
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRGD-modified dihydrolipoamide dehydrogenase conjugated to titanium dioxide nanoparticles – switchable integrin-targeted photodynamic treatment of melanoma cells†
Avraham Dayana,
Gideon Fleminger *a and
Osnat Ashur-Fabian*b
*a and
Osnat Ashur-Fabian*b
aThe School of Molecular Cell Biology and Biotechnology, George S. Wise Faculty of Life Sciences, Israel
bThe Department of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University, Ramat Aviv 69978, Israel. E-mail: gidifl@post.tau.ac.il; osnataf@gmail.com
First published on 1st March 2018
Abstract
The photocytotoxic effect of UVA-excited titanium dioxide (TiO2), which is caused by the generation of reactive oxygen species (ROS), is often used in medical applications, such as cancer treatment. Photodynamic-therapy (PDT) is applied in several cancer models including cutaneous melanoma (CM), however the lack of selectivity causing damage to surrounding healthy tissues limits its applicability and novel targeted-delivery approaches are required. As cancer cells often overexpress integrin receptors (e.g. αvβ3) on their cell surface, targeted delivery of TiO2 nanoparticles (NPs) via an Arg-Gly-Asp (RGD) motif would make PDT more selective. We have recently reported that the mitochondrial enzyme dihydrolipoamide dehydrogenase (DLDH) strongly and specifically conjugates TiO2 via coordinative bonds. In this work we have modified DLDH with RGD moieties (DLDHRGD), creating a molecular bridge between the integrin-expressing cancer cells and the photo-excitable TiO2 nanoparticles. Physicochemical assays have indicated that the hybrid-conjugated nanobiocomplex, TiO2–DLDHRGD, is producing controlled-release ROS under UVA illumination, with anatase NPs being the most photoreactive TiO2 form. This drug delivery system exhibited a cytotoxic effect in αvβ3 integrin-expressing mice melanoma cells (B16F10), but not in normal cells lacking this integrin (HEK293). No cytotoxic effect was observed in the absence of UV illumination. Our results demonstrate the feasibility of combining the high efficiency of TiO2-based PDT, with an integrin-mediated tumor-targeted drug delivery for nanomedicine.
1 Introduction
Titanium dioxide (TiO2) is frequently used in biomedical applications and food technologies due to its high chemical inertness, low toxicity, physical stability and commercial availability.1 Upon UV illumination, TiO2 is excited, leading to the production of toxic Reactive Oxygen Species (ROS)2,3 which play important roles in the photocatalytic reaction mechanism.4 Photodynamic-therapy (PDT) utilizes ROS production by TiO2 for the treatment of several proliferating skin diseases,5 such as vitiligo6 and psoriasis, using UVB and UVA radiation.7,8 UV illumination of TiO2 nanoparticles (NPs), has been also used for PDT in several types of cancer,9–14 including melanoma.15 Cutaneous melanoma (CM), which is one of the most rapidly growing cancers worldwide,16 is of a serious public health concern, given the substantial burden associated with the disease.17 Currently CM is treated by surgery, chemotherapy and radiation therapy as well as targeted- and immuno-therapies.18 In cases of advanced melanoma (stages III and IV) PDT may be used, often in combination with surgery19 and was shown to regress melanoma metastasis.20 However, the lack of specificity towards cancer cells limits the applicability of this approach due to potential damage to the surrounding healthy cells.21 This limitation may be overcome by preferentially directing the TiO2 NPs towards cancer cells.Integrins are a family of cell surface receptors which are over-expressed on tumor cells. Twenty-four integrin heterodimers were identified and consist of a combination between at least 18 α-subunits and 8 β-subunits.22 These receptors play a key role in the cross-talk between the cell and its surrounding stroma via an Arg-Gly-Asp (RGD) recognition motif and offer a docking site for endothelial, inflammatory and cancer cells.23 Integrin αvβ3 serves as an attractive and a rational target for anticancer drug delivery,24,25 including in melanoma.26,27 In order to increase the selectivity of PDT towards cancer cells, efforts on developing integrin-targeted drug-delivery tools using TiO2–RGD nanocomplexes were recently reported.28 We propose an alternative delivery approach, which is based on the tendency of TiO2 to adsorb to proteins.29 Our laboratory has identified the human protein dihydrolipoamide dehydrogenase (DLDH), which strongly and specifically binds TiO2 via coordinative bonds.30–32 Bioengineering this protein with RGD moieties and attaching it to TiO2 NPs led to the formation of a hybrid-conjugate nanobiocomplex (TiO2–DLDHRGD), offering integrin-targeted drug delivery. In this work we present a “switchable” nanomedicine which is activated by UVA illumination to produce ROS-depended cytotoxicity in integrin-expressing melanoma cancer cells, while normal cells with low integrin expression remain less affected (Scheme 1).
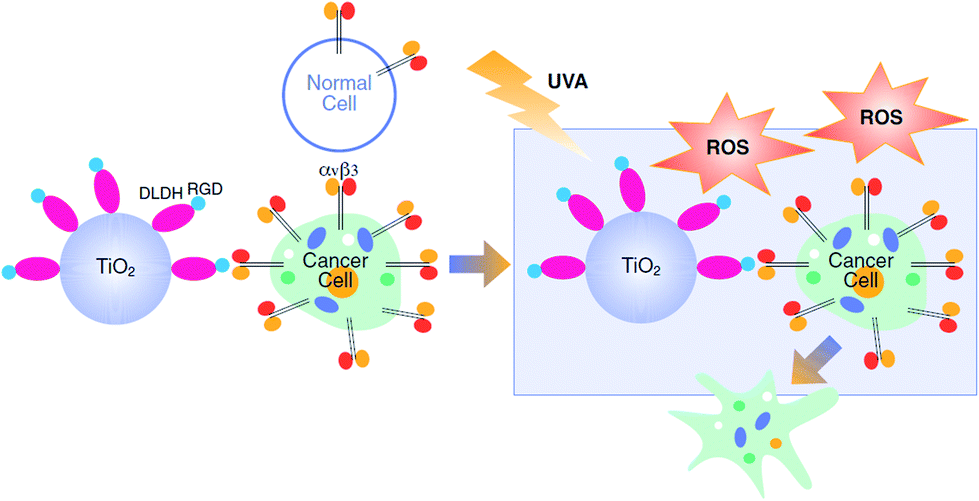 | ||
| Scheme 1 Integrin-targeted PDT by the nanobiocomplex is switched-on upon UVA illumination. | ||
2 Experimental section
2.1 Materials
All reagents and carriers were of analytical grade and were purchased from Sigma-Aldrich (St. Louise, MO), unless otherwise specified. P25 NPs were purchased from Degussa Chemicals (Hanau, Germany).Tissue culture media and cell lines were purchased from Biological Industries (Bet-Haemek, Israel), unless otherwise specified. B16F10 mouse cutaneous melanoma cells (ATCC, CRL-6475) and human normal embryonic kidney cells, HEK293 (ATCC CRL1573) were cultured in RPMI 1640 medium, supplemented with 10% (v/v) heat inactivated fetal calf serum, 2 mM L-glutamine and antibiotics (penicillin/streptomycin), in a humidified atmosphere of 5% CO2 at 37 °C. The cells were shown to be mycoplasma negative.
2.2 Cloning
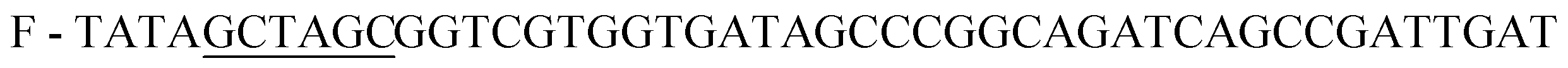 ;
; 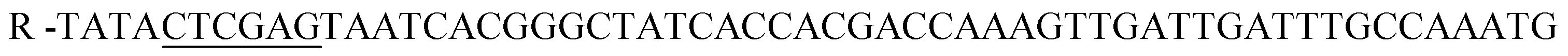 , restriction sites are underlined). The PCR products were purified, restricted with NheI and XhoI and ligated into a pET28a(+) expression vector (Novagen, Madison, Wi) using T4 DNA Ligase (New England BioLabs, MA). The plasmid contains an N-terminal hexahistidine (His6) tag, which serves as a purification tool. Next, the plasmids were transformed into CaCl2 E. coli competent cells. The bacterial strain, E. coli XL1-Blue, was used for all cloning work while E. coli BL21(DE3)pLysS strain was used for subsequent expression of the constructed plasmids. Plates of Lysogeny Broth medium and agar with kanamycin (30 μg ml−1) were used to grow single colonies of transformed E. coli at 37 °C for 16 h. DLDHRGD sequence was confirmed by DNA-sequencing as
, restriction sites are underlined). The PCR products were purified, restricted with NheI and XhoI and ligated into a pET28a(+) expression vector (Novagen, Madison, Wi) using T4 DNA Ligase (New England BioLabs, MA). The plasmid contains an N-terminal hexahistidine (His6) tag, which serves as a purification tool. Next, the plasmids were transformed into CaCl2 E. coli competent cells. The bacterial strain, E. coli XL1-Blue, was used for all cloning work while E. coli BL21(DE3)pLysS strain was used for subsequent expression of the constructed plasmids. Plates of Lysogeny Broth medium and agar with kanamycin (30 μg ml−1) were used to grow single colonies of transformed E. coli at 37 °C for 16 h. DLDHRGD sequence was confirmed by DNA-sequencing as  .
.2.3 Protein purification
The expressed His-tagged proteins were isolated by Immobilized Metal Affinity Chromatography (IMAC), on an Ni-NTA column.30 DLDHRGD concentrations were determined by the absorbance at 280 nm using extinction coefficient of 0.463 calculated from its amino acid compositions using the Expasy ProtParam Program (http://web.expasy.org/protparam).2.4 TiO2 binding
Preparation of TiO2 NPs-conjugated DLDHRGD was as described before for preparation of the TiO2–DLDH complex.302.5 Environmental scanning electron microscopy (ESEM)
ESEM was performed using a Quanta 20 High Resolution (HR) scanning electron microscope, equipped with a Field Emission Gun (FEG) and Energy Dispersive X-ray spectroscopy (EDS), Secondary Electron (SE) – Everhart-thornley and Back-Scattered Electron (BSE) detectors. The TiO2-NPs (rutile, P25 and anatase) preparations were dispersed in ethanol in an ultrasonic bath for 3 minutes and then applied as droplets to an adhesive C slices.2.6 Illumination
UVA exposures at 365 nm (Philips F15T8/BL 15 W lamp, Amsterdam, Netherland), were for 0–150 min equivalent to 2.7 J cm−2. Radiation flow was 1.5 mW cm−2 as determined by UVA light meter type YK-37UVSD, (Radiometer, Copenhagen, Denmark).2.7 Photodegradation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800g for 15 minutes. Optical densities of the supernatants (0.5 ml) were determined at 663 nm by a Genesys 10S UV-VIS spectrometer (Thermo Fisher Scientific, Waltham, MA).800g for 15 minutes. Optical densities of the supernatants (0.5 ml) were examined at 450 and 550 nm by a Genesys 10S UV-VIS spectrometer (Thermo Fisher, Waltham, MA, USA) to follow Fe3+Cyt-C reduction to Fe2+Cyt C, 1 mM reduced Cyt C = 23.65 OD 550 nm.33
800g for 15 minutes. Optical densities of the supernatants (0.5 ml) were determined at 663 nm by a Genesys 10S UV-VIS spectrometer (Thermo Fisher Scientific, Waltham, MA).800g for 15 minutes. Optical densities of the supernatants (0.5 ml) were examined at 450 and 550 nm by a Genesys 10S UV-VIS spectrometer (Thermo Fisher, Waltham, MA, USA) to follow Fe3+Cyt-C reduction to Fe2+Cyt C, 1 mM reduced Cyt C = 23.65 OD 550 nm.332.8 Cellular assays
000 cells per well), in presence of Draq5 (Biostatus, Loughborough, UK), 1μl per well. Normal HEK293 cells served as negative controls (50000 cells per well). Cells were exposed to UVA light (365 nm) for 1 h in the presence of TiO2 alone (5 mg ml−1), TiO2–DLDH (50 mg protein bound to 5 mg A-NPs) or TiO2–DLDHRGD (50 μg bound protein per 0–5 mg A-NPs). Control wells were covered with aluminum foil to create dark conditions. After 24 h of incubation, cells numbers were determined by automated cell counter and visualized by confocal microscopy.2.9 Statistical analysis
All data are expressed as means ± standard error of the mean. Significant differences were assessed by P value.3 Results and discussion
3.1 Anatase TiO2 NPs is the most photoreactive form
In this section we aimed to study the TiO2 physicochemical photocatalytic activity in order to define the optimal TiO2 forms which possess the highest surface area and ROS activity of the titanium dioxide molecules. For that, several types of TiO2 (anatase, rutile, amorphous) at different forms (disks, nanotubes, nano-particles) were examined. The different TiO2 preparations were assessed for ROS production using Methylene Blue (MB) photodegradation. Results indicate that the most active TiO2 form is the anatase nanoparticles (A-NPs) (Fig. S-1†).As nanoparticles possess the highest surface areas, and provide advantages due to their small size, permeability, high surface ratio and retention,34 we have focused on several TiO2 NPs and compared the photoreactivity of the A-NPs with two additional TiO2 forms, P-25 and rutile nanoparticles upon UVA illumination. For that, two methods were used to measure ROS production, MB photo degradation and cytochrome C (Cyt C) reduction. The highest rate of MB degradation (Fig. 1A) was observed with the A-NPs. Photoexcitability of rutile NPs was dramatically lower, while P25 showed an effect that was slightly lower compared to the A-NPs form. Illumination under UVA without Ti-NPs showed very low degree of photolysis. These results are in accord with reports of a higher surface activity of anatase, compared to rutile.35,36 To note, light at 365 nm wavelength was shown to penetrate dipper to the epidermis and dermis, compared to other UV rays37 and is therefore suitable for PDT treatments in melanoma cells.37–39
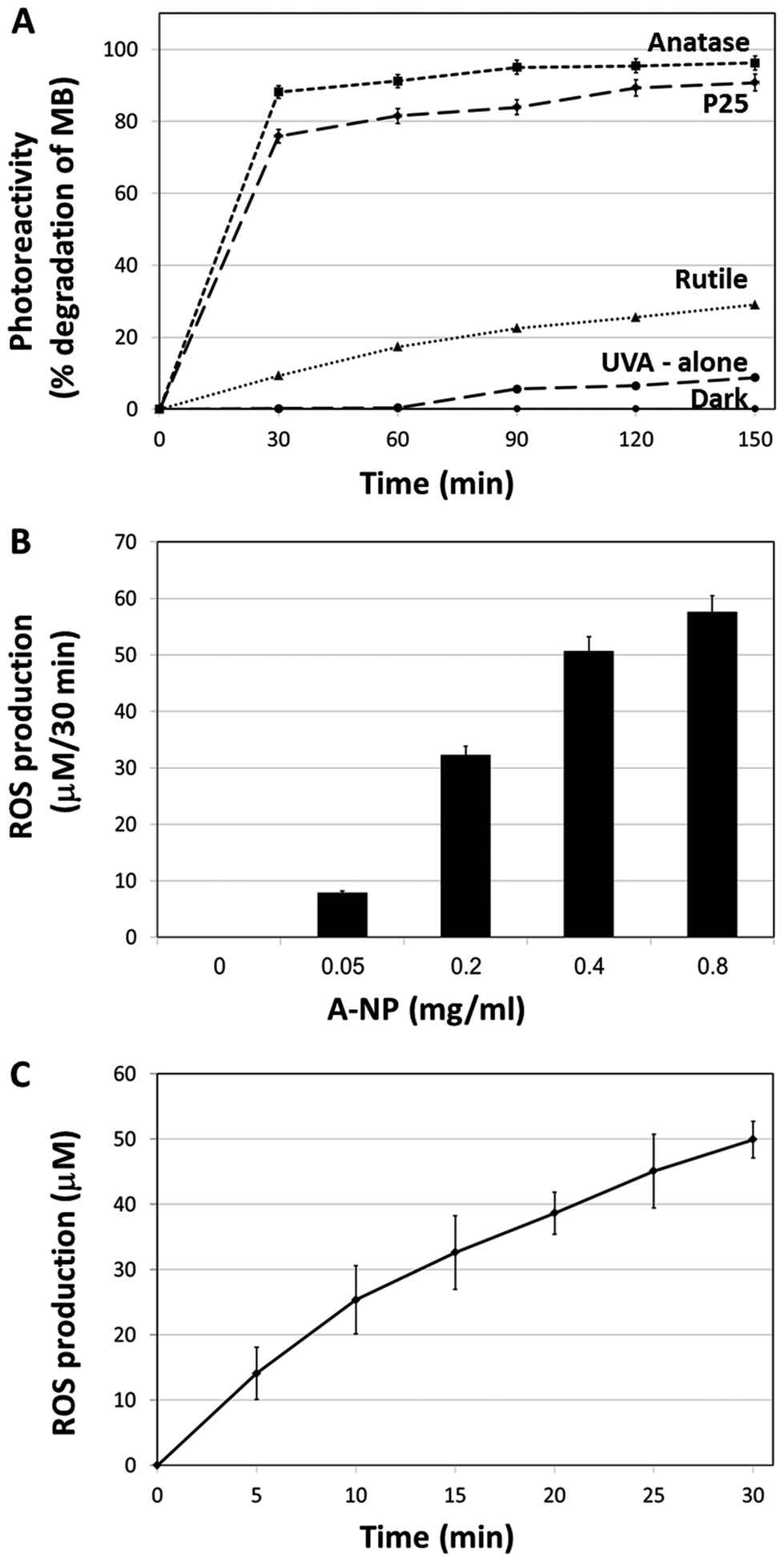 | ||
| Fig. 1 TiO2 nanoparticles photocatalytic activity. (A) Photodegradation of MB upon UVA illumination by the different preparations of TiO2. (B) ROS production by different amounts of A-NPs after 30 min of UVA illumination. (C) ROS production by a constant amount of A-NPs (0.4 mg ml−1) at different time intervals. Experiments were repeated three times in triplicates. | ||
Next, we set to measure the A-NPs ROS generation by Cyt C reduction assay under the same illumination conditions. Optimization of A-NPs concentration in the ROS generation assay for 30 min is shown in Fig. 1B. Based on this experiment 0.4 mg ml−1 of A-NPs was chosen to study the rate of ROS production over time (Fig. 1C). To note, similar ROS production was observed by A-NPs in different buffers, including cell culture media (Fig. S-2†).
3.2 DLDHRGD binding capabilities by different forms of TiO2 NPs
Many proteins have been found to attach to TiO2 by relatively weak reversible electrostatic\hydrophobic or hydrophobic bonds 29,40–42 which often results in failure in achieving strong attachment of cells and tissues.43,44In contrast, we have reported the ability of DLDH to bind TiO231,32 via strong coordinative bonds.30 These strong binding of DLDH to TiO2, has been thoroughly investigated by our group and was shown to be reversed only in the presence of high molar urea concentrations.30–32 We have modified DLDH monomer with GRGDSP moieties at the N- and C-termini, thus creating a dimer of DLDH with four RGD moieties (DLDHRGD). This GRGDSP sequence was shown to specifically interact with αvβ3 integrins.45,46 The RGD-modified DLDH is expected to serve as a molecular bridge and facilitate targeted-delivery of TiO2 to integrin-expressing cancer cells. Examination of the amounts of protein bound to TiO2 NPs revealed that comparable TiO2 binding capabilities were observed for DLDH before and after RGD modification.The RGD-modified DLDH is expected to serve as a molecular bridge and facilitate targeted-delivery of TiO2 to integrin-expressing cancer cells. Examination of the amounts of protein bound to TiO2 NPs revealed that comparable TiO2 binding capabilities were observed for DLDH before and after RGD modification. We intended to study which of the different TiO2 NPs (A-NPs, rutile and P25) bind DLDH most efficiently. In addition, we aimed to study the effect of the RGD-modification on the binding capabilities of DLDH. The NPs structure of anatase (Fig. 2A), rutile (Fig. 2B) and P25 (Fig. 2C) was demonstrated by ESEM, ranging between 20–90 nm. Examination of the amounts of protein (DLDH or DLDHRGD) bound to P25 NPs at different ratios of protein (DLDH, DLDHRGD) to P25 revealed (Fig. 2D) that comparable TiO2 binding capabilities were observed for DLDH before and after RGD modification. At a weight ratio of 10 mg DLDH or DLDHRGD per 1 g of P25 NPs, 90% binding was observed. Next, the binding of DLDH and DLDHRGD to the three TiO2 preparations, A-NP, rutile and P25, were compared at a 1 : 100 ratio of mg protein per mg TiO2 (Fig. 2E) and the amounts of bound non-modified DLDH or RGD-modified proteins were essentially the same. This indicates that DLDH retains its structural conformation and its TiO2 binding site is not hindered by RGD. It is pertinent to note that monomer form of DLDH and DLDHRGD binds TiO2 (data not shown). Previously, it was calculated that P25 binds DLDH with an apparent Kd of 9.43 ± 1.38 mg DLDH per gram TiO2 and a Bmax of 17.15 ± 0.67 mg bound DLDH per gram of TiO2 which represents about 2000 protein molecules (equivalent to 8000 RGD moieties) bound per μm2. Considering the molecular diameter of about 6.7 nm for the hDLDH dimer and the average pore size reported for P25 (8.3 nm), the protein is expected to bind mostly to the outer surface of the TiO2 particles.30Taken together, it was shown that A-NPs posess higher photocatalytic activity upon UVA illumination, while retaining DLDHRGD binding capabilities. As integrins are highly expressed on the surface of tumor cells, the A-NPs–DLDHRGD nanobiocomplex may serve as a novel integrin-drug delivery PDT strategy in this disease.
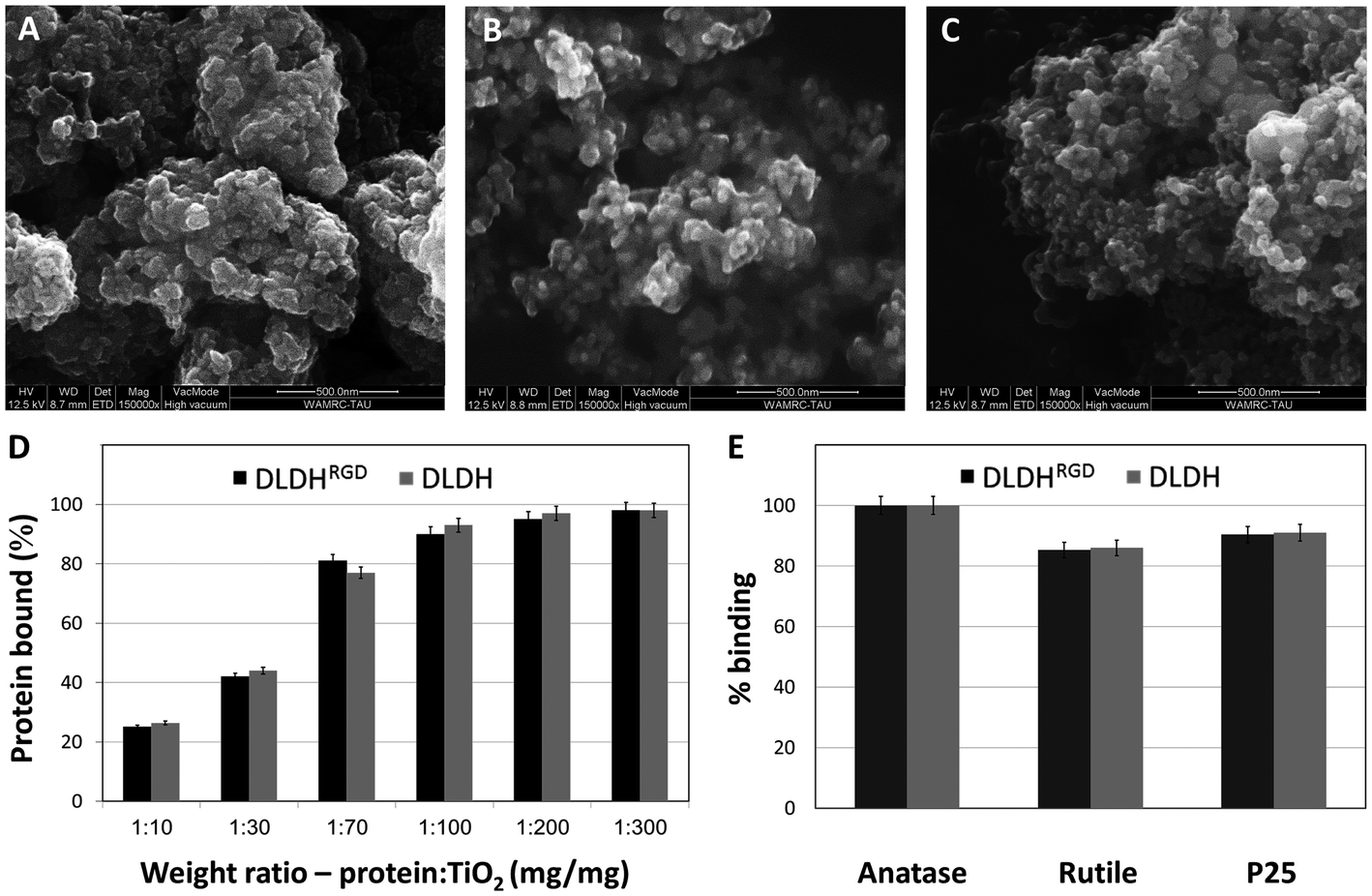 | ||
| Fig. 2 Structure of TiO2 NPs forms and their binding to DLDH and DLDHRGD. ESEM micrographs of (A) A-NPs (B) rutile NPs and (C) P25-NPs. (D) Comparative binding of DLDH (gray) and DLDHRGD (black) to different TiO2 NPs preparations. (E) Binding curve of DLDH (gray) and DLDHRGD (black) at a 1:100 protein to TiO2 weight ratio. Experiments were repeated three times in triplicates. Average ± stdev values are shown. | ||
3.3 Optimization of the A-NPs–DLDHRGD nanobiocomplex components in mice melanoma cells under UVA illumination or in the dark
Following the design and characterization of the TiO2–DLDHRGD nanobiocomplex, we aimed to optimize our formulation in vitro for PDT. We used mice cutaneous melanoma cells (B16F10) which highly express integrin αvβ3 on their surface.47 The cells (50 000 cells/24 well plates) were treated with different amounts of A-NPs (0−5 mg ml−1) conjugated to 50 μg DLDHRGD, with increasing TiO2–protein ratios. The cell nuclei are indicated by a red color (Draq5 stain). The cells were incubated with the sediment of the nanobiocomplex for 1h in the dark or under UVA illumination (365 nm), following an incubation for an overnight. The cells were incubated for an overnight and the next day were visualized using confocal microscopy. Results show that in the dark the cells remained intact (Fig. 3, upper panel). Following UVA illumination, a substantial amount of nuclear shredding, indicative of apoptosis, was shown with increasing nanobiocomplex ratio (Fig. 3, lower panel). This indicates that UVA illumination is essential for the activation of the nanobiocomplex and suggests that the cells were eliminated by the ROS produced by the photoexcited TiO2-NPs. As described before, excessive ROS can induce cell elimination through several death pathways. The highest cytotoxic effect was observed with 50 μg bound DLDHRGD to 5 mg A-NPs (1 : 100 ratio), a combination at which we have documented that 90% of the protein is bound to the TiO2 NPs. These results suggest that at this nanaobiocomplex ratio an optimal integrin-targeted delivery by the DLDHRGD of the TiO2 NPs is documented. Thus, subsequent experiments were conducted with this specific combination.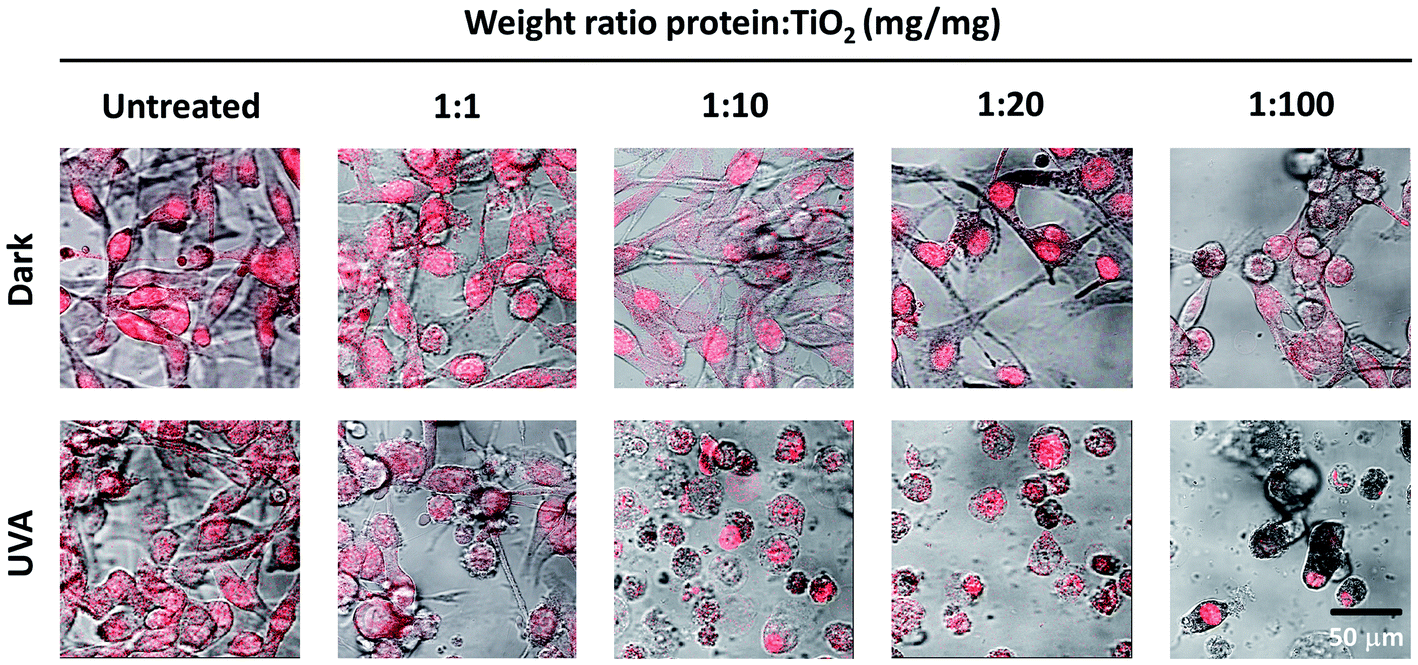 | ||
| Fig. 3 TiO2–DLDHRGD nanobiocomplex induces a cytotoxic effect on B16F10 mice melanoma cancer cells following UVA illumination. The numbers cited in the upper line represent the weight ratio used in the preparation stage. The actual ratio in the nanobiocomplex is presented in Fig. 2D The cell nuclei are stained by Draq5 (red). Merged images of light and red filter are shown using a 63× objective (fluorescence confocal microscope, Leica SP5). Experiments were repeated twice times in triplicates. | ||
3.4 The A-NPs–DLDHRGD nanobiocomplex produces significant cytotoxicity via integrin-targeted PDT
Lastly, we aimed to validate that the cytotoxic effect that was observed in the cells was directly via integrin-targeted delivery of the A-NPs by the DLDHRGD protein. For that we have treated B16F10 cells (50000 cells per 24 well plates) with A-NPs alone (5 mg ml−1) and compared this treatment to cells incubated with the optimized weight ratio (1:100) of A-NPs bound to native DLDH or modified DLDHRGD. The same experimental setting was conducted in normal cells which lack αvβ3 integrin expression (HEK293,48 Fig. S-3†). The treated cells were illuminated by UVA for an hour. As controls, cells treated in the same manner were kept in the dark. Following illumination, the cells were kept overnight in a biological incubator. The next day, the cells were stained with a nuclear dye (Draq5) and visualized using confocal microscopy. Results indicate that in the absence of photoactivation, no effect on cell density or morphology was observed for all detailed treatments in both the melanoma (Fig. 4A, upper panel) and control cells (Fig. 4A, lower panel). These results indicate that in the absence of illumination, conditions in which TiO2 is not activated, no cytotoxic outcome is anticipated.
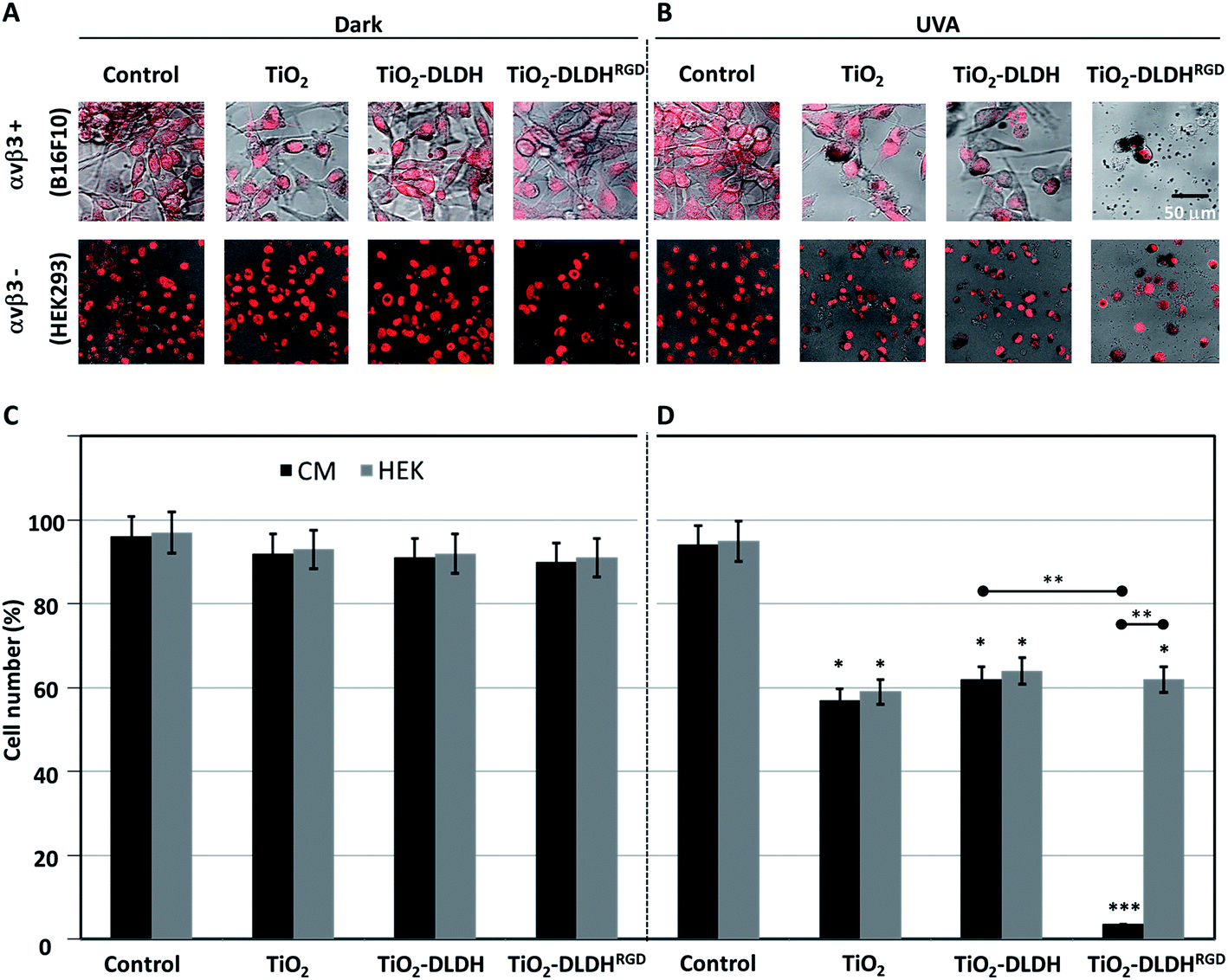 | ||
| Fig. 4 Cytotoxic effect of the nanobiocomplex (A-NPs–DLDHRGD) in the presence or absence of UVA illumination. B16F10 or HEK293 cells were treated with TiO2, TiO2–DLDH or TiO2–DLDHRGD and assessed after an overnight by confocal microscopy (Leica SP5) under (A) dark conditions or (B) UVA illumination (1 h, 365 nm). Cells treated in medium lacking TiO2 or the DLDH forms, served as controls. The cell nuclei were stained red (Draq5). Experiments were repeated twice in triplicates. Next the same experiment was analyzed for cell counts under (C) dark conditions or (D) UVA illumination. Experiments were repeated 3 times in triplicates. Significance (*p < 0.05, **p < 0.005, ***p < 0.001) from control cells after uva illumination. Significance between the experiments groups (±RGD, ±αvβ3) are indicated by an horizontal line. | ||
Following photoexcitation, A-NPs alone or bound to native DLDH produced a comparable cytotoxic effect in melanoma cells (Fig. 4B, upper panel). Notably, this cytotoxic effect by the A-NPs, was potently enhanced in the presence of the modified DLDHRGD. Regarding the effect observed in the normal control cells (Fig. 4B, lower panel), a comparable cytotoxic effect was documented by TiO2 alone or in the presence of native or RGD-modified DLDH. Next, the same experiment underwent analysis for absolute cell counts under dark conditions (Fig. 4C) or following illumination (Fig. 4D), results correlated with the microscopy data. In details, in the dark, the different TiO2 preparations, namely TiO2 alone, TiO2–DLDH or TiO2–DLDHRGD, did not affect the cell number of both B16F10 and HEK293 cells. However, illumination of TiO2 or TiO2–DLDH by UVA resulted in a comparable 50% reduction in cell number in both cell lines. In contrast, in the presence of TiO2–DLDHRGD a reduction of over 95% in the integrin positive cancer cells (B16F10) was shown following illumination. In the normal control cells, which express low integrin levels, 50% reduction in cell number was documented, which was similar to the effect observed by TiO2 or TiO2–DLDH. This suggests that the TiO2–DLDHRGD nanobiocomplex is ineffective in the absence of integrins. Taken together, we propose that the photosensitizing cytotoxic effect of TiO2 is greatly enhanced via integrin-targeted delivery.
Several in vitro and in vivo studies, in which TiO2 nanoparticles or nanotubes were assessed as potential anti-cancer treatments were published, mainly in cervical and skin cancers.10–14 In all of these works, however, cytotoxicity was induced in up to half of the cells, the value that was obtained by us with TiO2 nanoparticles alone. While using our novel TiO2–DLDHRGD nanobiocomplex, greatly enhanced the cytotoxic effect. In addition, the illumination time needed in our experimental settings to produce an effect was usually shorter. Lastly, it is pertinent to note that the approach of using TiO2 as a single agent is limited by the inability of the TiO2-emitted ROS to distinguish between normal and cancer cells.13,14
In recent years, studies on integrins which, in contrast to normal cells, are highly expressed on the surface of cancer cells, have ignited renewed interest in integrin-targeted-delivery, mainly via the specific recognition site for the three-peptide ligand RGD.49,50 By using TiO2 integrin-targeted-delivery, we have produced an effect that was cancer-specific. Similar attempts have been done to selectively deliver the TiO2 particles via an RGD-recognition site.28,51–55 Other approaches to target the TiO2 directly to cancer cells, sparing harmful effects in normal tissues, are under active research. Such targeted-delivery, for example, includes the attachment of TiO2 to an antibody recognizing colon-cancer specific antigen.56 However, most of these TiO2 conjugates require complex synthetic chemical steps.
4 Conclusions
The goal of this research was to develop an integrin-targeted nanomedicine for PDT. By modifying DLDH, a TiO2 binding protein, with RGD moieties, we have generated a nanobiocomplex that is UVA switchable, highly selective towards cancer cells and effective in producing cell death. We propose that this hybrid-conjugate TiO2-DLDHRGD may serve as a novel targeted therapeutic agent with relevance to several integrin-expressing tumor models.Conflicts of interest
The use of DLDH for cancer treatment is protected by a patent in which A. D., G. F. and O. A. F. are the inventors.Acknowledgements
This research was supported by the Israeli Ministry of Economics (Projects 52679 and 55869). The authors wish to express their gratitude to Dr Amir Toren for his valuable advices and assistance in this project and to thank Mrs Ronit Hollander for the graphic assistance and the table of contents illustration.Notes and references
- C. N. Elias, J. H. C. Lima, R. Valiev and M. A. Meyers, JOM, 2008, 60, 46–49 CrossRef CAS.
- I. Mfouo-Tynga and H. Abrahamse, Int. J. Mol. Sci., 2015, 16, 10228–10241 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Zaleska, Recent Pat. Eng., 2008, 2, 157–164 CrossRef CAS.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- B. M. El-Zawahry, D. A. Bassiouny, R. M. Sobhi, E. Abdel-Aziz, N. S. Zaki, D. F. Habib and D. M. Shahin, Photodermatol., Photoimmunol. Photomed., 2012, 28, 84–90 CrossRef CAS PubMed.
- T. Werfel, F. Holiangu, K. H. Niemann, O. Schmerling, F. Lullau, A. Zedler, H. D. Strater and M. Niebuhr, Br. J. Dermatol., 2015, 172, 746–753 CrossRef CAS PubMed.
- W. Lapolla, B. A. Yentzer, J. Bagel, C. R. Halvorson and S. R. Feldman, J. Am. Acad. Dermatol., 2011, 64, 936–949 CrossRef PubMed.
- H. Abrahamse and M. R. Hamblin, Biochem. J., 2016, 473, 347–364 CrossRef CAS PubMed.
- R. Cai, Y. Kubota, T. Shuin, H. Sakai, K. Hashimoto and A. Fujishima, Cancer Res., 1992, 52, 2346–2348 CAS.
- M. Kalbacova, J. M. Macak, F. Schmidt-Stein, C. T. Mierke and P. Schmuki, Phys. Status Solidi, 2008, 2, 194–196 CAS.
- J. W. Seo, H. Chung, M. Y. Kim, J. Lee, I. H. Choi and J. Cheon, Small, 2007, 3, 850–853 CrossRef CAS PubMed.
- K. C. Yoo, C. H. Yoon, D. Kwon, K. H. Hyun, S. J. Woo, R. K. Kim, E. J. Lim, Y. Suh, M. J. Kim, T. H. Yoon and S. J. Lee, Int. J. Nanomed., 2012, 7, 1203–1214 CAS.
- K. M. Ramkumar, C. Manjula, G. Gnanakumar, M. A. Kanjwal, T. V. Sekar, R. Paulmurugan and P. Rajaguru, Eur. J. Pharm. Biopharm., 2012, 81, 324–333 CrossRef CAS PubMed.
- Y. Cheng, Y. Chang, Y. Feng, N. Liu, X. Sun, Y. Feng, X. Li and H. Zhang, Small, 2017, 13 Search PubMed.
- V. Nikolaou and A. J. Stratigos, Br. J. Dermatol., 2014, 170, 11–19 CrossRef CAS PubMed.
- G. P. Guy, Jr., D. U. Ekwueme, F. K. Tangka and L. C. Richardson, Am. J. Prev. Med., 2012, 43, 537–545 CrossRef PubMed.
- H. R. Keller, X. Zhang, L. Li, H. Schaider and J. W. Wells, OncoTargets Ther., 2017 Search PubMed.
- I. Baldea and A. G. Filip, J. Physiol. Pharmacol., 2012, 63, 109–118 CAS.
- T. Tammela, A. Saaristo, T. Holopainen, S. Yla-Herttuala, L. C. Andersson, S. Virolainen, I. Immonen and K. Alitalo, Sci. Transl. Med., 2011, 3, 69ra11 Search PubMed.
- R. Cai, K. Hashimoto, K. Itoh, Y. Kubota and A. Fujishima, Bull. Chem. Soc. Jpn., 1991, 64, 1268–1273 CrossRef CAS.
- I. Dijkgraaf, C. B. Yim, G. M. Franssen, R. C. Schuit, G. Luurtsema, S. Liu, W. J. Oyen and O. C. Boerman, Eur. J. Nucl. Med. Mol. Imaging, 2011, 38, 128–137 CrossRef CAS PubMed.
- J. A. Askari, P. A. Buckley, A. P. Mould and M. J. Humphries, J. Cell Sci., 2009, 122, 165–170 CrossRef CAS PubMed.
- Y. Gilad, E. Noy, H. Senderowitz, A. Albeck, M. A. Firer and G. Gellerman, Biopolymers, 2015 DOI:10.1002/bip.22800.
- Y. Sun, C. Kang, F. Liu, Y. Zhou, L. Luo and H. Qiao, Drug Dev. Res., 2017 DOI:10.1002/ddr.21399.
- E. Petitclerc, S. Strömblad, T. L. von Schalscha, F. Mitjans, J. Piulats, A. M. Montgomery, D. A. Cheresh and P. C. Brooks, Cancer Res., 1999, 59, 2724–2730 CAS.
- I. M. Bachmann, R. G. Ladstein, O. Straume, G. N. Naumov and L. A. Akslen, BMC Cancer, 2008, 8, 362 CrossRef PubMed.
- H. Zhao, M. Wang, P. Zhou, Q. Wang, Z. Zhou, D. Wang, H. Yang and S. Yang, J. Mater. Sci., 2017, 52, 13356–13364 CrossRef CAS.
- Z. J. Deng, G. Mortimer, T. Schiller, A. Musumeci, D. Martin and R. F. Minchin, Nanotechnology, 2009, 20, 455101 CrossRef PubMed.
- A. Dayan, G. Babin, A. Ganoth, N. S. Kayouf, N. Nitoker-Eliaz, S. Mukkala, Y. Tsfadia and G. Fleminger, J. Mol. Recognit., 2017, 30, e2617 CrossRef PubMed.
- A. Siegmann, A. Komarska, Y. Betzalel, I. Brudo, S. Jindou, G. Mor and G. Fleminger, J. Mol. Recognit., 2008, 22, 138–145 CrossRef PubMed.
- G. Gertler, I. Brudo, R. Kenig and G. Fleminger, Materialwiss. Werkstofftech., 2003, 34, 1138–1144 CrossRef CAS.
- M. L. Cunningham, J. S. Johnson, S. M. Giovanazzi and M. J. Peak, Photochem. Photobiol., 1985, 42, 125–128 CrossRef CAS PubMed.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- T. Van der Meulen, A. Mattson and L. Österlund, J. Catal., 2007, 251, 131–144 CrossRef CAS.
- L. Liu, H. Zhao, J. M. Andino and Y. Li, ACS Catal., 2012, 2, 1817–1828 CrossRef CAS.
- A. Amaro-Ortiz, B. Yan and J. A. D'Orazio, Molecules, 2014, 19, 6202–6219 CrossRef PubMed.
- B. Breiner, K. Kaya, S. Roy, W.-Y. Yang and I. V. Alabugin, Org. Biomol. Chem., 2012, 10, 3974–3987 CAS.
- E. Gemenetzidis, O. Shavorskaya, Y.-Z. Xu and G. Trigiante, J. Dermatol. Treat., 2013, 24, 209–214 CrossRef CAS PubMed.
- Z. Xu, X.-W. Liu, Y.-S. Ma and H.-W. Gao, Environ. Sci. Pollut. Res., 2010, 17, 798–806 CrossRef CAS PubMed.
- F. Y. Oliva, L. B. Avalle, O. R. Cámara and C. P. De Pauli, J. Colloid Interface Sci., 2003, 261, 299–311 CrossRef CAS PubMed.
- S. Sousa, P. Moradas-Ferreira, B. Saramago, L. Viseu Melo and M. Barbosa, Langmuir, 2004, 20, 9745–9754 CrossRef CAS PubMed.
- D. Rivera-Chacon, M. Alvarado-Velez, C. Acevedo-Morantes, S. Singh, E. Gultepe, D. Nagesha, S. Sridhar and J. Ramirez-Vick, J. Biomed. Nanotechnol., 2013, 9, 1092–1097 CrossRef CAS PubMed.
- L. Vroman, Ann. N. Y. Acad. Sci., 1987, 516, 300–305 CrossRef CAS PubMed.
- P. Schaffner and M. Dard, Cell. Mol. Life Sci., 2003, 60, 119–132 CrossRef CAS PubMed.
- T. G. Kapp, F. Rechenmacher, S. Neubauer, O. V. Maltsev, E. A. Cavalcanti-Adam, R. Zarka, U. Reuning, J. Notni, H.-J. Wester and C. Mas-Moruno, Sci. Rep., 2017, 7 Search PubMed.
- R. Rathinam and S. K. Alahari, Cancer Metastasis Rev., 2010, 29, 223–237 CrossRef CAS PubMed.
- E. Shinderman-Maman, K. Cohen, C. Weingarten, D. Nabriski, O. Twito, L. Baraf, A. Hercbergs, P. J. Davis, H. Werner, M. Ellis and O. Ashur-Fabian, Oncogene, 2016, 35, 1977–1987 CrossRef CAS PubMed.
- F. Danhier, A. Le Breton and V. Preat, Mol. Pharmaceutics, 2012, 9, 2961–2973 CrossRef CAS PubMed.
- J. Bolley, Y. Lalatonne, O. Haddad, D. Letourneur, M. Soussan, J. Perard-Viret and L. Motte, Nanoscale, 2013, 5, 11478–11489 RSC.
- G. Valente, N. Depalo, I. de Paola, R. M. Iacobazzi, N. Denora, V. Laquintana, R. Comparelli, E. Altamura, T. Latronico, M. Altomare, E. Fanizza, M. Striccoli, A. Agostiano, M. Saviano, A. Del Gatto, L. Zaccaro and M. C. Curri, Nano Res., 2016, 9, 644–662 CrossRef CAS.
- M.-L. Yu, M.-F. Yu, L.-Q. Zhu, T.-T. Wang, Y. Zhou and H.-M. Wang, BioMed Res. Int., 2015, 2015 Search PubMed.
- F. Li, Y. Zhao, C. Mao, Y. Kong and X. Ming, Mol. Pharmaceutics, 2017, 9, 2961–2973 Search PubMed.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. S. Casalongue, D. Vinh and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS PubMed.
- H. Qin, K. Maruyama, T. Amano, T. Murakami and N. Komatsu, Mater. Res. Express, 2016, 3, 105049 CrossRef.
- J. Xu, Y. Sun, J. Huang, C. Chen, G. Liu, Y. Jiang, Y. Zhao and Z. Jiang, Bioelectrochemistry, 2007, 71, 217–222 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13777j |
| This journal is © The Royal Society of Chemistry 2018 |