
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular solar thermal systems – control of light harvesting and energy storage by protonation/deprotonation†
Martin Drøhse Kilde‡
 ,
Paloma Garcia Arroyo‡,
Anders S. Gertsen‡§
,
Kurt V. Mikkelsen and
Mogens Brøndsted Nielsen*
,
Paloma Garcia Arroyo‡,
Anders S. Gertsen‡§
,
Kurt V. Mikkelsen and
Mogens Brøndsted Nielsen*
Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark. E-mail: mbn@chem.ku.dk
First published on 8th February 2018
Abstract
Molecular solar thermal (MOST) systems that undergo photoisomerizations to long-lived, high-energy forms present one approach of addressing the challenge of solar energy storage. For this approach to mature, photochromic molecules which can absorb at the right wavelengths and which can store a sufficient amount of energy in a controlled time period have to be developed. Here we show in a combined experimental and theoretical study that incorporation of a pyridyl substituent onto the dihydroazulene/vinylheptafulvene photo-/thermoswitch results in molecules whose optical properties, energy-releasing back-reactions and energy densities can be controlled by protonation/deprotonation. The work thus presents a proof-of-concept for using acid/base to control the properties of MOST systems.
Introduction
Efficient exploitation of solar energy is a major challenge for meeting the increasing energy demands of the world. Development of organic molecules for photovoltaics has for this reason attracted considerable focus. Light-harvesting, energy storage, and ultimately heat release by photochromic molecules, in brief molecular solar thermal (MOST) systems, has been an alternative niche area, which in recent years, however, has attracted attention.1 MOST systems correspond to closed-energy cycles with no release of CO2 or other oxidation products. Photochromic molecules are interchangeable upon stimuli with light in at least one direction. The practical criteria for MOST systems are in particular sufficiently large energy densities (an upper limit is probably 1 MJ kg−1 corresponding to that of the norbornadiene/quadricyclane couple2) and the ability to release the stored energy in a controlled manner when needed.The yellow dihydroazulene (DHA; 2-phenyl-1,8a-dihydroazulene-1,1-dicarbonitrile)3 is a one-way photoswitch with a high quantum yield4 of photoisomerization. Upon irradiation at its lowest-energy absorption maximum (ca. 350 nm), DHA converts to its higher energy isomer, the red vinylheptafulvene (VHF; Scheme 1).3 Upon ring-opening, the VHF is formed as its s-cis conformer, which rapidly changes to the more stable s-trans conformer.5 As the back-reaction has to proceed through the s-cis VHF, its rate depends on both the position of the s-trans/s-cis pre-equilibrium and the activation energy of ring closure. Fine-tuning of the rate of the thermal back-reaction is possible with changes in solvent polarity6 and electronic character of substituent groups.7 The DHA/VHF system has many different positions to functionalize and with synthetic protocols in hand, the study of structural modifications at various positions has been possible providing both ultrafast ring closures and very stable VHFs.8 For example, bridging the DHA and the phenyl substituent by a –CH2CH2– linker as in the “dihydronaphthalene–DHA” (DHN–DHA) sketched in Scheme 1 results in almost instantaneous ring closure of the resulting VHF as it is locked in its reactive s-cis conformation.8a The parent DHA/VHF system only provides an energy density9 of 0.11 MJ kg−1 and a VHF half-life of 218 min (MeCN, 25 °C).6 Yet, even minor structural changes can greatly enhance the energy storage capacity.8b,10
 | ||
| Scheme 1 Photo/thermal switching of the dihydroazulene (DHA)/vinylheptafulvene (VHF) system. By incorporating a CH2CH2 bridge (indicated in blue color), a locked dihydronaphthalene–DHA (DHN–DHA) is obtained. Inset: pH controllable DHAs: pyridyl-DHA (Py-DHA, 1a) and the locked dihydroisoquinoline-DHA (DHIQ-DHA, 2a). | ||
Hecht and co-workers11 have recently demonstrated how acid can be used to trigger release of stored light energy in the thermal cycloreversion of a diarylethene.12 Previous work on the DHA/VHF system13 has employed protonation/deprotonation of an anilino substituent group to control switching properties, and we became interested to further explore the concept of acid/base control by incorporating a basic pyridine group. Here we demonstrate the synthesis, optical and switching properties and theoretical investigations of proof-of-concept systems, “pyridyl-DHA” 1a (Py-DHA) and “dihydroisoquinoline-DHA” 2a (DHIQ-DHA) (Scheme 1). The pyridine ring provides via the basic nitrogen a handle for controlling the VHF-to-DHA back-reaction by protonation as well as for tuning the absorption maxima and energy storage capacities. Our investigations were driven by an initial computational screening of potential interesting systems and based on the initial computational screening, relevant systems were selected for experimental work and more detailed calculations.
Results and discussion
Synthesis
Synthesis of 1a started with a Knoevenagel condensation between 4-acetylpyridine and malononitrile in HMDS/AcOH which furnished the adduct 4 (Scheme 2). This compound was thermally unstable, and prolonged heating as well as purification by column chromatography resulted in degradation products. Therefore, 4 was just used in the next step after aqueous work-up (with only minor impurities remaining). Addition of tropylium tetrafluoroborate and Et3N in MeCN at room temperature (rt) gave the VHF-precursor 5 in good yield, which easily could be obtained on large scale after dry-column vacuum chromatographic work-up. Oxidation of VHF-precursors has previously shown to be a tedious step,8 which was also the case in the present case. In a first attempt, tritylium tetrafluoroborate was used as a hydride abstractor in refluxing 1,2-dichloroethane. However, this attempt was fruitless, giving no desired product formation even after prolonged refluxing. Instead we turned to nitrosonium tetrafluoroborate (NOBF4) in MeCN at −40 °C followed by the addition of dilute pyridine in CH2Cl2; this in turn yielded the distinct red VHF (as observed on TLC), which after heating to reflux thermally cyclized to Py-DHA 1a. The pyridyl nitrogen serves as a convenient site for further chemistry. Thus, methylation of 1a was achieved by treatment with MeI in MeCN to yield the salt 6a in quantitative yield. | ||
| Scheme 2 Synthesis of Py-DHA 1a and further methylation at the pyridyl nitrogen to yield the salt 6a. HMDS = hexamethyldisilazane. | ||
To obtain the locked dihydroisoquinoline-DHA 2a, the commercially available 5,6,7,8-tetrahydroisoquinoline 7 was used as starting material in initial steps following literature protocols14 (Scheme 3). Oxidation with H2O2 in AcOH gave the N-oxide 8, which underwent Polonovski rearrangement in refluxing acetic anhydride to provide the acetoxy product 9. Hydrolysis of this compound in acidic media (10% aq. HCl) followed by oxidation led finally to the desired ketone 11 in an overall yield of 22% starting from 5.14 Then, Knoevenagel condensation of 11 with malononitrile and NH4OAc/AcOH buffer in toluene gave 12, which was turned into the corresponding VHF-precursor 13 by treatment with tropylium tetrafluoroborate and Et3N in MeCN at rt, with both reactions being high-yielding (87 and 80%, respectively). The obtained VHF-precursor 13 turned out to be even more challenging to oxidize than 4. First, we tried using the same conditions as for 4; 13 was treated with NOBF4 in MeCN at −40 °C followed by the addition of dilute pyridine in CH2Cl2, but no desired product could be detected or isolated. Instead, treating the tropylium cationic intermediate with the stronger base DBU gave DHIQ-DHN 2a in 6% yield, after ring closure of the intermediate VHF isomer 2b. Unfortunately, this procedure was not reproducible, and we could only isolate 2a on one occasion. On the other hand, if the VHF-precursor 13 was first protonated by trifluoroacetic acid (TFA) and then treated with the hydride abstractor (NOBF4) and DBU, we could isolate 2a in an improved yield of 13%. The overall yield of 2a from 7 is 2% while that of 1a is 8% from the starting material 3.
 | ||
| Scheme 3 Synthesis of DHIQ-DHN 2a. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. | ||
Multimode switching studies
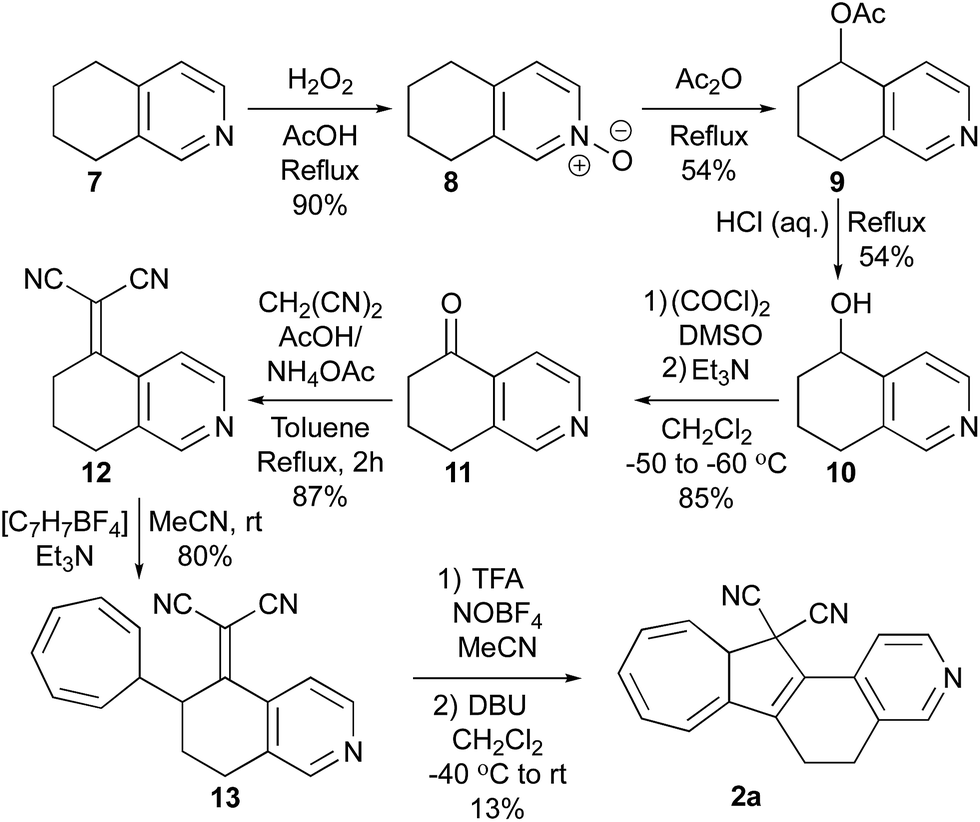
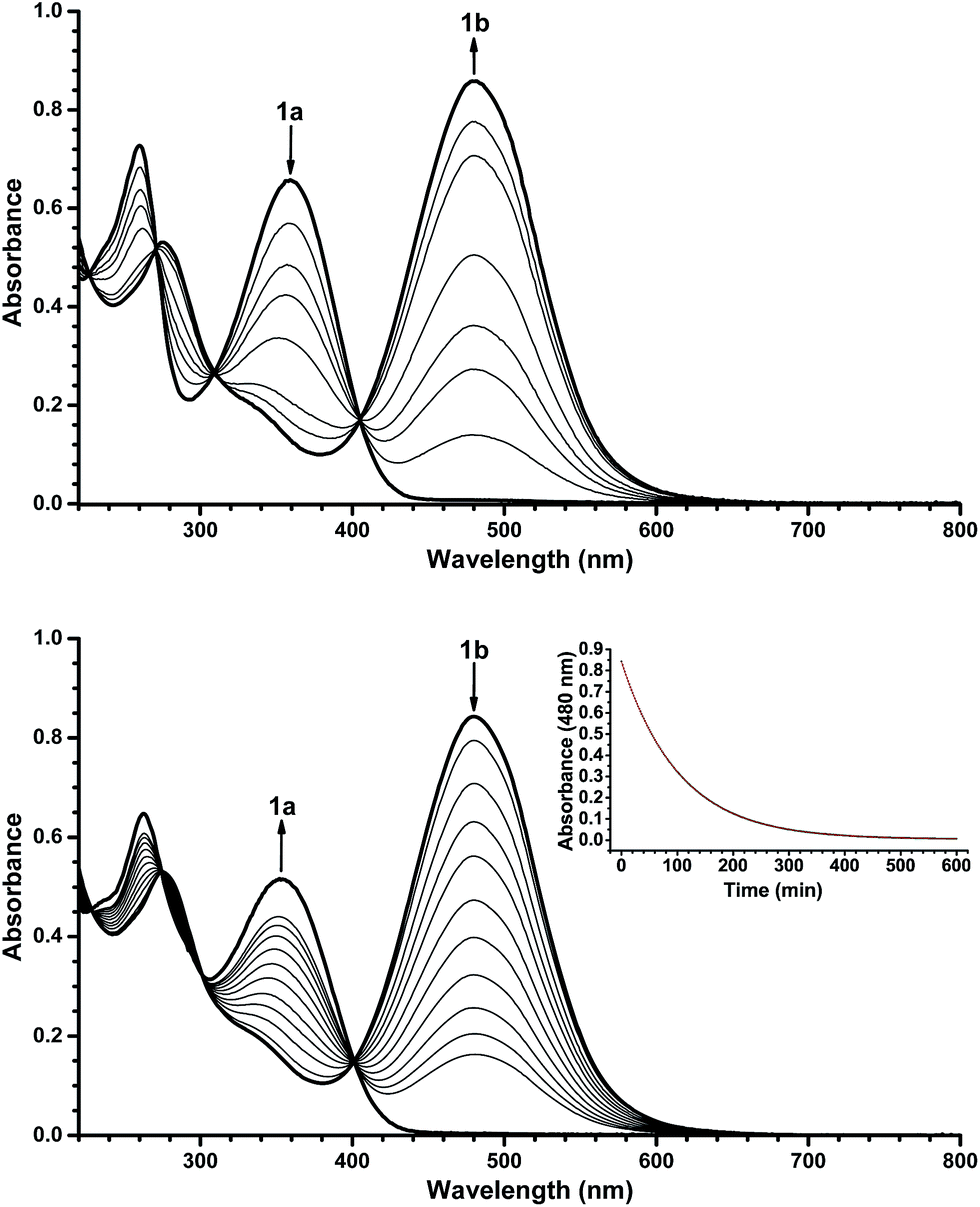
With the novel photoswitches Py-DHA 1a and DHIQ-DHA 2a in hand, their photophysical and photochemical properties were studied, including the influence of protonation of the basic nitrogens (Scheme 4). UV/Vis absorption spectroscopy studies of Py-DHA 1a were conducted in MeCN at 25 °C and the results compared in Table 1 to its respective carbon analogue (DHA,6 shown in Scheme 1). Py-DHA 1a could easily be converted to VHF 1b upon light irradiation (365 nm), seen by a distinctive redshifted absorption band appearing, going from 1a (λmax = 359 nm) to 1b (λmax = 480 nm) (Fig. 1, top). Then, 1b thermally relaxed to 1a (Fig. 1, bottom). By plotting the decay of the VHF absorbance at 480 nm against time (first-order kinetics), an exponential fit provided the rate constant. The pyridyl-substituent at 1b induced an increased rate of back-reaction compared to the carbon analogue (VHF); half-lives of t1/2 = 72 min and 218 min, respectively, as expected when incorporating an electron-withdrawing substituent.7 Next, we studied the influence of protonation of Py-DHA 1a with 1H-NMR and UV/Vis spectroscopies. Firstly, the gradual protonation of Py-DHA 1a was followed by 1H NMR spectroscopy using trifluoroacetic acid (TFA) in CDCl3 at 300 MHz (Fig. 2). Upon protonation, all proton resonances moved downfield, and not only those assigned to the pyridyl ring. Chemical shifts of 1a and 1aH+ are listed in Table 2. Secondly, gradual protonation with TFA in MeCN was easily followed with UV/Vis absorption spectroscopy in which isosbestic points were observed going from Py-DHA 1a (359) to more redshifted 1aH+ (λmax = 406 nm) (Fig. 3). Addition of Et3N to 1aH+ resulted in deprotonation and the initial 1a was obtained as determined by UV/Vis spectroscopy (Fig. S31, ESI†). The protonated species 1aH+ also showed photochromism, and the thermal back-reaction of 1bH+ was 8-fold faster than that of 1b (t1/2 = 9 min and 72 min, respectively). We also investigated whether the pyridyl could act as a handle for triggering the heat release when needed. Thus, Py-DHA 1a was turned into its corresponding VHF 1b, which was voluntarily allowed to return to 1a. After at least one half-life, TFA was added, resulting in a dramatic enhancement of the thermal back-reaction. After one additional half-life, Et3N was added, and the rate of back-reaction decreased again, albeit not to exactly the same rate as before the acid–base treatment (Fig. 4). It should be emphasized that some degradation also seems to occur in MeCN during one opening-closure cycle despite the isosbestic points. | ||
| Scheme 4 Multimode switch based on acid/base and light/heat stimuli. TFA = trifluoroacetic acid. | ||
| Solvent | DHA λmax [nm] | VHF λmax [nm] | Temp. [°C] | k [min−1] VHF → DHA | t1/2 [min] | |
|---|---|---|---|---|---|---|
| a Ref. 6.b Obtained by addition of 20 equiv. TFA and 20 equiv. Et3N.c Kinetics data obtained after addition of 20 equiv. TFA (absorption maxima: DHA 402 nm at 20 equiv. TFA and 406 nm at 200 equiv. TFA; VHF 483 nm at 20 equiv. TFA).d Ref. 8a. | ||||||
| DHA/VHFa | MeCN | 353 | 470 | 25 | 0.0032 | 218 |
| 1a/1b | MeCN | 359 | 480 | 25 | 0.0097 | 72 |
| 1a/1bb | MeCN | 359 | 480 | 25 | 0.0101 | 69 |
| 1aH+/1bH+c | MeCN | 406 | 483 | 25 | 0.0770 | 9 |
| 6a | MeCN | 407 | ||||
| DHN–DHA/DHN–VHFd | EtOH | 365 | 476 | −50 | 0.313 | 2.2 |
| 2a/2b | EtOH | 364 | 440 | −50 | 0.0079 | 88 |
| 2aH+/2bH+ | EtOH | 380 | — | Varying | — | — |
 | ||
| Fig. 1 UV/Vis absorption spectra recorded at 25 °C in MeCN. Top: Acquired during the ring-opening of Py-DHA 1a to 1b. Bottom: Acquired during the ring closure of 1b to 1a. Inset: Decay of VHF absorbance (at 480 nm) against time (min) (first-order kinetics). | ||
 | ||
| Fig. 2 Stacked 1H-NMR spectra recorded upon protonation of 1a (bottom) to 1aH+ (top) with TFA in CDCl3 (300 MHz); each spectrum corresponds to subsequent addition of 0.2 equiv. of TFA. | ||
| H | 1a δ [ppm] | 1aH+ δ [ppm] | Δδ |
|---|---|---|---|
| 8a | 3.81 | 3.89 | 0.08 |
| 8 | 5.83 | 5.85 | 0.02 |
| 7 | 6.34 | 6.40 | 0.06 |
| 6 | 6.55 | 6.66 | 0.11 |
| 5 | 6.61 | 6.68 | 0.07 |
| 4 | 6.46 | 6.64 | 0.18 |
| 3 | 7.09 | 7.37 | 0.28 |
| β-Py | 8.74 | 8.93 | 0.19 |
| α-Py | 7.57 | 8.00 | 0.03 |
 | ||
| Fig. 3 UV/Vis absorption spectra resulting from protonation of Py-DHA 1a (7.0 × 10−5 mol L−1) with TFA in MeCN. The spectra presented are in the range of 0 equiv. TFA to 200 equiv. TFA. For full details of protonation, see ESI.† | ||
 | ||
| Fig. 4 Natural logarithm (ln) of the VHF absorbance (at a wavelength of 480 nm for 1b and 1b + TFA + Et3N mixture and at 483 nm for 1b + TFA mixture) against time. The thermal heat release is activated by addition of TFA (steep slope) and the heat release is retarded again upon addition of Et3N. | ||
The N-methyl pyridinium salt 6a exhibits a characteristic absorption maximum at 407 nm in MeCN, close to that observed for the protonated form of 1a (λmax 406 nm). While this band slowly decreased upon irradiation (365 nm) and spectral changes occurred with isosbestic points, no characteristic VHF absorption band was observed. Rather than photoisomerization, prolonged irradiation thus seems instead to lead to a chemical conversion of 6a, possibly into the fully conjugated azulene by elimination of HCN (supported by 1H-NMR spectroscopy by disappearance of the characteristic signal for the 8a proton), but this was not investigated in further detail.
The isoquinoline moiety in the DHIQ-DHA 2a/2b system forces the VHF to be in the s-cis conformation. Hence, the thermal back-reaction is very fast, and we had to retard it by cooling when acquiring the absorption spectrum of the VHF 2b in EtOH. Thus, DHIQ-DHA 2a was cooled to −50 °C and upon light irradiation (365 nm), it turned into the VHF 2b, seen by a distinctive redshifted absorption band appearing, going from 2a (λmax = 364 nm) to 2b (λmax = 440 nm) (Fig. 5, top). Then, 2b thermally relaxed to 2a (Fig. 5, bottom). By plotting the decay of VHF, 2b, absorbance at 440 nm against time (first-order kinetics) an exponential fit provided the half-life to be 88 min at −50 °C in EtOH. Interestingly, when comparing the half-life of 2b to that of the related DHN–DHA system, the half-life of 2b is 40-fold longer. Currently, we cannot rationalize this result, since the electron-withdrawing pyridyl is expected to enhance the ring closure (as seen for 1b). Density functional theory calculations also suggest that this intuition is correct, yielding thermal back-reaction barriers for the 2b ring closure that are consistently 5 kJ mol−1 lower than those of the DHN–VHF (vide infra).
 | ||
| Fig. 5 UV/Vis absorption spectra recorded at −50 °C in EtOH. Top: Acquired during the ring-opening of DHIQ-DHA 2a to 2b. Bottom: Acquired during the ring closure of 2b to 2a. Inset: Decay of VHF absorbance (440 nm) against time (min) (first-order kinetics). | ||
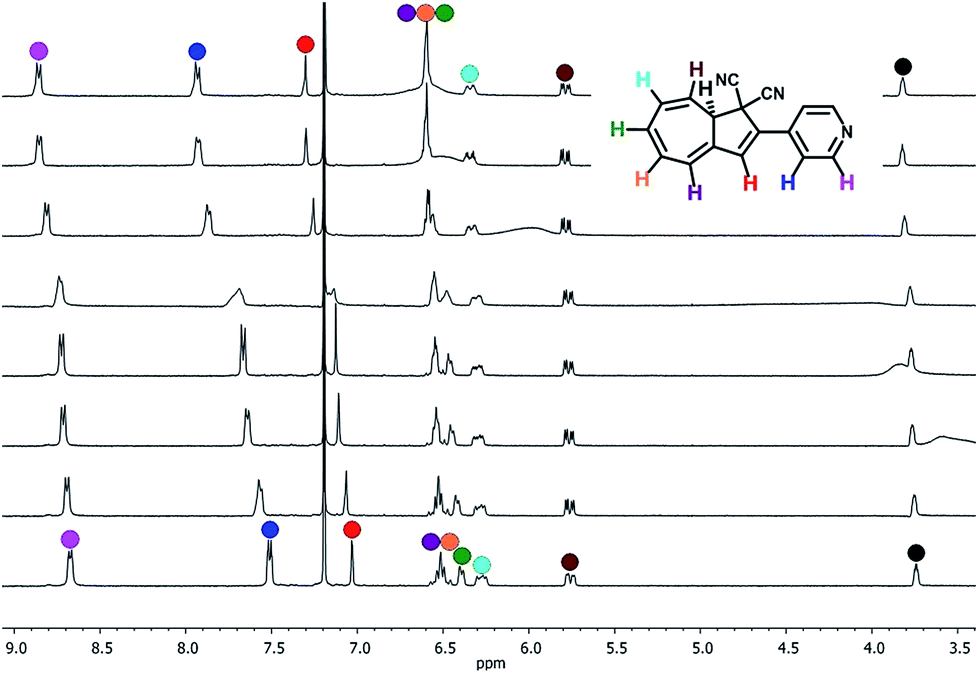
Protonation of 2a with TFA in abs. EtOH at 25 °C was easily followed with UV/Vis absorption spectroscopy, revealing a redshift in the characteristic DHA absorption (Fig. 6). Irradiation (365 nm) of 2aH+ (at various temperatures in the interval from −70 to 25 °C) gave no changes in the absorption spectrum. Instead, the cuvette took upon a blue fluorescent color (Fig. 7, inset). The emission spectra of 2aH+ and the non-protonated 2a are shown in Fig. 7. We cannot exclude an ultrafast back-reaction of 2bH+, if formed, but the apparent lack of photoisomerization of 2aH+ may partly be accounted for by its stronger fluorescence than that of the unprotonated 2a. No apparent fluorescence is visible by the naked eye for 1a and 1aH+ at room temperature.
 | ||
| Fig. 6 UV/Vis absorption spectra resulting from protonation of DHIQ-DHA 2a (6.6 × 10−5 mol L−1) with TFA in EtOH. The spectra presented are in the range of 0 equiv. TFA to 400 equiv. TFA. For full details of protonation, see ESI.† | ||
 | ||
| Fig. 7 UV/Vis absorption (broken line) and emission (full line) spectrum of 2a (blue) and 2aH+ (with 20 equiv. TFA; red) in abs. EtOH at 25 °C. Inset: Photo of the cuvette of 2aH+ upon light irradiation (365 nm) inside the cryostat at −50 °C. | ||
Calculations
A computational study was performed in order to shed light on the thermal back-reactions. All calculations were performed in Gaussian 16 (ref. 15) or Gaussian 09 (ref. 16) using density functional theory (DFT). In correspondence with suggestions from previously published benchmark studies,9 the M06-2X global exchange–correlation functional was employed together with the 6-311++G(d,p) basis-set.17 The solvent effects in this work are considered both using explicit solvent molecules (hence the inclusion of both diffuse and polarization functions on hydrogens in the basis-set in order to capture specific effects of hydrogen bonding) and the integral equation formalism of the polarizable continuum model (IEFPCM) as well as a combination hereof. In the case of the ethanol solvent (EtOH), hydrogen bonds are believed to be crucial for the observed tendencies, and we chose to include calculations with one ethanol molecule hydrogen-bonded to the pyridine nitrogen (OH⋯N) or its protonated analogue (O⋯HN+) in addition to the pure continuum model treatments. We thus considered vacuum and six different media: toluene, CH2Cl2, EtOH, and MeCN using IEFPCM, an explicit EtOH molecule in vacuum, and an explicit EtOH molecule combined with EtOH modelled by IEFPCM.Initially, geometry optimizations of the (8aR) stereoisomers of the DHAs and their corresponding VHFs as well as the protonated analogues of all of these were performed and confirmed as minima by harmonic frequency analyses (see ESI† for structures). In correspondence with the literature,10a there are two types of VHF → DHA ring closure transition states (TSP and TST) which were located and confirmed as first-order saddle points by harmonic frequency analyses. All systems were then subjected to the solvation schemes described above. The thermochemical data for these are calculated at a temperature of 203.15 K and a pressure of 1 atm and presented in Table 3. In this table, we report six different thermochemical properties:
| Vacuum | PhMe | CH2Cl2 | EtOH | MeCN | Vacuum + EtOHa | EtOH + EtOHa | ||
|---|---|---|---|---|---|---|---|---|
| a One molecule of ethanol was included in the structure, with an OH⋯N(pyridine) or O⋯HN+(pyridine) hydrogen bond in the case of the pyridine derivatives. | ||||||||
| 1a/1b | ΔΔGTS | −8.003 | −9.195 | −7.937 | * | * | −7.821 | * |
| ΔΔGVHF | 6.629 | 7.288 | 7.610 | 7.567 | 7.590 | 6.569 | 9.110 | |
| K | 0.069 | 0.053 | 0.046 | 0.047 | 0.047 | 0.071 | 0.025 | |
| ΔGTBR | 103.9 | 94.62 | 87.62 | 85.68 | 85.34 | 104.3 | 85.74 | |
| ΔG′TBR | 110.7 | 102.0 | 95.34 | 93.36 | 93.05 | 111.1 | 94.91 | |
| ΔHstorage | 36.46 | 31.52 | 27.01 | 25.52 | 25.24 | 35.69 | 25.49 | |
| En. dens. | 0.142 | 0.122 | 0.105 | 0.099 | 0.098 | 0.139 | 0.099 | |
| 1aH+/1bH+ | ΔΔGTS | −9.974 | −7.756 | * | * | * | −10.95 | * |
| ΔΔGVHF | −6.291 | −3.004 | 3.418 | 4.797 | 5.078 | −4.135 | 4.752 | |
| K | 12.649 | 3.359 | 0.252 | 0.144 | 0.129 | 5.302 | 0.147 | |
| ΔGTBR | 87.83 | 85.00 | 78.42 | 78.19 | 78.15 | 89.46 | N/A | |
| ΔG′TBR | 88.02 | 85.64 | 82.39 | 83.32 | 83.53 | 89.88 | N/A | |
| ΔHstorage | 59.74 | 48.82 | 37.74 | 34.14 | 33.45 | 55.37 | 29.86 | |
| En. dens. | 0.231 | 0.189 | 0.146 | 0.132 | 0.129 | 0.214 | 0.116 | |
| 2a/2b | ΔΔGTS | −7.089 | −8.966 | * | * | * | −9.313 | * |
| ΔGTBR | 93.10 | 79.62 | 69.44 | 66.67 | 66.14 | 90.60 | 65.75 | |
| ΔHstorage | 66.52 | 64.39 | 61.71 | 60.66 | 60.43 | 68.44 | 61.64 | |
| En. dens. | 0.235 | 0.227 | 0.218 | 0.214 | 0.213 | 0.241 | 0.217 | |
| 2aH+/2bH+ | ΔΔGTS | −13.00 | −9.82 | * | * | * | −14.07 | * |
| ΔGTBR | 67.84 | 60.58 | 56.29 | 55.52 | 55.37 | 70.85 | 58.84 | |
| ΔHstorage | 83.62 | 79.20 | 72.58 | 70.27 | 69.84 | 79.51 | 68.65 | |
| En. dens. | 0.295 | 0.279 | 0.256 | 0.248 | 0.246 | 0.280 | 0.242 | |
| DHN–DHA/DHN–VHF | ΔΔGTS | −3.741 | −6.782 | * | * | * | −0.163 | * |
| ΔGTBR | 97.06 | 85.03 | 75.27 | 72.22 | 71.64 | 97.71 | 72.69 | |
| ΔHstorage | 64.28 | 61.08 | 57.73 | 56.53 | 56.29 | 63.71 | 56.18 | |
| En. dens. | 0.228 | 0.217 | 0.205 | 0.201 | 0.200 | 0.226 | 0.200 | |
(1) The relative transition state stability ΔΔGTS which is calculated as the difference in Gibbs free energy of the TSP and TST transition states, ΔΔGTS = ΔGTS(P) − ΔGTS(T).
(2) The relative s-cis-VHF/s-trans-VHF stability ΔΔGVHF which is calculated as the difference in Gibbs free energy of the s-cis-VHF and the s-trans-VHF, ΔΔGVHF = ΔGs-cis-VHF − ΔGs-trans-VHF (only relevant for the 1b and 1bH+ systems) and the corresponding equilibrium constants K.
(3) The thermal back-reaction barrier ΔGTBR calculated as the difference in Gibbs free energy of the s-cis-VHF and the lowest transition state.
(4) The corrected thermal back-reaction barrier ΔG′TBR which is corrected for the pre-equilibrium between the s-cis-VHF and the s-trans-VHF by modifying the rate constant (obtained using the Eyring equation) of the uncorrected back-reaction by a factor K/(K + 1) where K is the equilibrium constant of the s-cis/s-trans conformational change and from this converted back to a corrected Gibbs free energy through the Eyring equation (only relevant for the 1a/1b and 1aH+/1bH+ systems).
(5) The energy storage capacity ΔHstorage calculated as the difference in enthalpy between the s-trans-VHF and the (8aR)-DHA of the 1a/1b and 1aH+/1bH+ systems and as the difference in enthalpy between the s-cis-VHF and the (8aR)-DHA of the 2a/2b, 2aH+/2bH+, and DHN–DHA/DHN–VHF systems.
(6) The energy density calculated as the energy storage capacity divided by the molecular weight of the system.
First, we note that the energy density is significantly increased for the bridged system (2a/2b) relative to the unbridged one (1a/1b), independently of the medium. Secondly, for both systems, protonation increases the energy density – in vacuum from 0.142 MJ kg−1 (1a/1b) to 0.231 MJ kg−1 (1aH+/1bH+) and from 0.235 MJ kg−1 (2a/2b) to 0.295 MJ kg−1 (2aH+/2bH+). The energy densities are smaller in polar media, while just adding one hydrogen-bonded ethanol molecule to the pyridine-based systems in vacuum has little effect (hydrogen bonding to the pyridine N or N+H).
In regard to kinetics, we see that, indeed, the locked system 2b has a lower back-reaction barrier (ΔGTBR) than the unlocked system 1b, in agreement with experimental findings (vide supra). In addition, protonation leads to a very significant lowering of the barrier for both systems, which was also found experimentally for 1b. For the thermal back-reaction of 2b we find a lower barrier than for DHN–VHF regardless of the solvent environment. This contrasts, as mentioned and discussed above, the experimental findings, and we have at the moment no explanation for this discrepancy. We note, however, that the calculations are in correspondence with the general influence of electron-withdrawing groups that we previously have observed experimentally.7
For both 1a and 2a, protonation was found experimentally to result in significantly redshifted longest-wavelength absorption maxima. Calculations reveal that the HOMO and LUMO become more localized at each end of the molecule for 1aH+ and 2aH+ (Fig. 8), and the transition thus has slightly more charge-transfer character for the protonated species, explaining the observed absorption redshifts.
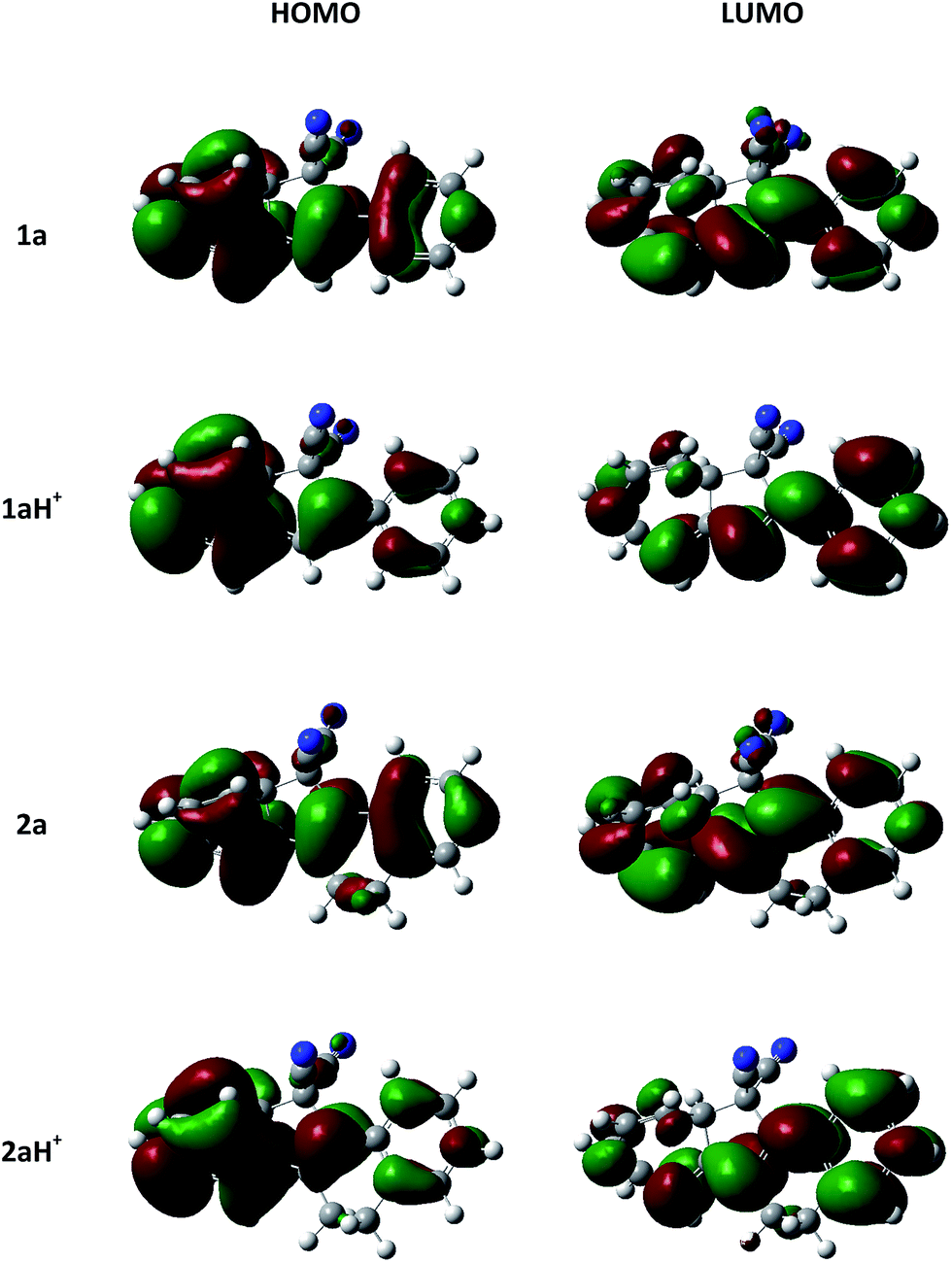 | ||
| Fig. 8 Computationally determined HOMO and LUMO of 1a, 1aH+, 2a, and 2aH+ with the M06-2X global exchange–correlation functional and the 6-311++G(d,p) basis-set visualised with an isovalue (contour threshold) of 0.02 au. | ||
Calculated UV-Vis absorption spectra of both protonated and non-protonated DHAs 1a and 2a provided the same red-shifting influence of protonation as observed in experiment. In Fig. 9, calculated absorption spectra in MeCN for 1a/1aH+ and in EtOH for 2a/2aH+ can be found, both in full qualitative correspondence with experiments (cf. Fig. 3 and 6, respectively). The ESI includes spectra in other media. All spectra are determined using linear response TD-DFT at the M06-2X/6-311++G(d,p) level of theory based on the 298.15 K geometries by calculating the 30 lowest energy excited states for each compound. Gaussians with a standard deviation of 0.4 eV were fitted to the oscillator strengths in order to produce the absorption curves. It should be noted that M06-2X has previously been shown to slightly underestimate excitation wavelengths of DHAs,9 and the observation of this tendency for these compounds too is thus expected.
 | ||
| Fig. 9 Calculated absorption spectra of 1a/1aH+ in MeCN (top) and 2a/2aH+ in EtOH (bottom) (linear response TD-DFT, M06-2X/6-311++G(d,p)). | ||
In general, the observed red-shift of DHA absorptions following protonation decreases with increasing solvent polarity for both systems. This is due to the low-energy absorption peaks of the protonated species blue-shifting with increasing solvent polarity, while the low-energy absorption peaks of the non-protonated species remain at almost the same values regardless of solvent. The energies of the low-energy absorption peaks in vacuum and all solvents for VHFs 1b/1bH+ and 2b/2bH+ can be found alongside those of the DHAs 1a/1aH+ and 2a/2aH+ in ESI, Table S2.† These all comply with the above. To summarize, the experimental findings are thus substantiated, and the calculations provide strong indications that protonation of pyridyl-DHAs can serve to red-shift their absorptions toward high solar photon flux spectral ranges without hampering their energy storage capabilities.
Conclusion
Incorporation of a pyridyl substituent onto the dihydroazulene/vinylheptafulvene photo-/thermoswitch was synthetically rather challenging, but we managed to prepare two such derivatives. The pyridine unit acts as a convenient site for controlling properties by protonation/deprotonation. Thus, protonation leads to a significant redshift in the characteristic DHA absorption maximum as the transition gets more charge-transfer character. Protonation can also serve as a method to promote the vinylheptafulvene back-reaction (triggering energy release), and calculations reveal how the energy density is also increased significantly by protonation. Locking the VHF in its s-cis conformation enhances the energy density, but at the same time, the back-reaction becomes undesirably fast. This may be accounted for by other means; for example, suitable substitution of the core structure can be used to further tune the rate of ring closure.7 Despite some stability issues (during isolation and during switching cycles), the pyridyl substituent presents in all a useful structural modification, which in combination with other modifications may ultimately render the dihydroazulene/vinylheptafulvene system suitable for real MOST systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
University of Copenhagen is acknowledged for financial support. Dr Marco Santella is thanked for assistance regarding fluorescence measurements.Notes and references
- (a) H.-D. Scharf, J. Fleischhauer, H. Leismann, I. Ressler, W. Schleker and R. Weitz, Angew. Chem., Int. Ed. Engl., 1979, 18, 652 CrossRef; (b) T. J. Kucharski, Y. Tian, S. Akbulatov and R. Boulatov, Energy Environ. Sci., 2011, 4, 4449 RSC; (c) K. Moth-Poulsen, in Organic Synthesis and Molecular Engineering, ed. M. B. Nielsen, Wiley, Hoboken, USA, 2014, pp. 179–196 Search PubMed; (d) A. Lennartson, A. Roffey and K. Moth-Poulsen, Tetrahedron Lett., 2015, 56, 1457 CrossRef CAS.
- Z.-I. Yoshida, J. Photochem., 1985, 29, 27 CrossRef CAS.
- (a) J. Daub, T. Knöchel and A. Mannschreck, Angew. Chem., Int. Ed. Engl., 1984, 23, 960 CrossRef; (b) T. Mrozek, A. Ajayaghosh and J. Daub, Optoelectronic Molecular Switches Based on Dihydroazulene–Vinylheptafulvene (DHA–VHF), in Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, 2001, pp. 63–106 Search PubMed; (c) S. L. Broman and M. B. Nielsen, Phys. Chem. Chem. Phys., 2014, 16, 21172 RSC.
- (a) H. Görner, C. Fischer, S. Gierisch and J. Daub, J. Phys. Chem., 1993, 97, 4110 CrossRef; (b) H. Görner, C. Fischer and J. Daub, J. Photochem. Photobiol., A, 1995, 85, 217 CrossRef.
- O. Schalk, S. L. Broman, M. Å. Petersen, D. V. Khakhulin, R. Y. Brogaard, M. B. Nielsen, A. E. Boguslavskiy, A. Stolow and T. I. Sølling, J. Phys. Chem. A, 2013, 117, 3340 CrossRef CAS PubMed.
- S. L. Broman, S. L. Brand, C. R. Parker, M. Å. Petersen, C. G. Tortzen, A. Kadziola, K. Kilså and M. B. Nielsen, ARKIVOC, 2011, ix, 51 Search PubMed.
- S. L. Broman, M. Jevric and M. B. Nielsen, Chem.–Eur. J., 2013, 19, 9542 CrossRef CAS PubMed.
- (a) S. L. Broman, O. Kushnir, M. Rosenberg, A. Kadziola, J. Daub and M. B. Nielsen, Eur. J. Org. Chem., 2015, 4119 CrossRef; (b) M. Cacciarini, A. B. Skov, M. Jevric, A. S. Hansen, J. Elm, H. G. Kjaergaard, K. V. Mikkelsen and M. B. Nielsen, Chem.–Eur. J., 2015, 21, 7454 CrossRef CAS PubMed.
- S. T. Olsen, J. Elm, F. E. Storm, A. N. Gejl, A. S. Hansen, M. H. Hansen, J. R. Nikolajsen, M. B. Nielsen, H. G. Kjaergaard and K. V. Mikkelsen, J. Phys. Chem. A, 2015, 119, 896 CrossRef CAS PubMed.
- (a) M. H. Hansen, J. Elm, S. T. Olsen, A. N. Gejl, F. E. Storm, B. N. Frandsen, A. B. Skov, M. B. Nielsen, H. G. Kjaergaard and K. V. Mikkelsen, J. Phys. Chem. A, 2016, 120, 9782 CrossRef CAS PubMed; (b) A. S. Gertsen, S. T. Olsen, S. L. Broman, M. B. Nielsen and K. V. Mikkelsen, J. Phys. Chem. C, 2017, 121, 195 CrossRef CAS; (c) M. Koerstz, J. Elm and K. V. Mikkelsen, J. Phys. Chem. A, 2017, 121, 3148 CrossRef CAS PubMed.
- J. Gurke, M. Quick, N. P. Ernsting and S. Hecht, Chem. Commun., 2017, 53, 2150 RSC.
- For a recent review on the control of diarylethene photoswitches by external stimuli, see: S.-Z. Pu, Q. Sun, C.-B. Fan, R.-J. Wang and G. Liu, J. Mater. Chem. C, 2016, 4, 3075 RSC.
- (a) L. Gobbi, P. Seiler and F. Diederich, Angew. Chem., Int. Ed., 1999, 38, 674 CrossRef CAS; (b) L. Gobbi, P. Seiler, F. Diederich, V. Gramlich, C. Boudon, J.-P. Gisselbrecht and M. Gross, Helv. Chim. Acta, 2001, 84, 743 CrossRef CAS; (c) M. Å. Petersen, S. L. Broman, K. Kilså, A. Kadziola and M. B. Nielsen, Eur. J. Org. Chem., 2011, 1033 CrossRef CAS.
- The steps towards 11 follow known procedures: (a) J. Epsztajn and A. Bieniek, J. Chem. Soc., Perkin Trans. 1, 1985, 213 RSC; (b) D. R. Boyd, R. J. H. Davies, L. Hamilton and J. J. McCullough, J. Chem. Soc., 1992, 31 RSC; (c) C.-Y. Cheng, L.-W. Hsin and J.-P. Liou, Tetrahedron, 1996, 52, 10935 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis protocols, NMR spectra, switching studies and computational data. See DOI: 10.1039/c7ra13762a |
| ‡ These authors contributed equally to the article. |
| § Present address: Department of Energy Conversion and Storage, Technical University of Denmark, Frederiksborgvej 399, DK-4000 Roskilde, Denmark. |
| This journal is © The Royal Society of Chemistry 2018 |