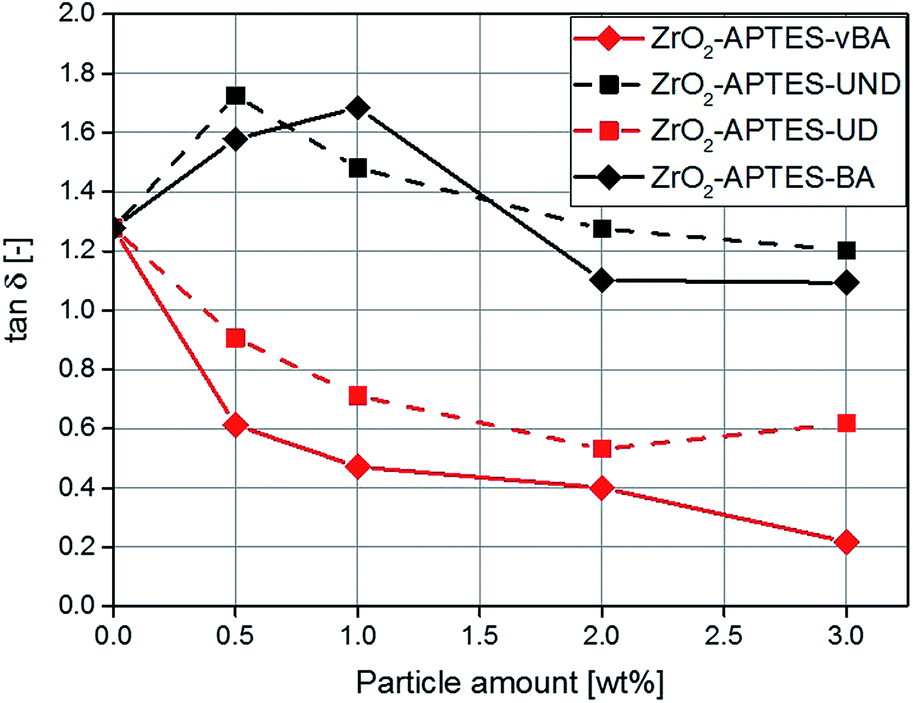 | ||
| Fig. 15 tanδ for NCs with different contents of the modified particles. | ||
For the potentially reactive particle modifications, an increase of the filler content leads to a decrease of tanδ. This effect is very significant for the NCs with –UD terminated particles (decrease by 54% for 3 wt%), and even greater for the -vBA terminated particles (decrease by 83% for 3 wt%). Since this ratio describes the interaction between the particles and the matrix, a lower value of tanδ is attributed to stronger interactions, higher cross-linking density or entanglement between the particles and the matrix.53 The –UD terminated particles therefore clearly show an increase in the interaction compared to the –UND terminated particles, which can be assigned to a covalent linkage between the particles and the matrix. A similar behavior can be observed for the particles terminated with –vBA which is in agreement with the results for the Young's modulus. Because of a strong binding between the particles and the matrix, the polymer chains are immobilized and cannot move as freely as in systems with non-reactive terminal groups. This probably induces a linkage between different polymer chains with the particles as cross-linkers, as the particles carry a large number of reactive groups (Fig. 5 and 6), explaining the strong effects observed in the mechanical properties. Polymer chains that are immobilized in this way would enhance effects like crack pinning or blunting, since cracks need more energy for progression or formation than in a system of freely moving polymer chains.
Although a direct influence of terminal double bonds on the mechanical properties can be seen and is directly correlated with the interaction between the particle and the matrix, secondary effects on the polymer matrix like a shift in the molecular weight or different tacticity might also contribute to the resulting mechanic properties, although the molar ratio of functional terminal groups to styrene is relatively low and no substantial differences are expected. Since the aim of this study is to comparably monitor the influence of the nanoparticle modification on the mechanical properties, the elucidation of the specific mechanism of reinforcement will be objective of our future work.
Conclusions
In this contribution, the effect of the particle–matrix interaction of nanoparticle fillers on the properties of polystyrene-based nanocomposites was studied systematically. In a first step, zirconia nanoparticles were successfully modified via a two-step approach to covalently bind two possibly reactive and two inert ligands to the particle surface. The successful surface modification was shown by FT-IR and TGA, whereby also the presence of the reactive groups was proven. Afterwards, the modified particles were processed to nanocomposites using styrene as a monomer and a commercially available curing agent. The distribution of the particles in the polystyrene matrix was characterized via TEM and SAXS. Here, it became clear that the distribution of the differently modified particles was very similar in all samples, with 6 nm-sized primary particles forming agglomerates of 27–34 nm. Furthermore, the successful linkage of the particles featuring reactive terminal groups with the matrix could be monitored by rheology, whereas no such linkage was found for particles featuring inert groups. The influence of the chemical affinity as well as the influence of the particle–matrix linkage was examined by comparison of the aliphatic with the aromatic and of the reactive with the inert modifications, respectively. The chemical affinity of the nanoparticle modification showed a stronger influence on the bulk properties (the Young's modulus) than on the surface properties like the scratch resistance. Hereby it can be seen that a high affinity and chemical resemblance of the particle modification with the polystyrene matrix leads to greater mechanical stability. A linkage between the particles and the matrix also leads to a larger increase in the Young's modulus, but also has a significant impact on the scratch resistance. Thereby, with an amount of 3 wt% of incorporated ZrO2-APTES-vBA particles that feature both high chemical affinity and a reactive group, a reduction of the scratch depth by 50% and an increase of the Young's modulus by 27% were achieved.Overall, it became obvious that aside from the distribution of the particles in the matrix, the particle surface and therefore the particle–matrix interface is of greatest importance for the mechanical performance of nanocomposites. For highly disperse nanoparticles, the covalent grafting as well as the chemical affinity of the particles to the matrix can lead to a great rise in favourable mechanical properties, even for small amounts of particles. For a practical use of optimized nanocomposite systems, it is of high importance to consider and optimize the surface chemistry of the nanofillers, ideally using rational chemical approaches to achieve a specially designed, strong interaction with the matrix and enable a highly homogeneous distribution of the particles, to ensure large enhancement in composite properties even for small contents of the inorganic filler.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was partially financed by the Deutsche Forschungsgemeinschaft within the Research Unit FOR 2021 “Acting Principles of Nano-Scaled Matrix Additives for Composite Structures” (grant GA 1492/10-2). The authors would like to thank Mr Bilal Temel for TEM measurements, and the Laboratory for Nano and Quantum Engineering (LNQE) at Leibniz University Hannover for the TEM instrument.References
- B. J. Ash, R. W. Siegel and L. S. Schadler, Macromolecules, 2004, 37(4), 1358–1369 CrossRef CAS.
- T. A. Cheema, A. Lichtner, C. Weichert, M. Böl and G. Garnweitner, J. Mater. Sci., 2012, 47(6), 2665–2674 CrossRef CAS.
- B. Schönstedt, G. Garnweitner, N. Barth, A. Mühlmeister and A. Kwade, Langmuir, 2011, 27, 8396–8403 CrossRef PubMed.
- Z. Guo, T. Pereira, O. Choi, Y. Wang and H. T. Hahn, J. Mater. Chem., 2006, 16(27), 2800–2808 RSC.
- M. Z. Rong, M. Q. Zhang and W. H. Ruan, Mater. Sci. Technol., 2006, 22(7), 787–796 CrossRef CAS.
- G. Armstrong, Eur. J. Phys., 2015, 36(6), 063001 CrossRef.
- S. Kango, S. Kalia, A. Celli, J. Njuguna, Y. Habibi and R. Kumar, Prog. Polym. Sci., 2013, 38(8), 1232–1261 CrossRef CAS.
- T.-C. Mo, H.-W. Wang, S.-Y. Chen and Y.-C. Yeh, Ceram. Int., 2008, 34(7), 1767–1771 CrossRef CAS.
- D. Schadler, D. Ma, T. A. Hugener, R. W. Siegel, A. Christerson, E. Mårtensson, C. Önneby and S. Linda, Nanotechnology, 2005, 16(6), 724 CrossRef.
- X. Huang, Y. Zheng, P. Jiang and Y. Yin, IEEE Trans. Dielectr. Electr. Insul., 2010, 17(2), 635–643 CrossRef CAS.
- S. Singha and M. Thomas, IEEE Trans. Dielectr. Electr. Insul., 2008, 15(1), 12–23 CrossRef CAS.
- A. Laachachi, E. Leroy, M. Cochez, M. Ferriol and J. M. L. Cuesta, Polym. Degrad. Stab., 2005, 89(2), 344–352 CrossRef CAS.
- C.-H. Chen, J.-Y. Jian and F.-S. Yen, Composites, Part A, 2009, 40(4), 463–468 CrossRef.
- H. Wang, P. Xu, W. Zhong, L. Shen and Q. Du, Polym. Degrad. Stab., 2005, 87(2), 319–327 CrossRef CAS.
- P. N. Manoudis, I. Karapanagiotis, A. Tsakalof, I. Zuburtikudis and C. Panayiotou, Langmuir, 2008, 24(19), 11225–11232 CrossRef CAS PubMed.
- M. Sangermano, A. Perrot, A. Gigot, P. Rivolo, F. Pirri and M. Messori, Macromol. Mater. Eng., 2016, 301(1), 93–98 CrossRef CAS.
- A. M. Rabea, M. Mohseni, S. M. Mirabedini and M. H. Tabatabaei, Appl. Surf. Sci., 2012, 258(10), 4391–4396 CrossRef CAS.
- Q. F. Xu, J. N. Wang and K. D. Sanderson, ACS Nano, 2010, 4(4), 2201–2209 CrossRef CAS PubMed.
- H. Kakehi, M. Muira, N. Isu and H. Sawada, Polym. J., 2008, 40(11), 1081–1086 CrossRef CAS.
- E. Ritzhaupt-Kleissl, J. Boehm, J. Hausselt and T. Hanemann, Mater. Sci. Eng., C, 2006, 26(5–7), 1067–1071 CrossRef CAS.
- A. H. Yuwono, B. Liu, J. Xue, J. Wang, H. I. Elim and W. Ji, J. Mater. Chem., 2004, 14(20), 2978–2987 RSC.
- X.-W. Du, Y.-S. Fu, J. Sun, X. Han and J. Liu, Semicond. Sci. Technol., 2006, 21(8), 1202 CrossRef CAS.
- O. Asi, Compos. Struct., 2010, 92(2), 354–363 CrossRef.
- D. Ciprari, K. Jacob and R. Tannenbaum, Macromolecules, 2006, 39(19), 6565–6573 CrossRef CAS.
- S. Zhao, J. Zhang, S. Zhao, W. Li and H. Li, Compos. Sci. Technol., 2003, 63(7), 1009–1014 CrossRef CAS.
- R. Siegel, S. Chang, B. Ash, J. Stone, P. Ajayan and R. Doremus, Scr. Mater., 2001, 44(8–9), 2061–2064 CrossRef CAS.
- Y. Wang, S. Lim, J. L. Luo and Z. H. Xu, Wear, 2006, 260(9–10), 976–983 CrossRef CAS.
- C. B. Ng, L. S. Schadler and R. W. Siegel, Nanostruct. Mater., 1999, 12(1–4), 507–510 CrossRef.
- S. Mallakpour and E. Khadem, Prog. Polym. Sci., 2015, 51, 74–93 CrossRef CAS.
- G. V. Kozlov, Phys.-Usp., 2015, 58(1), 33 CrossRef.
- M. Nadler, J. Werner, T. Mahrholz, U. Riedel and W. Hufenbach, Composites, Part A, 2009, 40(6), 932–937 CrossRef.
- M. Zhang and R. P. Singh, Mater. Lett., 2004, 58(3–4), 408–412 CrossRef CAS.
- D. Balaraman, P. M. Raj, L. Wan, I. R. Abothu, S. Bhattacharya and S. Dalmia, J. Electroceram., 2004, 13(1–3), 95–100 CrossRef CAS.
- M. J. Owen, Coupling agents: Chemical bonding at interfaces A2, Elsevier Science, Amsterdam, 2002 Search PubMed.
- B. Wetzel, P. Rosso, F. Haupert and K. Friedrich, Eng. Fract. Mech., 2006, 73(16), 2375–2398 CrossRef.
- S. Zokaei, M. R. Lesan Khosh and R. Bagheri, Mater. Sci. Eng., A, 2007, 445–446, 526–536 CrossRef.
- B. B. Johnsen, A. J. Kinloch, R. D. Mohammed, A. C. Taylor and S. Sprenger, Polymer, 2007, 48(2), 530–541 CrossRef CAS.
- H. Jiang, R. Browning and H.-J. Sue, Polymer, 2009, 50(16), 4056–4065 CrossRef CAS.
- J. W. Krumpfer, T. Schuster, M. Klapper and K. Müllen, Nano Today, 2013, 8(4), 417–438 CrossRef CAS.
- G. Kickelbick, Prog. Polym. Sci., 2003, 28(1), 83–114 CrossRef CAS.
- A. Kockmann, J. Hesselbach, S. Zellmer, A. Kwade and G. Garnweitner, RSC Adv., 2015, 5(75), 60993–60999 RSC.
- T. A. Cheema and G. Garnweitner, CrystEngComm, 2014, 16, 3366–3375 RSC.
- P. Stolzenburg and G. Garnweitner, React. Chem. Eng., 2017, 2, 337–348 CAS.
- A. Bergmann, G. Fritz and O. Glatter, J. Appl. Crystallogr., 2000, 33(5), 1212–1216 CrossRef CAS.
- Z. Wu, H. Xiang, T. Kim, M.-S. Chun and K. Lee, J. Colloid Interface Sci., 2006, 304(1), 119–124 CrossRef CAS PubMed.
- M. A. Schiavon, S. Redondo, S. Pina and I. Yoshida, J. Non-Cryst. Solids, 2002, 304(1–3), 92–100 CrossRef CAS.
- C.-D. Varganici, A. Durdureanu-Angheluta, D. Rosu, M. Pinteala and B. C. Simionescu, J. Anal. Appl. Pyrolysis, 2012, 96, 63–68 CrossRef CAS.
- I. K. Tonle, E. Ngameni, D. Njopwouo, C. Carteret and A. Walcarius, Phys. Chem. Chem. Phys., 2003, 5(21), 4951–4961 RSC.
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34(2), 595–598 CrossRef CAS PubMed.
- M. A. Meyers and K. K. Chawla, Mechanical behavior of materials, Cambridge University Press, New York, Cambridge, 2nd edn, 2009 Search PubMed.
- J. Kodavaty and A. Deshpande, Def. Sci. J., 2014, 64(3), 222–229 CrossRef CAS.
- C. Zhou, Q. Wu, Y. Yue and Q. Zhang, J. Colloid Interface Sci., 2011, 353(1), 116–123 CrossRef CAS PubMed.
- D. Calvet, J. Y. Wong and S. Giasson, Macromolecules, 2004, 37(20), 7762–7771 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00052b |
| This journal is © The Royal Society of Chemistry 2018 |