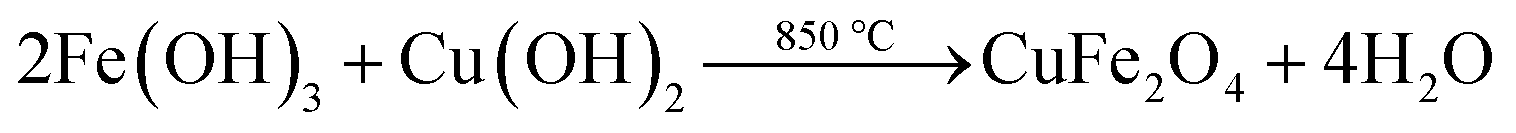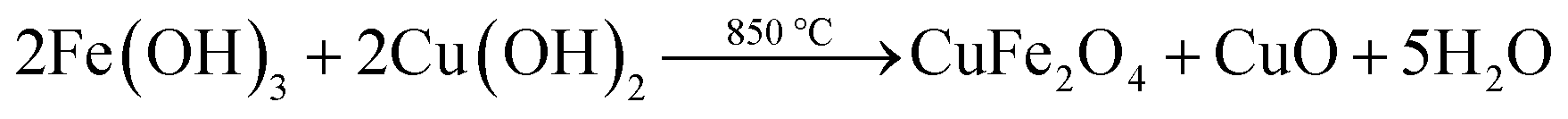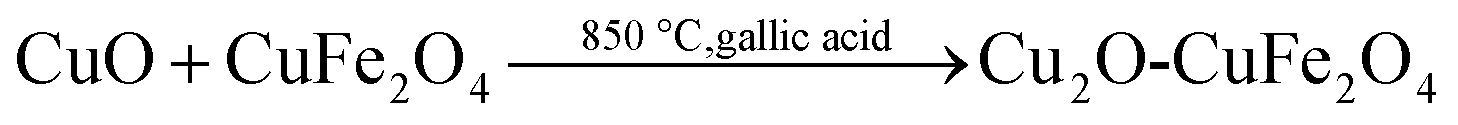DOI:
10.1039/C7RA13608K
(Paper)
RSC Adv., 2018,
8, 5740-5748
Synthesis of Cu2O–CuFe2O4 microparticles from Fenton sludge and its application in the Fenton process: the key role of Cu2O in the catalytic degradation of phenol
Received
23rd December 2017
, Accepted 30th January 2018
First published on 2nd February 2018
Abstract
This paper presents the key role of Cu2O in Fenton catalysis using Cu2O–CuFe2O4 magnetic microparticles, which were prepared using Fenton sludge as an iron source. The catalytic activity of the as-prepared Cu2O–CuFe2O4 and CuFe2O4 microparticles was evaluated in a heterogeneous Fenton system for the degradation of recalcitrant phenol. The Cu2O–CuFe2O4 microparticles demonstrated relatively superior catalytic performance as compared to CuFe2O4 microparticles when used as a Fenton catalyst. The relatively higher catalytic activity of Cu2O–CuFe2O4 for phenol degradation during the Fenton process could be attributed to the availability of both monovalent [Cu(I)] and divalent [Cu(II)] as well as Fe(II)/Fe(III) redox pairs, which could react quickly with H2O2 to generate hydroxyl radicals (HO˙). An electron bridge was formed between Cu(I) and Fe(III), which accelerates the formation of Fe(II) species in order to boost the reaction rate. Highly reactive and excessively available Cu(I) species for as prepared Cu2O–CuFe2O4 microparticles could be considered to be rather crucial for the generation of highly reactive HO˙ radical species. In addition, the as-prepared Cu2O–CuFe2O4 magnetic microparticles exhibited sound stability and reusability.
1. Introduction
Fenton oxidation technology, which is well known as advanced oxidation processes (AOPs), has been extensively used to remove various recalcitrant or non-biodegradable organic pollutants from industrial wastewater.1,2 During Fenton process, a powerful oxidant, hydroxyl radicals (HO˙) can be generated efficiently at near-ambient temperature and pressure by the reaction between Fe2+ and H2O2.3 The distinct properties of generated hydroxyl radicals (HO˙) such as strong oxidation potential (E0 = 2.8 V) and non-selective reactivity are considered to be responsible for the effectiveness of AOPs in the field of organic pollutant elimination. These generated radicals can react with a variety of organic pollutants, causing efficient degradation, even complete mineralization.4,5 However, in spite of the simplicity of Fenton process, bulk quantity of iron sludge generated during neutralization after Fenton oxidation restricts its implementation on large scale due to the increasing cost for Fenton sludge treatment and disposal to avoid environment deterioration.6
Two approaches have been implemented to reduce the Fenton sludge generation yield, i.e., development of heterogeneous iron bearing catalyst and reuse of Fenton sludge as iron source. Various heterogeneous catalysts have been synthesized, such as nano scale zero-valent iron,7 iron-containing clays,8 natural minerals,9 iron exchanged zeolite,10 and some solid support immobilized by iron.11,12 Immobilization of iron within the interlayer of heterogeneous catalysts results in relatively high oxidation performance. Moreover, heterogeneous catalysts have been proven to be superior to classical homogeneous catalysts i.e., Fe2+, because they are easily separated from wastewater treatment system through applied magnetic field or simple sedimentation. Yoo et al., suggested the recycling of iron incorporated sludge produced in Fenton oxidation system as coagulant during coagulation process.13 Through the recycling of Fenton sludge as coagulant, coagulant dosage could be lowered by 50% and sludge disposal could be decreased by 50%. Recycling of iron incorporated sludge also follows the 3R's rules regarding integrated solid waste management, i.e., reduce, reuse and recycle. Iron sludge–graphene composite with low amount of graphene were synthesized as a heterogeneous Fenton catalyst by using iron sludge as the iron precursor. The as-prepared catalyst showed wide pH operating range, excellent stability, and was efficient for the degradation of acid red G and metronidazole.14 Zhang et al. synthesized magnetic biochar catalyst by using ferric sludge and biological sludge via hydrothermal process. This catalyst showed excellent characteristics to promote a heterogeneous Fenton reaction in methylene blue treatment.15 These methods hold promise for ferric sludge waste reclamation and heterogeneous Fenton catalyst fabrication.
In addition to the recycling of iron incorporated sludge, the use of low cost heterogeneous catalyst in Fenton treatment could be another alternative for the reduction of hazardous ferric sludge. Recently, different approaches have been investigated to boost up the performance of synthesized heterogeneous Fenton catalyst, e.g., to reduce the catalyst size up to nano-scale in order to increase surface energy and available active sites, to embed appropriate transition metal (Cu, Ni, V, Ti, Cr, Zn, Mn and Co, etc.) into the skeleton of Fe3O4 to improve the catalyst performance.16 The improvement of heterogeneous catalyst performance as a result of transition metals embedment into Fe3O4 structure could be attributed to the synergetic effect between newly inserted transition metal and Fe3+. This synergetic effect could be described by formation of galvanic cell between embedded transition metal and Fe3+. The improvement in catalyst activity could be ascribed to formation of redox pairs Fe3+/Fe2+ due to rapid electron flow as a result of transition metal insertion into Fe3O4 structure. A newly developed material can be implemented in different fields if it holds the properties of higher reactivity, sound stability and easy separation from reaction medium.
Mixed iron oxides or ferrites have drawn much more attention of scientist and researcher due to their ability to apply in different fields such as microwave devices,17 catalysis or catalyst,18 magnetic fluids19 and gas sensors.20 Recently, various ferrites such as ZnFe2O4 (ref. 21) and CuFe2O4,22 and iron–copper bimetallic nanoparticles embedded within ordered mesoporous carbon composite,23 have been utilized as the catalyst for heterogeneous Fenton like process to remove organic contaminants from wastewater. In our previous study, NiFe2O4 were synthesized through co-precipitation method followed by sintering at 800 °C.24 In the Fenton system using NiFe2O4 as heterogeneous catalyst, phenol removal efficiency as high as 95 ± 3.4% could be achieved, indicating excellent catalytic performance of NiFe2O4 in the heterogeneous Fenton process. Roonasi and Nezhad carried out a comparative study to investigate the performance of synthetic M-ferrite nanoparticles (M = Cu, Zn, Fe or Mn) in heterogeneous catalysis. Among these ferrite nanoparticles, CuFe2O4 was found to be the best catalyst for phenol removal, as 78% of 100 mg L−1 phenol could be efficiently removed within 175 min.25 In addition, the effectiveness of Cu(I) species as compare to Cu(II) species for the activation of H2O2 in order to generate HO˙ radicals for the degradation of bisphenol has been confirmed in literature.26 Recently, the nanocomposite CuO-CuFe2O4 has been synthesized in a single stage by one pot polyol method. The electrical, structural and optical properties of the synthesized CuO-CuFe2O4 were investigated as a function of different annealing temperatures ranges from 200–1000 °C.27 Furthermore, a novel and recyclable magnetic catalyst Cu2O/nano-CuFe2O4 has been reported for the coupling of carbonyl compounds–alkynes–amines in order to synthesize propargylamines under solvent-free condition.28 These studies gave us a direction to synthesize Cu2O–CuFe2O4 particles with both Cu(I) and Cu(II) species, resulting in the fast activation of H2O2 to get hydroxyl radicals HO˙ for the elimination of recalcitrant organic pollutant such as phenol.
In this study, we have explored a facile and novel method to fabricate Cu2O–CuFe2O4 microparticles using Fenton sludge as iron source, which was further used as a heterogeneous catalyst in Fenton process for the elimination of recalcitrant phenol. Thus, the aims of the present study were (1) to fabricate and characterize Cu2O–CuFe2O4, (2) to assess the catalytic activity of fabricated Cu2O–CuFe2O4 in Fenton oxidation, (3) to investigate the synergetic effect of Cu(I)/Cu(II) as well as Fe(III)/Fe(II) redox pairs in Fenton reaction, and (4) to propose possible catalytic mechanism involved in efficient Fenton reaction at the presence of Cu2O–CuFe2O4.
2. Materials and methods
2.1 Characteristics of the Fenton sludge used
The characteristics of iron-containing sludge obtained from neutralization process after Fenton oxidation could be described as follows. As a rich iron source, total iron content of Fenton sludge used for the synthesis of Fenton catalyst was as high as 8.65 ± 0.78 g L−1. Chemical oxygen demand (COD) concentration of the Fenton sludge used was about 6700 ± 375 mg L−1. The higher COD value exhibited the presence of abundant organics in Fenton sludge. Moreover, the Fenton sludge used in this study could be fluidized in nature, having total solid as well as water content of about 87.48% ± 0.97% and 12.52 ± 0.97%, respectively.
2.2 Synthesis of CuFe2O4 microparticles
CuFe2O4 microparticles were prepared by co-precipitation technique, i.e., CuSO4·5H2O was dissolved into ultrapure water and then the prepared CuSO4 solution was mixed with Fenton sludge at Fe/Cu molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Thereafter, sodium hydroxide solution (5 mol L−1) was added into the mixture of Fenton sludge and CuSO4 drop by drop under continuous stirring to raise the pH value to 10.0. The mixture was stirred continuously at temperature of 65 °C for 2 h. The obtained precipitate was then separated by centrifugation and washed with ultrapure water until the pH value of the filtrate reached 7.0. The resulting product was kept in an oven at temperature of 105 °C for 3 h to make it dry and then finally sintered at 850 °C for 3 h under nitrogen atmosphere. The formation of CuFe2O4 microparticles could be described by eqn (1)–(3):
:1. Thereafter, sodium hydroxide solution (5 mol L−1) was added into the mixture of Fenton sludge and CuSO4 drop by drop under continuous stirring to raise the pH value to 10.0. The mixture was stirred continuously at temperature of 65 °C for 2 h. The obtained precipitate was then separated by centrifugation and washed with ultrapure water until the pH value of the filtrate reached 7.0. The resulting product was kept in an oven at temperature of 105 °C for 3 h to make it dry and then finally sintered at 850 °C for 3 h under nitrogen atmosphere. The formation of CuFe2O4 microparticles could be described by eqn (1)–(3):| | |
Fe3+ + 3(OH−) → Fe(OH)3
| (1) |
| | |
Cu2+ + 2(OH−) → Cu(OH)2
| (2) |
| |
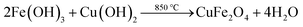 | (3) |
2.3 Synthesis of Cu2O–CuFe2O4 microparticles
Cu2O–CuFe2O4 microparticles were prepared through modified hydrothermal technique.29 In this case, CuSO4·5H2O solution and Fenton sludge were mixed together at Fe/Cu molar ratio of 1:1. Sodium hydroxide solution (5 mol L−1) was added drop wise into the mixture of Fenton sludge and CuSO4 under continuous stirring to raise the pH value to 10.0 to get colloidal suspension. After this, 0.97 g gallic acid was blended with the suspension. The obtained mixture was sonicated for 15 min and then was poured into a 200 mL Teflon-line stainless steel autoclave. The closed autoclave was then allowed to keep at 200 °C for almost 12 h and finally cooled normally to room temperature. During hydrothermal process the gallic acid attached with chemical substances as organic ligands. The obtained product was separated and washed several times with ultrapure water, then dried at 105 °C and finally sintered at 850 °C for 3 h under nitrogen atmosphere. The calcination temperature was chosen as 850 °C because higher temperature causes lower reactivity and lower temperature favors severe leaching from catalysts.30 Moreover, for comparison purpose, the Cu2O microparticles was synthesized in a similar way by using CuSO4·5H2O individually as a precursor material. The formation of Cu2O–CuFe2O4 microparticles could be described by eqn (4) and (5):| |
 | (4) |
| |
 | (5) |
2.4 Catalytic degradation experiment
Catalytic degradation of phenol was carried out in a series of 100 mL centrifuge tubes. 50 mL phenol solution at initial concentration of 250 mg L−1 and initial pH of 4.0 was added into these centrifuge tubes. Fenton reaction was initiated by adding H2O2 and magnetic heterogeneous Fenton catalyst into phenol solution. The initial concentration of H2O2 and magnetic heterogeneous Fenton catalyst was set at 80 mmol L−1 and 2 g L−1, respectively. The Fenton reaction was carried out on a rotary shaker at 30 °C and 220 rpm. The heterogeneous Fenton catalyst was recovered from solution after the reaction by applying external magnetic field for expected reuse. The control experiments carried out with Fenton catalyst but without H2O2 or with H2O2 but without Fenton catalyst were performed in the same manner.
2.5 Characterization and analytical methods
Total iron content of Fenton sludge, copper and iron leaching were analyzed through Inductive Couple Plasma Optical Emission Spectrometer (ICP-OES) (Optima 7000DV, PerkinElmer, USA). A mixture of 2.5 mL HClO4, 2.5 mL HNO3 (69%) and 10 mL HF (40%) was used for wet digestion of Fenton sludge. The COD concentration of Fenton sludge was analyzed by the standard potassium dichromate method. SEM (JSM-6380, JEOL, Japan) and HRTEM (FEI Philips CM300 UT/FEG) were applied to characterize the surface morphology, size and shape of as prepared CuFe2O4 and Cu2O–CuFe2O4 microparticles. BET (Micromeritics, ASAP 2020, USA) surface area was quantified by nitrogen adsorption data at 77 K. For the investigation of crystal structure of as-prepared materials, XRD (D8 Advance, Burker, Germany) analysis was performed. X-Ray Photoelectron Spectroscopy (XPS, ESCALAB 250) technique was implemented to determine the synthesized materials elemental composition and different oxidation states of corresponding elements. The magnetic strength was tested on a vibrating sample magnetometer (VSM) (Lake Shore 7410, Lake Shoe Cryotronics, Inc. USA). Phenol identification and quantification was carried out by high performance liquid chromatography (HPLC) (Waters 2996, Waters Incorporation, USA). Identification of intermediates formed during phenol degradation was confirmed by GC-MS as well as HPLC.
3. Results and discussion
3.1 Characterization of the synthesized microparticles
In this study, the CuFe2O4 and Cu2O–CuFe2O4 microparticles were synthesized by co-precipitation and modified hydrothermal method followed by sintering under nitrogen atmosphere. Fig. 1 showed the SEM and high resolution TEM images of CuFe2O4 and Cu2O–CuFe2O4 microparticles. It was obvious that only one type of specific shape agglomerated particles could be observed in case of synthesized CuFe2O4 microparticles (Fig. 1(a)). However, for Cu2O–CuFe2O4 microparticles, two different types of agglomerated particles could be observed and the new types of agglomerated particles were pointed out with red arrows in SEM image (Fig. 1(b)). Similar evidence was revealed by high resolution TEM images (Fig. 1(c) and (d)). Two different particles were existed in case of Cu2O–CuFe2O4 microparticles as compared to CuFe2O4 microparticles, the results were in agreement with SEM images. Through comparison between both synthesized particles and literature review, it was speculated that the microparticles with the shape like chrysanthemum might be Cu2O, which should be further clarified by XRD analysis. The Cu/Cu2O/CuO@C catalyst particles recently synthesized by Zhao et al., which had excellent catalytic performance, was also similar to chrysanthemum as in our case.31 BET surface area analysis was also carried out for the synthesized heterogeneous catalysts, as indicated in Table 1. The BET surface area (1.08 m2 g−1) for CuFe2O4 particles was smaller than that of Cu2O–CuFe2O4 particles (1.65 m2 g−1), which might be directed towards the generation of additional chrysanthemum shaped Cu2O particles along with CuFe2O4 particles at the presence of gallic acid. The presence of puffy ball-like structure particles might result in higher BET surface area of Cu2O–CuFe2O4 particles. For synthesized Cu2O micro particles the BET surface area was 1.50 m2 g−1 which was higher than that of CuFe2O4 (1.08 m2 g−1). It could be referred to puffy ball like shape of Cu2O particles.
 |
| | Fig. 1 SEM (a, b) and high resolution TEM (c, d) images of CuFe2O4 and Cu2O–CuFe2O4. | |
Table 1 Catalyst or catalysts physicochemical properties, their catalytic performance and metals ions leaching
| Catalyst |
BET S. area (m2 g−1) |
Average particle size (μm) |
TOC removal (%) |
Phenol removal (%) |
Iron leaching (mg L−1) |
Copper leaching (mg L−1) |
| CuFe2O4 |
1.08 |
2–3 |
47.6 ± 1.0 |
57.8 ± 3.6 |
1.16 |
30.18 |
| Cu2O–CuFe2O4 |
1.65 |
0.5–2 |
85.6 ± 0.7 |
97.2 ± 0.4 |
1.53 |
28.60 |
| Cu2O |
1.50 |
2–3.5 |
36.5 ± 1.5 |
61.9 ± 2.3 |
— |
— |
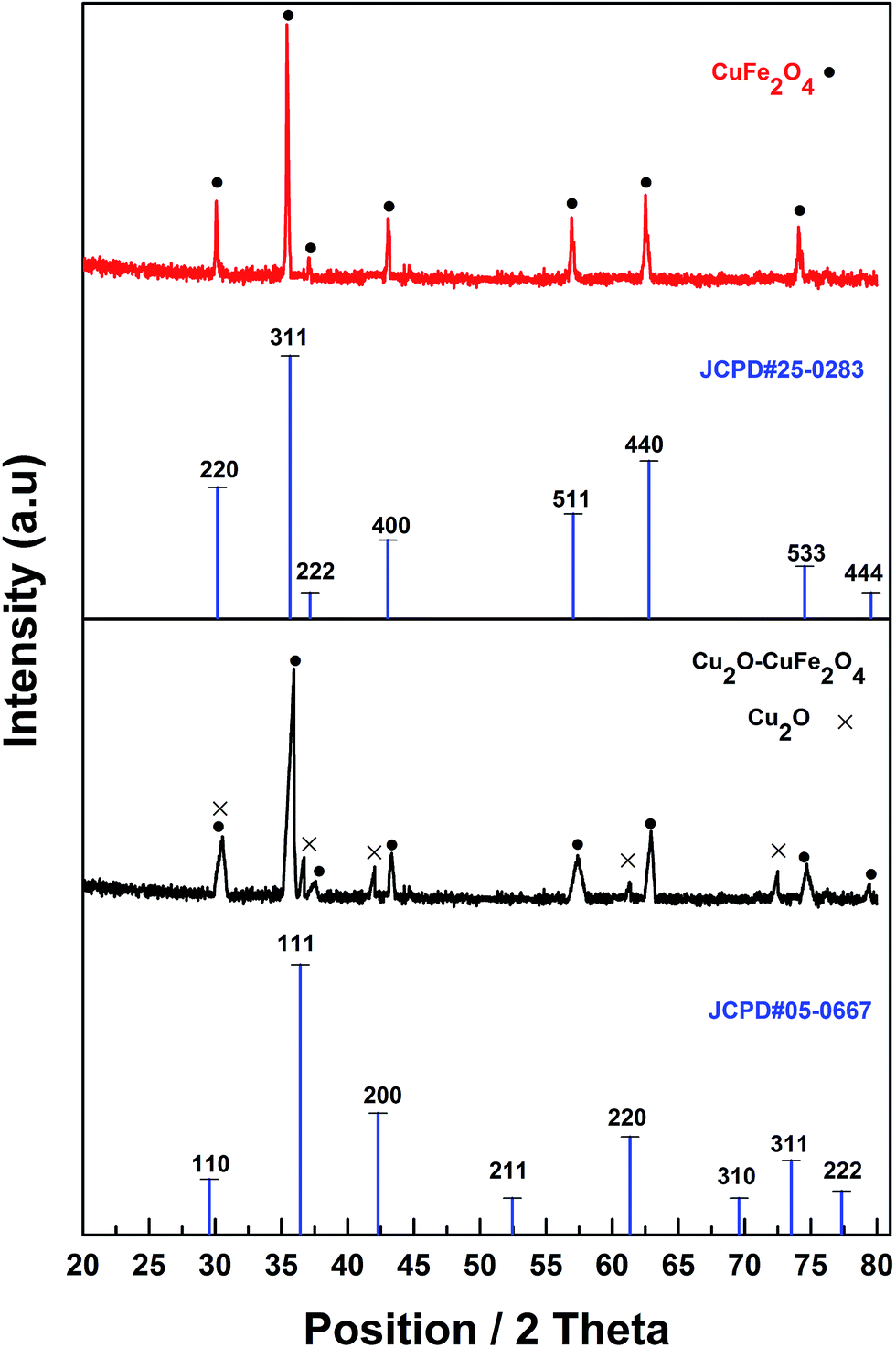
XRD analysis was carried out to examine the crystal phase and structure of synthesized products (Fig. 2). The diffraction peaks appeared at 2θ of 18.5° (111), 30.2° (220), 35.6° (311), 37.2° (222), 43.0° (400), 57.1° (511), 62.8° (440) and 74.5° (533) could be assigned to pure cuprospinel CuFe2O4 (JCPDS#25-0283). In comparison with pure cuprospinel CuFe2O4, newly originated diffraction peaks positioning at 2θ of 36.7° (111), 42.5° (200), 61.6° (220) and 73.7° (311) could be attributed to rhombic dodecahedral crystal of Cu2O (JCPDS#05-0667) in cubic phase, which confirmed the existence of Cu2O in as-synthesized Cu2O–CuFe2O4.
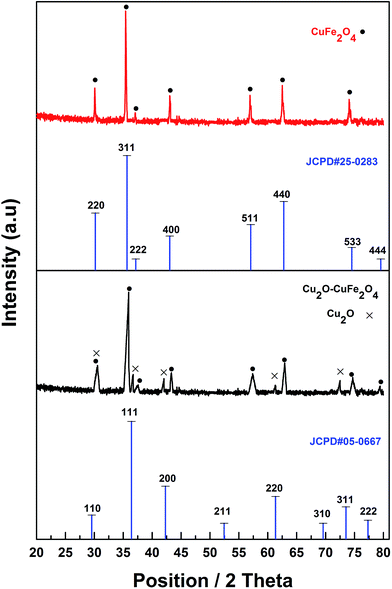 |
| | Fig. 2 XRD of CuFe2O4 and Cu2O–CuFe2O4. | |
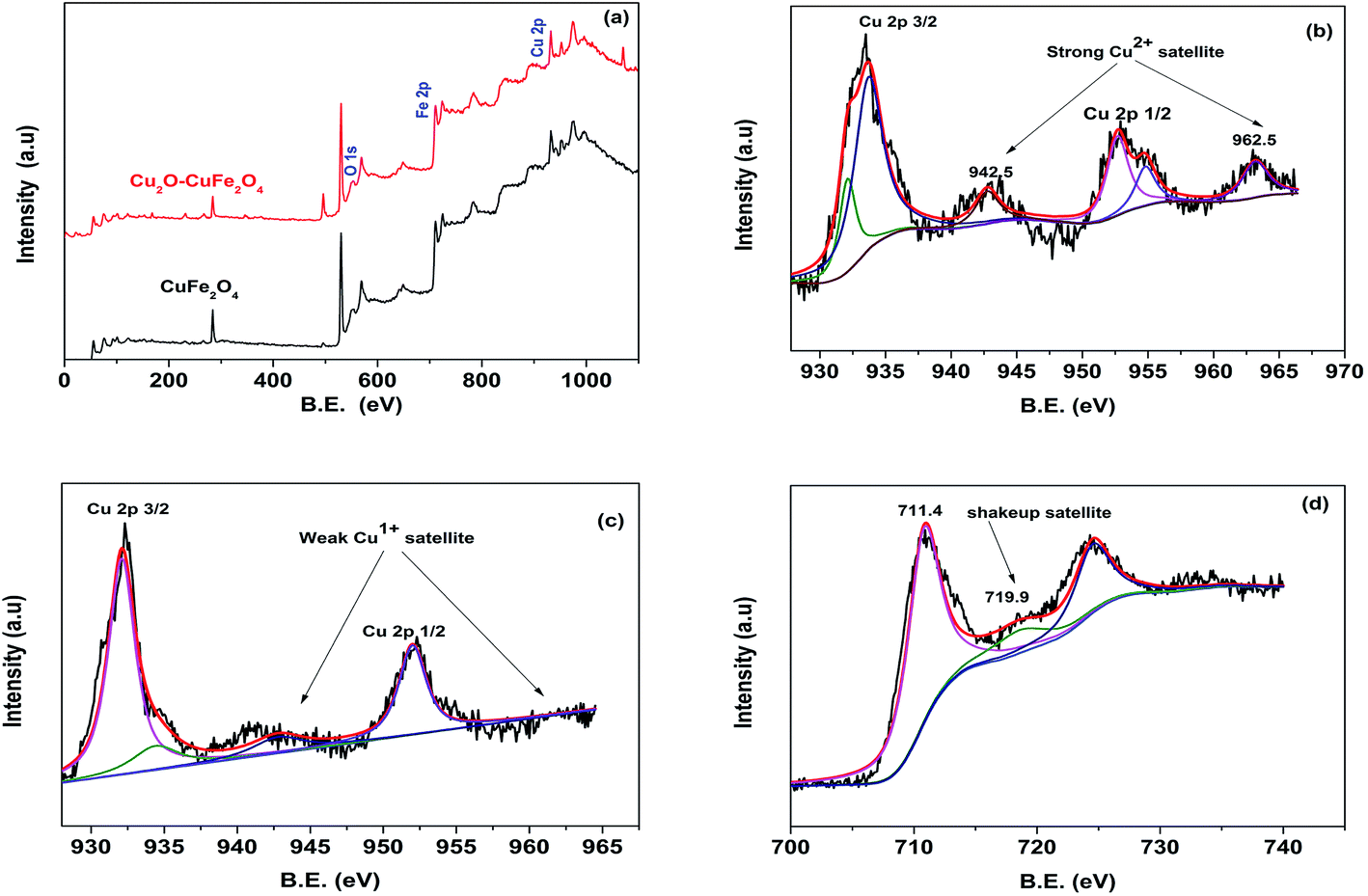
XPS technique was applied for thorough study of chemical composition and different oxidation states of constituent elements of synthesized materials. From XPS wide spectrum in Fig. 3(a), it was clear that both microparticles were comprised of same parent constituents, such as Fe, Cu and O elements. Someone could differentiate between materials having the same parent constituents on the behalf of different oxidation states of existed constituent elements. For example, existence of Cu2+ in CuFe2O4 and the presence of Cu+ in Cu2O–CuFe2O4 materials could be confirmed on the behalf of different binding energy of Cu2+ and Cu+, respectively. As shown in Fig. 3(b), the detail XPS spectra of Cu 2p reveals that strong Cu2+ satellites could be observed at binding energy of 942.5 eV and 962.5 eV along with peak at 934.3 eV, which confirmed the presence of Cu2+ in CuFe2O4.32,33 In addition, on the basis of the relatively weak peak at 932.1 eV,34 there were Cu+ species on the surface of CuFe2O4, which might come from the reduction of Cu2+ during calcination process due to the abundant organics in Fenton sludge. In contrast, for Cu2O–CuFe2O4, there was a relatively strong peak at 932.1 eV in Fig. 3(c), which could be referred to Cu2O. Similar result was found for Cu/Cu2O/CuO@C catalyst particles.31 In addition, peak at 934.3 eV and weak satellite peaks at 942.5 eV and 962.5 eV confirm the formation of Cu2+ along with CuFe2O4 particles for as-prepared Cu2O–CuFe2O4 microparticles. Moreover, for wide scan of Fe 2p spectra, the peaks raised at 711.4 eV along with shakeup satellite pointed at 719.9 eV corresponded to the existence of Fe3+ cations. However, XPS spectrum for Fe 2p and O 1s for both CuFe2O4 and Cu2O–CuFe2O4 were similar, as shown in Fig. 3(a) and (d).
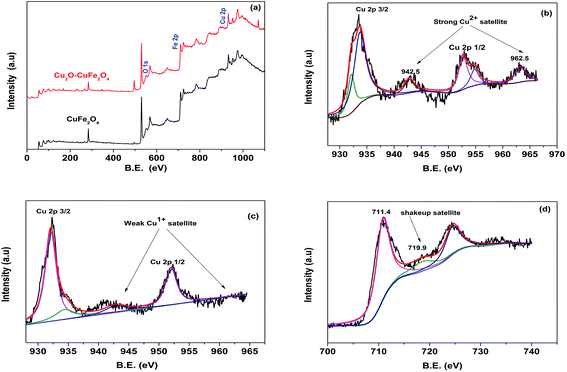 |
| | Fig. 3 XPS spectra for CuFe2O4 and Cu2O–CuFe2O4 (a), strong Cu2+ satellite for CuFe2O4 (b), weak Cu1+ satellite for Cu2O–CuFe2O4 (c), similar Fe 2p region for both CuFe2O4 and Cu2O–CuFe2O4 (d). | |
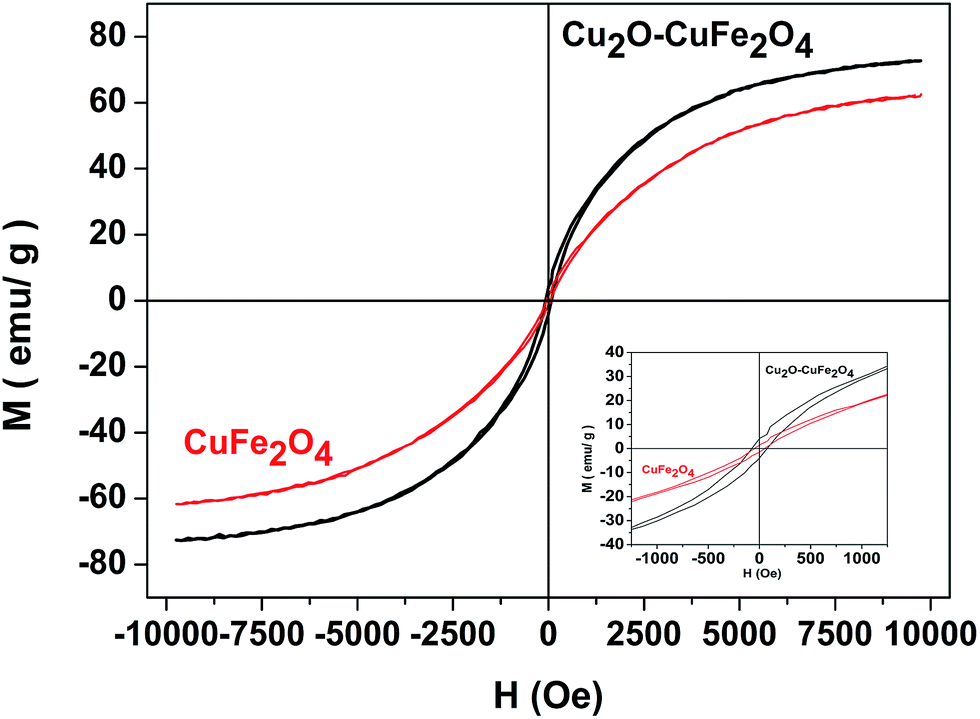
The magnetic property of the as-prepared Cu2O–CuFe2O4 and CuFe2O4 was determined by applied field of ±10000 Oe. As shown in Fig. (4), the saturation magnetization and coercivity values for as-prepared Cu2O–CuFe2O4 and CuFe2O4 were 71 emu g−1 and 73 Oe, 61 emu g−1 and 50 Oe, respectively. The magnetic hysteresis loop exhibited better ferromagnetic behavior of Cu2O–CuFe2O4 as compared to CuFe2O4. The material having magnetic features could be easily separated by external applied magnetic field for possible reuse. CuFe2O4 nanoparticles synthesized by Phuruangrat et al. through microwave-hydrothermal method showed a maximum saturation magnetization of about 56 emu g−1.35 Higher saturation magnetization values of Cu2O–CuFe2O4 observed in this study could be attributed to the presence of additional Cu2O in CuFe2O4 particles. Previous study about the magnetic property of Cu2O has confirmed that Cu2O could act as a diamagnetic to ferromagnetic depending upon the role of native defects in bulk Cu2O.36
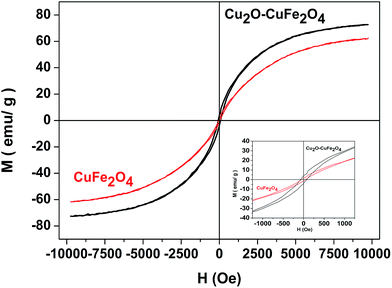 |
| | Fig. 4 Hysteresis curve for Cu2O–CuFe2O4 and CuFe2O4. | |
3.2 Heterogeneous Fenton performance of Cu2O–CuFe2O4
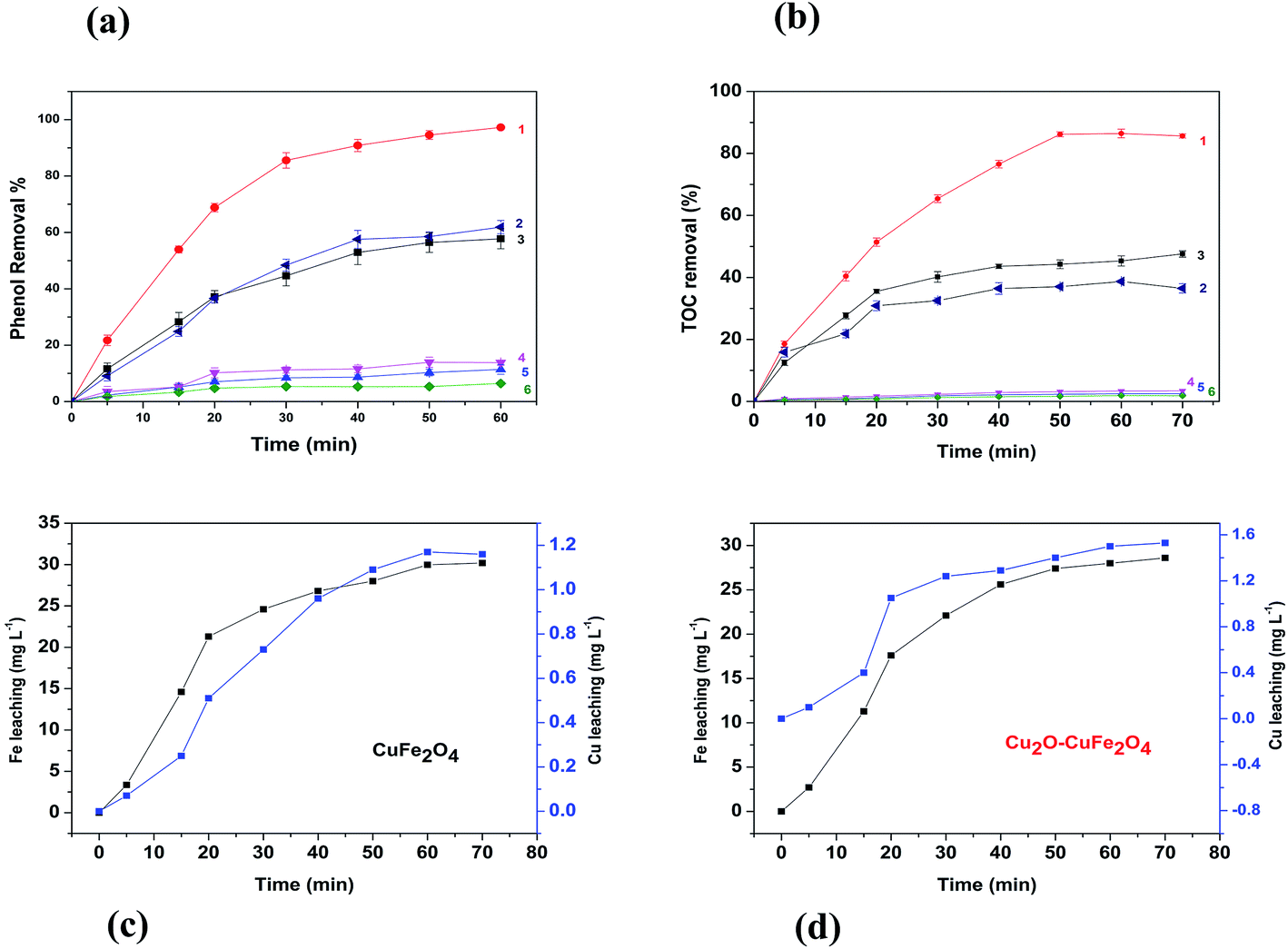
Heterogeneous Fenton performance of the catalyst prepared was mainly dependent on the potential to generate hydroxyl radical (OH˙) through H2O2 decomposition. As shown in Fig. 5(a), in the system with H2O2 alone but without CuFe2O4 or Cu2O–CuFe2O4, phenol removal efficiency within 60 min was as low as 6.4 ± 0.6%, indicating the poor oxidation ability of H2O2 alone toward phenol. In the control experiments without H2O2 but with CuFe2O4 and Cu2O–CuFe2O4, phenol removal efficiencies were 11.4 ± 1.7% and 13.8 ± 1.0%, respectively, which could be attributed to the adsorption of phenol by as-prepared CuFe2O4 and Cu2O–CuFe2O4. When CuFe2O4 and Cu2O were utilized in the presence of H2O2, phenol removal efficiency within 60 min was significantly increased to 57.8 ± 3.6% and 61.9 ± 2.3%, respectively, indicating the positive role of CuFe2O4 and Cu2O in phenol oxidation by H2O2. In case of Cu2O, slightly higher phenol removal efficiency as compared to CuFe2O4 could be attributed to more active site availability due to higher surface area and relatively more generation of HO˙ radical than HO2˙ as a result of monovalent copper Cu(I). Moreover, When Cu2O–CuFe2O4 was used as Fenton catalyst, phenol removal efficiency increased sharply within 30 min to 85.5 ± 2.8% and further increased to 97.2 ± 0.4% within 60 min. When Cu2O–CuFe2O4 was used as the catalyst for phenol oxidation by H2O2, a remarkable increase in terms of phenol removal indicated better catalysis performance of Cu2O–CuFe2O4 than CuFe2O4 and Cu2O.
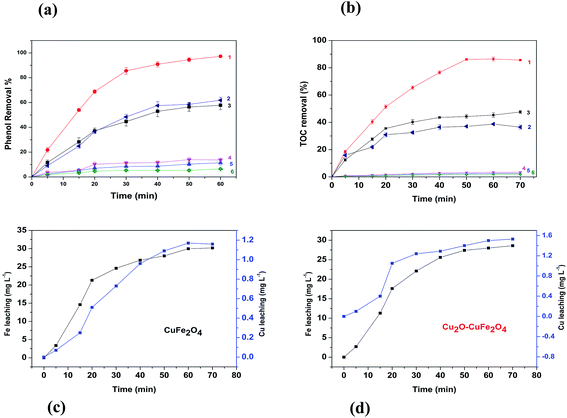 |
| | Fig. 5 Catalysts performance for phenol removal (a), TOC removal (b), iron and copper leaching for CuFe2O4 (c) and Cu2O–CuFe2O4 (d). (1): Cu2O–CuFe2O4 + H2O2; (2): Cu2O + H2O2 (3): CuFe2O4 + H2O2; (4): Cu2O–CuFe2O4 alone; (5) CuFe2O4 alone; (6) H2O2 alone. | |
Moreover, TOC removal observed in heterogeneous Fenton system followed the similar trend. As indicated in Fig. 5(b), in the Fenton system with H2O2 alone, with CuFe2O4 alone and with Cu2O–CuFe2O4 alone, TOC removal efficiencies within 60 min were as low as 1.8 ± 0.1%, 2.5 ± 0.1%, 3.4 ± 0.1%, respectively. However, in Fenton system catalyzed by Cu2O, CuFe2O4, and Cu2O–CuFe2O4, TOC removal efficiencies within 60 min were as high as 36.5 ± 1.5%, 47.6 ± 1.0% and 85.6 ± 0.7%, respectively. Nevertheless, no remarkable increase of TOC removal efficiencies could be observed when reaction time prolonged to 70 min. TOC removal performance confirmed the amazing catalytic activity of CuFe2O4 and Cu2O–CuFe2O4 towards H2O2 oxidation, especially for Cu2O–CuFe2O4.
Fenton and Cu2+ coupled system has been studied for the enhanced mineralization of phenol, with phenol removal efficiency as high as 94% was achieved in Fenton-Cu2+ system at initial phenol concentration of 100 mg L−1.24,37 In addition, Stoia et al. prepared MnFe2O4 nanoparticles, which was used for the oxidative degradation of phenol at initial concentration of about 50 mg L−1.38 Phenol removal efficiency of 90% could be achieved at pH of 3.0–3.5 under the catalyst dosage of 3 g L−1. In this study, phenol removal efficiency as high as 97.2 ± 0.4% could be achieved within 60 min at the presence of Cu2O–CuFe2O4, even at initial phenol concentration as high as 250 mg L−1. The relatively higher catalytic role of Cu2O–CuFe2O4 obtained in this study could be attributed to the coexistence of both monovalent copper [Cu(I)] and divalent copper [Cu(II)] in the structure of Cu2O–CuFe2O4.
3.3 Sound stability and reusability of Cu2O–CuFe2O4
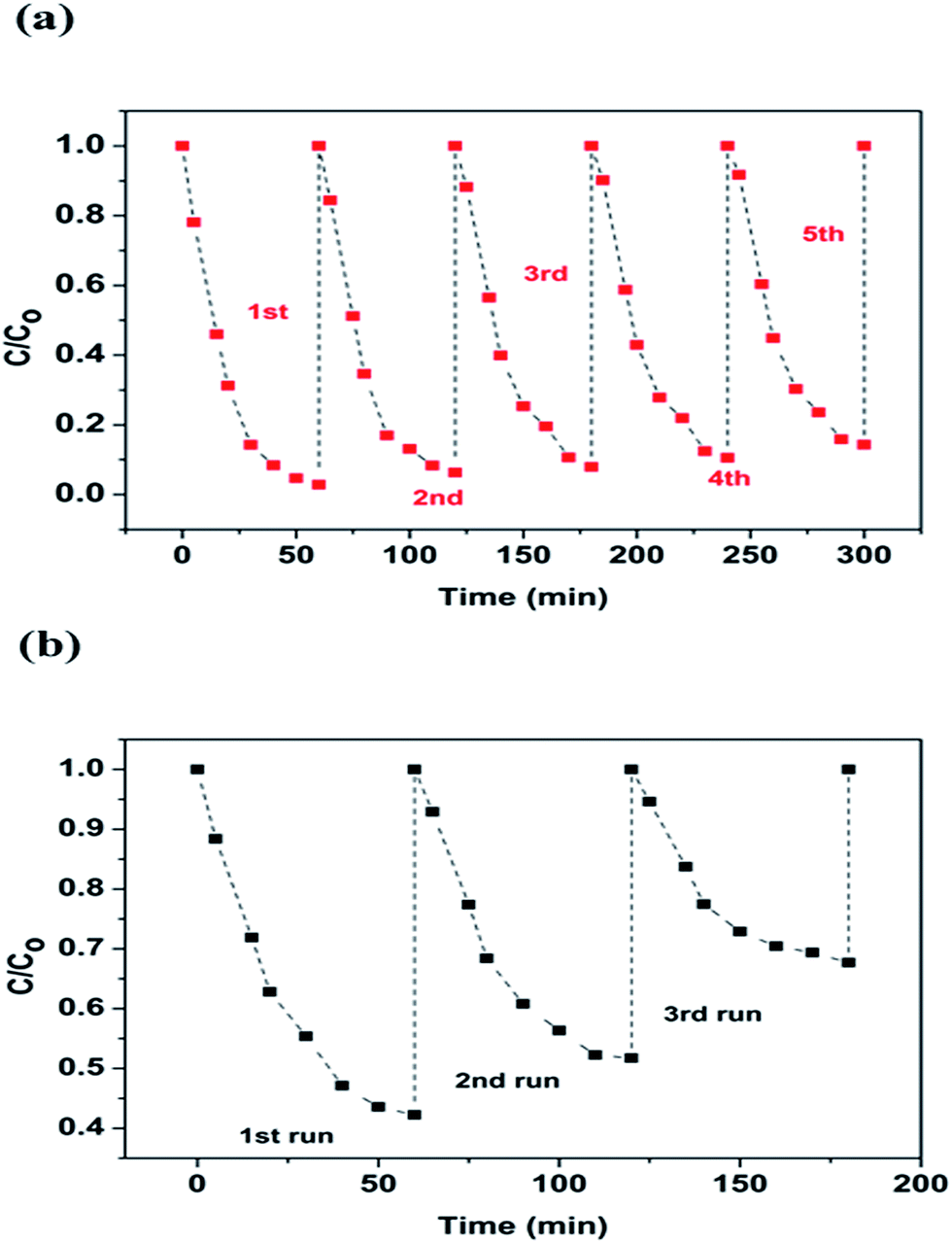
Leaching study for the as-prepared CuFe2O4 and Cu2O–CuFe2O4 was carried out to investigate their stability and reusability, as shown in Fig. 5(c) and (d). The concentration of leached iron at pH 4.0 for CuFe2O4 and Cu2O–CuFe2O4 was 30.18 mg L−1 and 28.60 mg L−1, accounted for 3.22% and 4.77% of the total iron contents at applied catalyst dosage of 2.0 g L−1. Moreover, for CuFe2O4 and Cu2O–CuFe2O4, the observed copper leaching was 1.16 mg L−1 and 1.53 mg L−1, accounted for 0.22% and 0.15% of total copper contents at applied catalyst dosage of 2.0 g L−1. Although the iron leaching for Cu2O–CuFe2O4 was a little higher than that of CuFe2O4, the observed copper leaching for Cu2O–CuFe2O4 was lower than that of CuFe2O4. Therefore, Cu2O–CuFe2O4 microparticles show good stability as compared to CuFe2O4 considering the low leaching of copper. In addition, these values for iron and copper leaching observed in this study were relatively lower than those reported in the literature. For example, the iron leaching accounted for 5.08% of the total iron contents at applied catalyst dosage of 0.24 g L−1 has been reported in literature, where ferrite particles were applied as heterogeneous Fenton catalyst for the degradation of Rhodamine B.39 Fontecha-Cámara et al. applied mixed iron oxide as Fenton catalyst in order to remove gallic acid from aqueous solutions.40 The observed copper leaching for copper ferrite was 2.85 mg L−1 at pH 4.3, which accounted for 21.50% of the total copper contents at applied catalyst dosage of 0.05 g L−1. As for the reusability, Cu2O–CuFe2O4 exhibited a slight decrease in phenol degradation efficiency (from 97.2 ± 0.4% to 85.7 ± 0.5%) within five consecutive runs, as shown in Fig. 6(a). However, in case of CuFe2O4, a sharp decrease of phenol removal efficiency (from 57.77 ± 3.55% to 32.32 ± 1.55%) was observed even after three cycles, as indicated in Fig. 6(b). The decrease in phenol degradation efficiency was due to vanishing of available active site on CuFe2O4 surface after reuse, which could be overcome through the incorporation of Cu2O in CuFe2O4 structure.
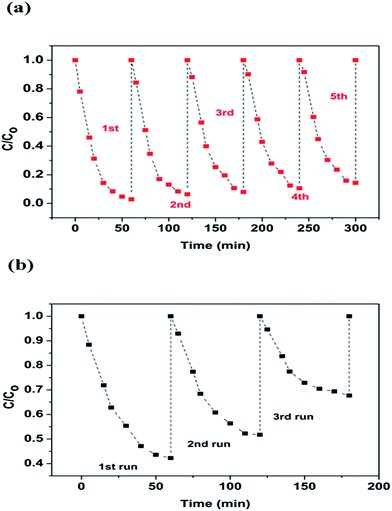 |
| | Fig. 6 Reusability study for Cu2O–CuFe2O4 (a) and CuFe2O4 (b). | |
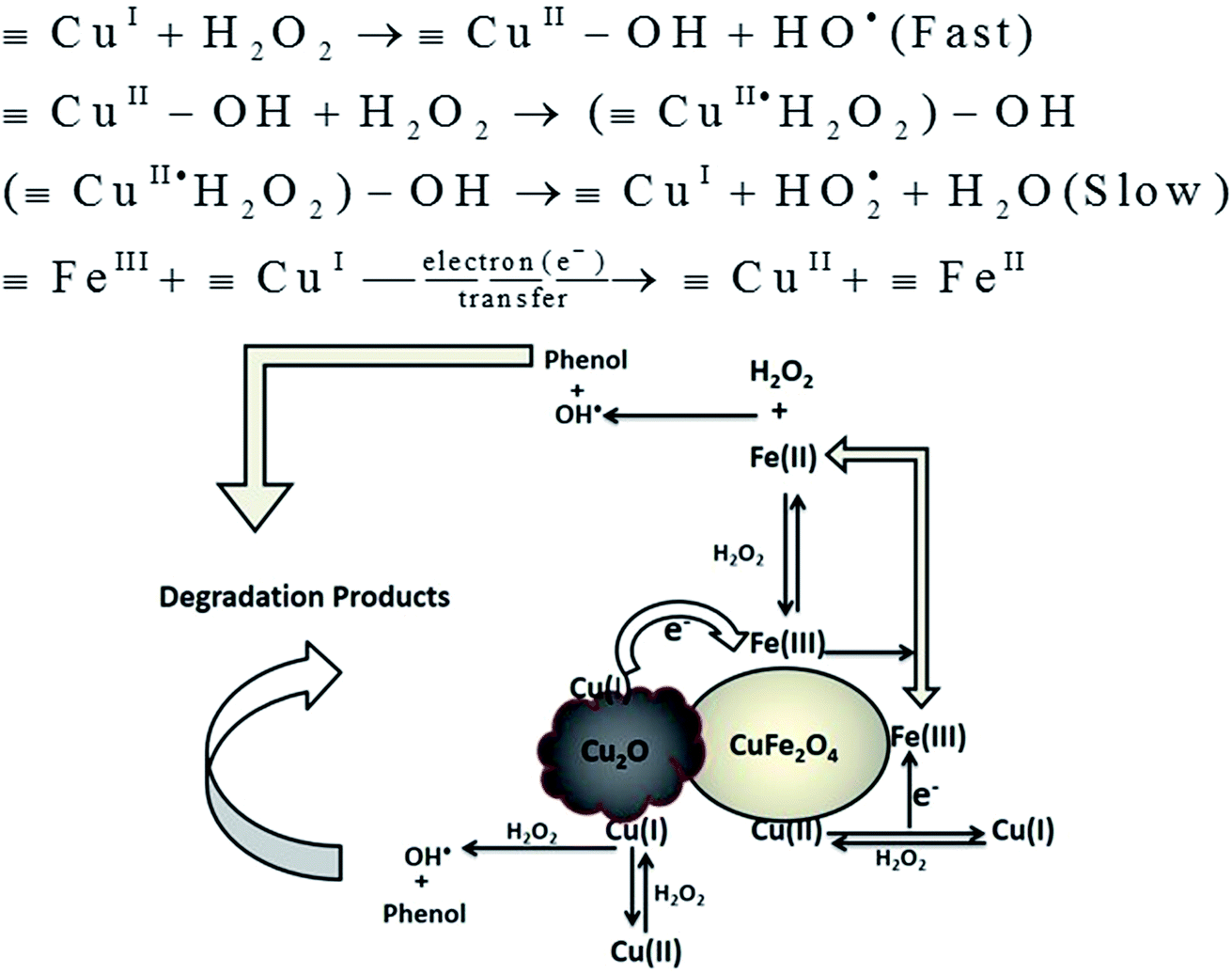
3.4 The possible mechanism involved in Fenton reaction by Cu2O–CuFe2O4
Based on the above results, the possible mechanism involved in Fenton reaction by Cu2O–CuFe2O4 could be described in Fig. 7. In our case, the available Fe(III) on both CuFe2O4 and Cu2O–CuFe2O4 surface was firstly reduced into Fe(II) due to its reaction with adsorbed H2O2 on the surface of both microparticles. The desired HO˙ radical was produced when the generated Fe(II) further reacted with H2O2 in a similar way, which was known as Haber–Weiss process.41 Literature study reveals that conversion of Fe(III) to Fe(II) was very slow when compared with the conversion of Fe(II) to Fe(III).42 The presence of transition metal copper in the skeleton of spinel catalyst may enhance phenol degradation due to availability of Cu(II)/Cu(I) redox pairs, which causes acceleration of the redox cycle of Fe(III)/Fe(II) ions at room temperature. Zhang et al. prepared CuFeO2 microparticles for the degradation of bisphenol A, confirming the effectiveness of Cu(I) species as compare to Cu(II) species for the activation of H2O2 to generate HO˙ radicals.26 As indicated in literature study, the reaction rate constant (k) between monovalent copper [Cu(I)] and H2O2 was as high as 1.0 × 104 M−1 S−1, as compared to that between divalent copper [Cu(II)] and H2O2, which was as low as 4.6 × 102 M−1 S−1.43 In our study, the generation of reactive HO˙ radical may start by ligand displacement between the hydrous surface of FeIII–OH/CuII–OH and H2O2, with the generation of FeIII·H2O2 and CuII·H2O2. Then initially generated FeIII·H2O2 and CuII·H2O2 species can produce HO2˙ and regenerate FeII and CuI by intramolecular electron transfer, which subsequently produced HO˙ and degraded phenol. The hydroxyl radicals HO˙ were considered as major oxidizing species for phenol mineralization and HO2˙ radicals as minor oxidizing species. The generation route for major and minor oxidizing species, and proposed mechanism for phenol degradation could be described by eqn (6)–(15):| |
(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) FeIII–OH) + H2O2 → (FeIII–OH)·(H2O2) FeIII–OH) + H2O2 → (FeIII–OH)·(H2O2)
| (6) |
| | |
(FeIII·H2O2)–OH → FeII + HO2˙ + H2O
| (7) |
| | |
FeII + H2O2 → FeIII–OH + HO˙
| (8) |
| | |
FeIII–OH + HO2˙ → FeII + H2O + O2
| (9) |
| | |
FeIII + CuI → CuII + FeII
| (10) |
| | |
CuI + H2O2 → CuII–OH + HO˙
| (11) |
| | |
CuII–OH + H2O2 → (CuII·H2O2)–OH
| (12) |
| | |
(CuII·H2O2)–OH → CuI + HO2˙ + H2O
| (13) |
| | |
CuII–OH + HO2˙ → CuI + H2O + O2
| (14) |
| | |
Phenol + HO˙ → intermediates → degradation products
| (15) |
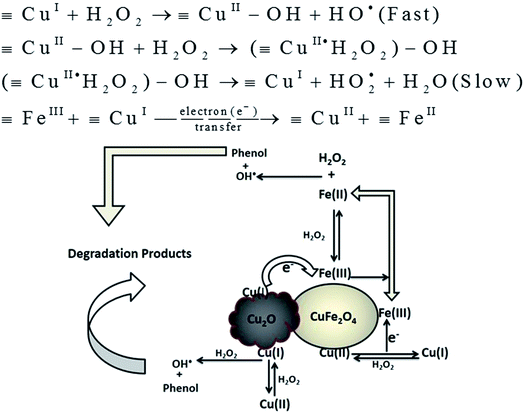 |
| | Fig. 7 Possible mechanisms for phenol degradation in Fenton system based on Cu2O–CuFe2O4. | |
The higher degradation efficiency of phenol for the as-prepared Cu2O–CuFe2O4 could be attributed to the presence of both monovalent copper [Cu(I)] and divalent copper [Cu(II)]. The existence of redox pairs Cu(I)/Cu(II) is likely considered as the key source for the activation of H2O2 in order to generate HO˙ radicals. The coexistence of Cu(I) and Cu(II) in Cu2O–CuFe2O4 resulted in faster reaction with H2O2 as compared to CuFe2O4 microparticles containing divalent copper [Cu(II)] alone in the structure. The faster reaction might be due to rapid electron (e−) transfer or formation of galvanic cell or electron bridge between [Cu(I)] and Fe(III) along with the production of highly reactive radical HO˙ as compared to HO2˙ species. For the mineralization of carbamazepine, Ding et al. prepared recyclable CuFeO2 microparticles for heterogeneous activation of peroxymonosulfate (PMS) in order to generate sulfate radicals (SO4˙−). It was concluded that enhanced activation of PMS by micro-CuFeO2 referred to synergetic effect of surface Cu(I) and Fe(III).44 The higher Fenton activity by Cu2O–CuFe2O4 might also be attributed to faster reaction rate between Cu(I) and H2O2 due to presence of additional Cu2O particles, which results in faster reduction of H2O2 in order to generate highly reactive radical species HO˙. The faster reduction of H2O2 due to presence of Cu2O additional particles in case of Cu2O–CuFe2O4 microparticles causes the relatively efficient consumption of H2O2 due to more available and highly reactive Cu(I) species.
4. Conclusion
The present work focus on the synthesis of a heterogeneous Fenton catalyst, namely Cu2O–CuFe2O4, for the reuse of iron incorporated in Fenton sludge. In comparison to CuFe2O4, much higher phenol catalytic degradation was found for Cu2O–CuFe2O4, indicating the key role of Cu2O in Fenton reaction. The rapid electron transfer built up galvanic cell between Cu(I) and Fe(III), which favored the formation of relatively excess Fe(II) species. The highly reactive Fe(II) species interaction with adsorbed H2O2 gave abundant HO˙ radicals for phenol degradation. The higher Fenton catalytic activity of Cu2O–CuFe2O4 could be attributed to the synergetic effect between Cu(I)/Cu(II) and Fe(II)/Fe(III) redox pairs. The as prepared Cu2O–CuFe2O4 was stable, recoverable and reusable, offering a promising potential as a heterogeneous Fenton catalyst.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This research is financed by Natural Science Foundation of Jiangsu Province for Distinguished Young Scholars (BK20170038), National Natural Science Foundation of China (51708293) and Natural Science Foundation of Jiangsu Province (BK20170842).
References
- J. Xu, Y. Long, D. Shen, H. Feng and T. Chen, J. Hazard. Mater., 2017, 323, 674–680 CrossRef CAS PubMed.
- D. Gümüş and F. Akbal, Process Saf. Environ. Prot., 2016, 103, 252–258 CrossRef.
- J. He, X. Yang, B. Men and D. Wang, J. Environ. Sci., 2016, 39, 97–109 CrossRef PubMed.
- W. M. Wang, J. Song and X. Han, J. Hazard. Mater., 2013, 262, 412–419 CrossRef CAS PubMed.
- E. Mousset, L. Frunzo, G. Esposito, E. D. van Hullebusch, N. Oturan and M. A. Oturan, Appl. Catal., B, 2016, 180, 189–198 CrossRef CAS.
- X. J. Ma and H. L. Xia, J. Hazard. Mater., 2009, 162, 386–390 CrossRef CAS PubMed.
- Y. Li, Y. Zhang, J. Li and X. Zheng, Environ. Pollut., 2011, 159, 3744–3749 CrossRef CAS PubMed.
- J. Feng, X. Hu, P. L. Yue and S. Qiao, Sep. Purif. Technol., 2009, 67, 213–217 CrossRef CAS.
- E. Garrido-Ramírez, B. Theng and M. Mora, Appl. Clay Sci., 2010, 47, 182–192 CrossRef.
- M. Tekbaş, H. C. Yatmaz and N. Bektaş, Microporous Mesoporous Mater., 2008, 115, 594–602 CrossRef.
- M. Aleksić, H. Kušić, N. Koprivanac, D. Leszczynska and A. L. Božić, Desalination, 2010, 257, 22–29 CrossRef.
- W. Wang, M. Zhou, Q. Mao, J. Yue and X. Wang, Catal. Commun., 2010, 11, 937–941 CrossRef CAS.
- H.-C. Yoo, S.-H. Cho and S.-O. Ko, J. Environ. Sci. Health, Part A: Environ. Sci. Eng., 2001, 36, 39–48 CrossRef CAS.
- S. Guo, N. Yuan, G. Zhang and J. C. Yu, Microporous Mesoporous Mater., 2017, 238, 62–68 CrossRef CAS.
- H. Zhang, G. Xue, H. Chen and X. Li, Chemosphere, 2018, 191, 64–71 CrossRef CAS PubMed.
- R. C. Costa, M. d. F. F. Lelis, L. C. Oliveira, J. D. Fabris, J. D. Ardisson, R. R. Rios, C. N. Silva and R. M. Lago, Catal. Commun., 2003, 4, 525–529 CrossRef CAS.
- T. Giannakopoulou, L. Kompotiatis, A. Kontogeorgakos and G. Kordas, J. Magn. Magn. Mater., 2002, 246, 360–365 CrossRef CAS.
- M. Rashad and O. Fouad, Mater. Chem. Phys., 2005, 94, 365–370 CrossRef CAS.
- E. Hasmonay, J. Depeyrot, M. Sousa, F. Tourinho, J.-C. Bacri, R. Perzynski, Y. L. Raikher and I. Rosenman, J. Appl. Phys., 2000, 88, 6628–6635 CrossRef CAS.
- N. Rezlescu, N. Iftimie, E. Rezlescu, C. Doroftei and P. Popa, Sens. Actuators, B, 2006, 114, 427–432 CrossRef CAS.
- F. Zhang, C. Wei, Y. Hu and H. Wu, Sep. Purif. Technol., 2015, 156, 625–635 CrossRef CAS.
- Y. Ding, L. Zhu, S. Wang and H. Tang, Appl. Catal., B, 2013, 129, 153–162 CrossRef CAS.
- Y. Wang, H. Zhao and G. Zhao, Appl. Catal., B, 2015, 164, 396–406 CrossRef CAS.
- H. Zhang, J. Liu, C. Ou, Faheem, J. Shen, H. Yu, Z. Jiao, W. Han, X. Sun, J. Li and L. Wang, J. Environ. Sci., 2017, 53, 1–8 CrossRef PubMed.
- P. Roonasi and A. Y. Nezhad, Mater. Chem. Phys., 2016, 172, 143–149 CrossRef CAS.
- X. Zhang, Y. Ding, H. Tang, X. Han, L. Zhu and N. Wang, Chem. Eng. J., 2014, 236, 251–262 CrossRef CAS.
- K. Ramachandran, S. Chidambaram, B. Baskaran, A. Muthukumarasamy and G. M. Kumar, Mater. Lett., 2016, 175, 106–109 CrossRef CAS.
- F. Nemati, A. Elhampour, H. Farrokhi and M. B. Natanzi, Catal. Commun., 2015, 66, 15–20 CrossRef CAS.
- X. Qiu, M. Liu, K. Sunada, M. Miyauchi and K. Hashimoto, Chem. Commun., 2012, 48, 7365–7367 RSC.
- J. Du, J. Bao, X. Fu, C. Lu and S. H. Kim, Sep. Purif. Technol., 2016, 11, 145–152 CrossRef.
- X. Zhao, Y. Tan, F. Wu, H. Niu, Z. Tang, Y. Cai and J. P. Giesy, Sci. Total Environ., 2016, 571, 380–387 CrossRef CAS PubMed.
- L. Huang, F. Peng and F. S. Ohuchi, Surf. Sci., 2009, 603, 2825–2834 CrossRef CAS.
- Z. Ai, L. Zhang, S. Lee and W. Ho, J. Phys. Chem. C, 2009, 113, 20896–20902 CAS.
- C. D. Wagner and G. Muilenberg, Handbook of X-ray photoelectron spectroscopy, Perkin-Elmer, 1979 Search PubMed.
- A. Phuruangrat, B. Kuntalue, S. Thongtem and T. Thongtem, Mater. Lett., 2016, 167, 65–68 CrossRef CAS.
- X. Yu, X. Zhang, S. Wang and G. Feng, Curr. Appl. Phys., 2015, 15, 1303–1311 CrossRef.
- J. Maekawa, K. Mae and H. Nakagawa, J. Environ. Chem. Eng., 2014, 2, 1275–1280 CrossRef CAS.
- M. Stoia, C. Muntean and B. Militaru, J. Environ. Sci., 2016, 53, 269–277 CrossRef PubMed.
- T. R. Giraldi, C. C. Arruda, G. M. da Costa, E. Longo and C. Ribeiro, J. Sol–Gel Sci. Technol., 2009, 52, 299–303 CrossRef CAS.
- M. A. Fontecha-Cámara, C. Moreno-Castilla, M. V. López-Ramón and M. A. Álvarez, Appl. Catal., B, 2016, 196, 207–215 CrossRef.
- S.-S. Lin and M. D. Gurol, Environ. Sci. Technol., 1998, 32, 1417–1423 CrossRef CAS.
- K. Li, Y. Zhao, M. J. Janik, C. Song and X. Guo, Appl. Surf. Sci., 2017, 396, 1383–1392 CrossRef CAS.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- Y. Ding, H. Tang, S. Zhang, S. Wang and H. Tang, J. Hazard. Mater., 2016, 317, 686–694 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*