
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIminoxyl radicals vs. tert-butylperoxyl radical in competitive oxidative C–O coupling with β-dicarbonyl compounds. Oxime ether formation prevails over Kharasch peroxidation†
I. B. Krylov,
S. A. Paveliev,
N. S. Shumakova,
M. A. Syroeshkin,
B. N. Shelimov,
G. I. Nikishin and
A. O. Terent'ev *
*
N. D. Zelinsky Institute of Organic Chemistry of the Russian Academy of Sciences, 47 Leninsky Prosp., Moscow 119991, Russian Federation. E-mail: alterex@yandex.ru
First published on 5th February 2018
Abstract
Oxidative coupling of oxime and β-dicarbonyl compounds dominates in a β-dicarbonyl compound/oxime/Cu(II)/t-BuOOH system; in the absence of oxime, oxidative coupling of t-BuOOH and a β-dicarbonyl compound (Kharasch peroxidation) takes place. The proposed conditions for oxidative coupling of oximes with dicarbonyl compounds require only catalytic amounts of copper salt and t-BuOOH serves as a terminal oxidant. The C–O coupling reaction proceeds via the formation of tert-butoxyl, tert-butylperoxyl and iminoxyl radicals. Apparently, tert-butylperoxyl radicals oxidize oxime into iminoxyl radical faster than they react with β-dicarbonyl compounds forming the Kharasch peroxidation product. Iminoxyl radicals are responsible for the formation of the target C–O coupling products; the yields are up to 77%.
Introduction
The development of selective processes of C–H functionalization and oxidative coupling (cross-dehydrogenative coupling, CDC) is one of the most rapidly developing areas of modern organic chemistry.1–19 While cross-coupling reactions involving functional groups (–Hal, –B(OR)2, –SnR3, –ZnHal, MgHal, etc.) have already become classic methods for the formation of C–C and C–heteroatom bonds (heteroatom = S, N, O, P), cross-dehydrogenative coupling is a relatively new direction in the methodology of coupling, which meets such principles of green chemistry as atom-economy and step-economy.3 CDC allows one to form the target chemical bond between molecules owing to selective cleavage of C–H and heteroatom–H bonds in the reagents without carrying out prefunctionalization.4–19One of the most important problems in the realization of CDC is the selection of an oxidant that selectively cleaves certain C–H and heteroatom–H bonds. Stoichiometric or excess amounts of transition metal salts,8–12 hypervalent iodine compounds13–17 and DDQ20,21 are often used as oxidants. The use of such oxidants increases the waste formation and cost of the product thus limiting the scalability of synthetic process. Important fundamental task in the development of CDC methods is the changeover from stoichiometric reagents to catalytic systems based on accessible and environmentally friendly oxidants, such as molecular oxygen and peroxides.18,19 The use of low-cost and less-toxic transition metal salts of the 3d series as catalysts is preferable. An example of the implementation of these principles is the Cu(II)/t-BuOOH system, which was applied for the radical peroxidation of β-dicarbonyl compounds and their heteroanalogues through the formation of tert-butylperoxyl radicals (Kharasch reaction, Scheme 1, route A).22,23 In the present work, it has been shown that a selective cross-dehydrogenative C–O coupling of β-dicarbonyl compounds with oximes occurs in the oxime/Cu(II)/t-BuOOH system (Scheme 1, route B) with an almost complete suppression of the competitive peroxidation process.
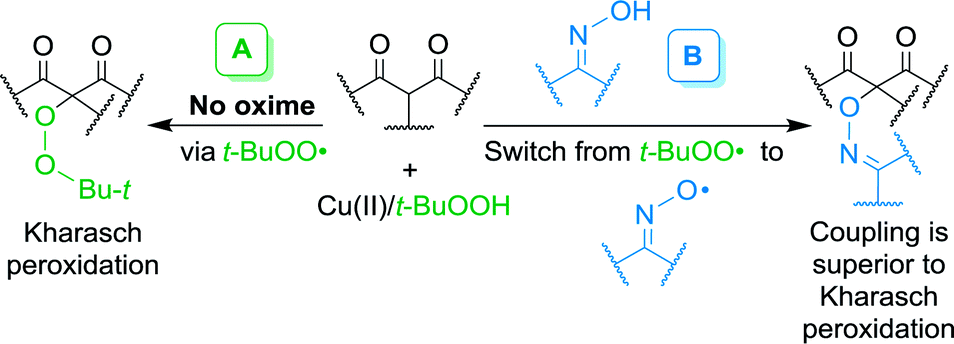 | ||
| Scheme 1 Kharasch peroxidation (A) and the discovered cross-dehydrogenative C–O coupling of β-dicarbonyl compounds with oximes (B). | ||
The proposed system is a convenient source of oxime radicals, which have found wide application in various processes of cyclization24–38 and C–O coupling recently.8,39 It should be noted that oxime radicals remain one of the least studied class of N-oxyl radicals, whereas amine-N-oxyl radicals and imide-N-oxyl radicals are used in numerous fields of chemistry, including oxidation methodology,40–42 living radical polymerization,40,43 spin-labeling,44,45 organic magnetic materials design.46
Oximes are difficult substrates for CDC, since under the action of oxidants they give a complex mixtures of products, including dimers of oxime radicals,47 or ketones.48 Previously, the oxidative C–O coupling of oximes with β-dicarbonyl compounds and their heteroanalogues was carried out only with the use of stoichiometric amounts of metal-containing oxidants (Scheme 2).8,9
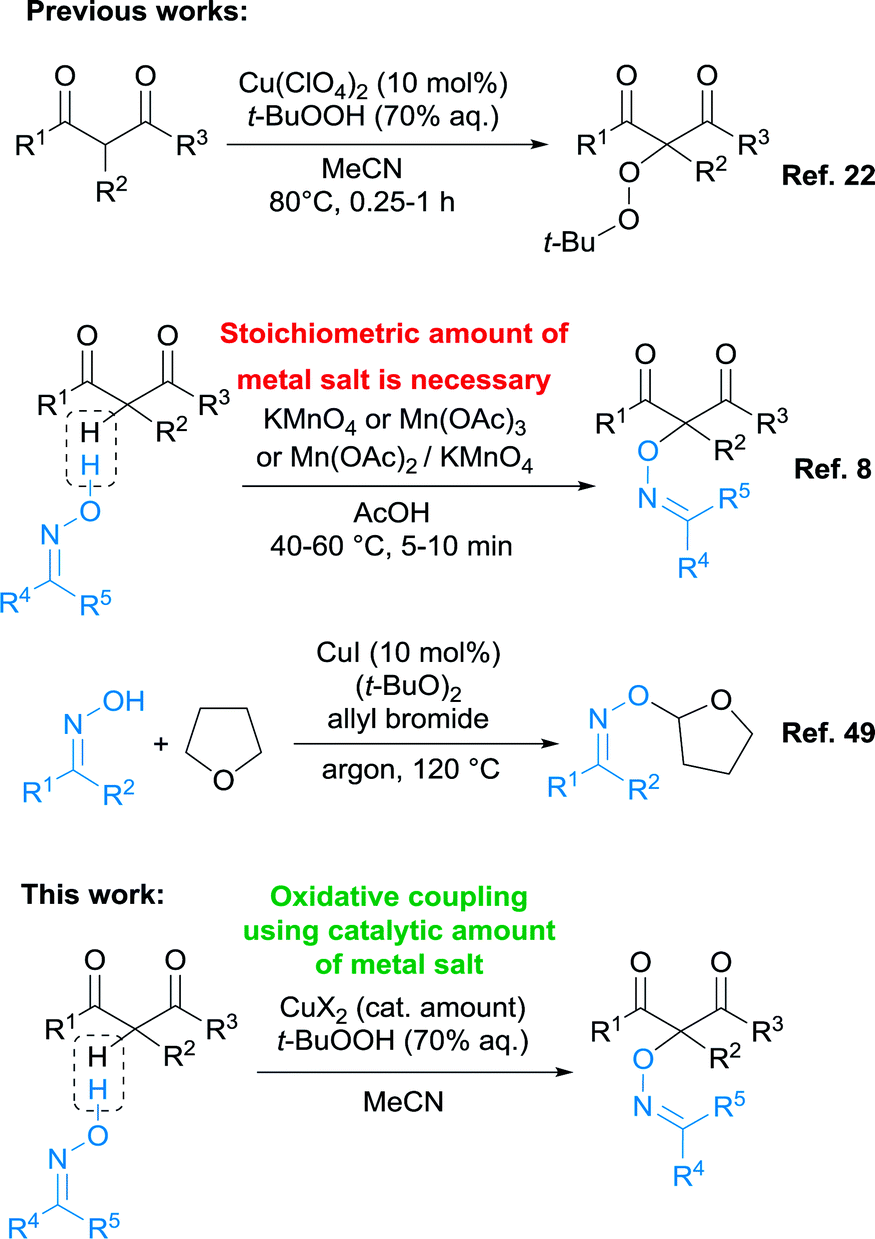 | ||
| Scheme 2 The present work is in the context of the development of a methodology for oxidative C–O coupling involving β-dicarbonyl compounds and oximes. | ||
CDC processes of oximes with isochromanes20 and derivatives of 1-phenylpropene21 in the presence of stoichiometric amounts of DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) were also reported. The only example of a catalytic cross-dehydrogenative coupling involving oximes is CuI-mediated CDC with THF in the presence of an excess of allyl bromide and di-tert-butyl peroxide as a oxidant (Scheme 2).49 The method is limited to the single structure of the CH-reagent (THF), which also plays the role of solvent.
The development of a convenient and green oxidative system for C–O coupling reactions involving oximes as O-reagents opens the short way to the synthesis of compounds containing a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–O fragment; such structures exhibit a wide range of biological activity and are valuable intermediates for organic synthesis and medical chemistry.50–52
N–O fragment; such structures exhibit a wide range of biological activity and are valuable intermediates for organic synthesis and medical chemistry.50–52
Results and discussion
In the model coupling reaction of ethyl 2-methylacetoacetate 1a with 3-(hydroxyimino)pentan-2,4-dione 2a Cu(I, II), Mn(II, III), Fe(III) and Ni(II) salts were tested as catalysts. Molecular oxygen, organic and inorganic peroxides were used as oxidants for the reaction (Table 1).
|
|||
|---|---|---|---|
| Entry | Metal salt | Oxidant (molar ratio: mol per mol of 1a) | Yield of 3ab (%) |
| a General reaction conditions: β-keto ester 1a (1 mmol), oxime 2a (1 mmol), metal salt (0.1 mmol, 10 mol%), oxidant (2–3 mmol), MeCN (5 mL) at 80 °C for 1 h.b Yield of isolated product.c MeCN–H2O (5 mL; v/v = 3/2) mixture was used as solvent.d AcOH (5 mL) was used as solvent.e β-keto ester 1a (1 mmol), oxime 2a (1.5 mmol), Mn(OAc)3·2H2O (0.1 mmol, 10 mol%), t-BuOOH (70% aq.) (3 mmol), MeCN (2.5 mL), 20–25 °C, 48 h. t-BuOOH was used as 70% aqueous solution. H2O2 was used as 34% aqueous solution. n.d. – not detected. | |||
| 1 | Cu(BF4)2·6H2O | O2 | 14 |
| 2 | Cu(BF4)2·6H2O | H2O2 (2) | 38 |
| 3c | Cu(BF4)2·6H2O | K2S2O8 (2) | 46 |
| 4 | Cu(BF4)2·6H2O | Oxone (2) | 28 |
| 5 | Cu(BF4)2·6H2O | t-BuOOH (3) | 64 |
| 6 | Cu(BF4)2·6H2O | (t-BuO)2 (2) | 39 |
| 7 | Cu(ClO4)2·6H2O | O2 | 24 |
| 8 | Cu(ClO4)2·6H2O | H2O2 (2) | 21 |
| 9c | Cu(ClO4)2·6H2O | K2S2O8 (2) | 54 |
| 10c | Cu(ClO4)2·6H2O | (NH4)2S2O8 (2) | 56 |
| 11 | Cu(ClO4)2·6H2O | t-BuOOH (3) | 51 |
| 12 | CuCl2 | t-BuOOH (3) | 58 |
| 13 | CuCl | t-BuOOH (3) | 40 |
| 14c | CuSO4·5H2O | K2S2O8 (2) | 43 |
| 15 | CuSO4·5H2O | t-BuOOH (3) | 11 |
| 16c | Cu(OAc)2 | (NH4)2S2O8 (2) | Trace |
| 17 | Cu(OAc)2 | t-BuOOH (3) | 27 |
| 18 | Cu(OTf)2 | t-BuOOH (3) | 50 |
| 19d | Mn(OAc)3·2H2O | t-BuOOH (3) | 26 |
| 20e | Mn(OAc)3·2H2O | t-BuOOH (3) | 20 |
| 21 | Mn(ClO4)2·6H2O | t-BuOOH (3) | n.d. |
| 22 | Fe(ClO4)3·11H2O | t-BuOOH (3) | 34 |
| 23 | Ni(OAc)2·4H2O | t-BuOOH (3) | n.d. |
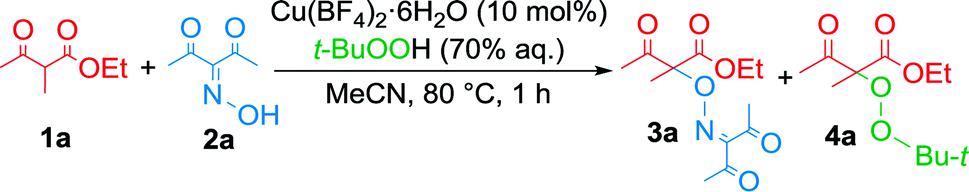
The best results were obtained with Cu(BF4)2·6H2O as catalyst (Table 1, entries 1–6). O2, H2O2 and Oxone were ineffective oxidants in combination with this salt, the yield of 3a did not exceed 40% (Table 1, entries 1, 2 and 4).
Possible reasons for the relatively low yields include oxidative cleavage of C–C bonds in the starting 1a,53–57 as well as hydroxylation of 1a at α-position.58–60 Satisfactory yield of 3a (46%) were observed with K2S2O8 (Table 1, entry 3).
The highest yield of 3a (up to 64%) was achieved using t-BuOOH (Table 1, entry 5). Di-tert-butyl peroxide did not show sufficient oxidizing capacity in this reaction (Table 1, entry 6), probably because its optimal temperature range of activation lies above the boiling point of acetonitrile.
Cu(ClO4)2·6H2O, CuCl2 and Cu(OTf)2 showed a moderate activity (Table 1, entries 7–12 and 18) that was comparable to the activity of Cu(BF4)2·6H2O. The other copper salts (Table 1, entries 13–17), as well as Mn(II), Mn(III), Fe(III) and Ni(II) salts (Table 1, entries 19–23) in combination with peroxydisulfates and t-BuOOH were not sufficiently effective. A combination of Mn(OAc)3·2H2O, t-BuOOH and MeCN was previously used to generate t-BuOO˙ radicals at rt.61 Using this combination 3a was obtained with yield of only 20% (entry 20) and conversion of β-keto ester 1a was about 27%.
It was found, that the product yields were substantially dependent on the molar ratio of the starting reagents 1a, 2a and t-BuOOH (Table 2). The yield of product 3a increased in the case of a 1.5-2-fold excess of the oxime 2a with respect to β-keto ester 1a (Table 2, entries 3–6). Presumably, the excess of oxime enhanced the trapping of the peroxyl radicals generated in the system of Cu(II) and t-BuOOH. When the reaction was carried out with an excess of β-keto ester 1a, in addition to the main product 3a, significant quantities of peroxidation by-product 4a were observed (Table 2, entries 7–9).
|
|||
|---|---|---|---|
| Entry | Molar ratio of 1a, 2a and t-BuOOH | Yield of 3ab (%) | Yield of 4ab (%) |
| a General reaction conditions: β-keto ester 1a (1–2 mmol), oxime 2a (1–3 mmol), Cu(BF4)2·6H2O (0.1 mmol, 10 mol%), t-BuOOH (70% aq.) (2–3 mmol), MeCN (5 mL) at 80 °C for 1 h.b Yields of 3a and 4a were determined based on 1H NMR using p-methoxyacetophenone as an internal standard; in the entries 2 and 3 isolated yields of 3a based on β-dicarbonyl compound are given in parenthesis. n.d. – not detected. | |||
| 1 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 :1:2 |
61 | Trace |
| 2 | 1:1:3 |
70 (64) | Trace |
| 3 | 1:1.5:2 |
81 (77) | n.d. |
| 4 | 1:2:2 |
76 | Trace |
| 5 | 1:3:2 |
60 | n.d. |
| 6 | 1:3:3 |
57 | Trace |
| 7 | 1.5:1:2 |
74 | 18 |
| 8 | 1.5:1:3 |
76 | 20 |
| 9 | 2:1:3 |
51 | 34 |
Thus, the optimal reaction conditions under which the yield of the target product 3a reached 77%, are the molar ratio of β-keto ester 1a:oxime 2a:t-BuOOH = 1:1.5:2 in the presence of 10 mol% of Cu(BF4)2·6H2O in MeCN at 80 °C (Table 2, entry 3). These conditions were used for coupling of a number of β-dicarbonyl compounds with various oximes (Table 3).
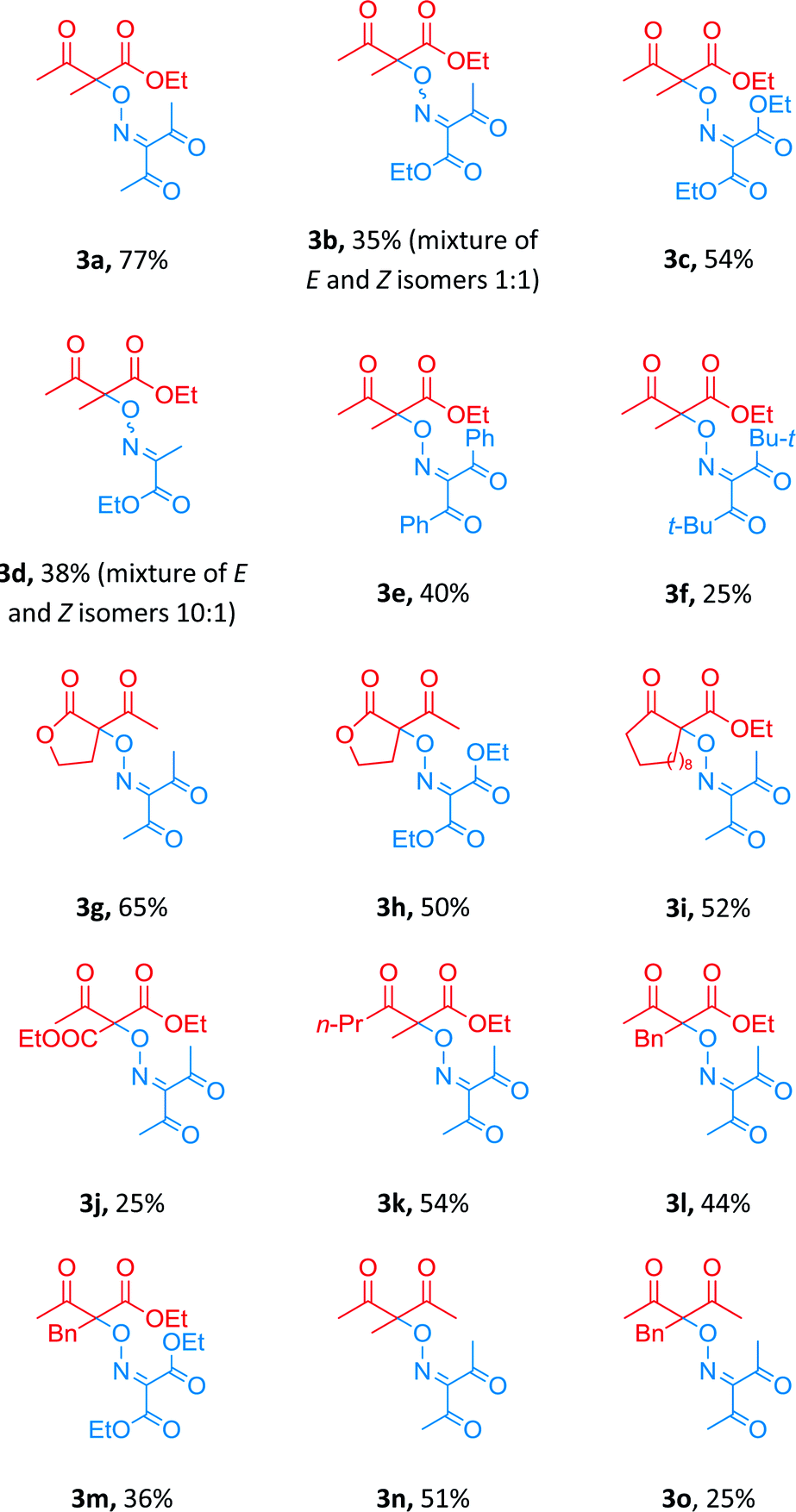
The cross-coupling reaction proceeded with oximes bearing one (compound 3d, yield 38%) or two (3a–c, 3e–o, yields 25–77%) electron-withdrawing ester- or keto-groups.
Diacetyloxime 1a (product 3a in comparison with 3b–f) proved to be the most effective OH-substrate in the coupling reaction, probably owing to increased stability of corresponding iminoxyl radical.39 Coupling with β-diketones proceeded with yields lower than those in the case of β-keto esters (products 3n and 3o in comparison with 3a and 3l).
Proposed reaction mechanism
Based on the literature data the mechanism of cross-dehydrogenative coupling of β-dicarbonyl compounds with oximes under the action of Cu(II)/t-BuOOH system was proposed. It consists of three key parts: formation of alkoxyl and peroxyl radicals I and II derived from t-BuOOH in the presence of Cu2+,22,23,62–65 oxidation of oxime III leading to the radical IV (ref. 8 and 47) and coupling of β-dicarbonyl compound V with radicals I and IV giving products VII and VIII (Scheme 3).22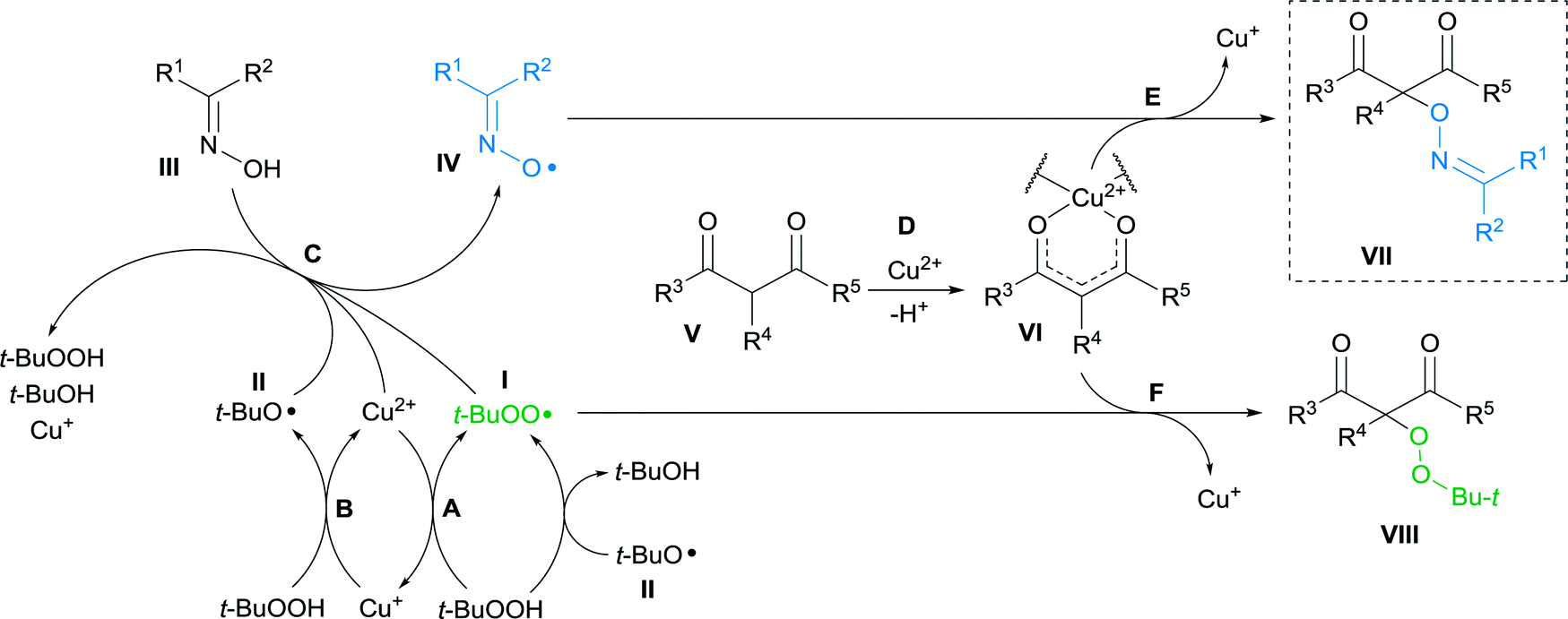 | ||
| Scheme 3 Proposed mechanism of oxidative C–O coupling of β-dicarbonyl compounds with radicals generated in the oxime/Cu(II)/t-BuOOH system. | ||
In stage A, tert-butylhydroperoxide interacts with Cu2+ to form Cu+ and tert-butylperoxyl radical I.62–65 t-BuOOH oxidizes Cu+ with the formation of Cu2+ and tert-butoxyl radical II (stage B).22,23,62–65 Furthermore, generation of tert-butylperoxyl radical I can occur via abstraction of hydrogen atom from t-BuOOH by t-BuO˙ radical II.63,65,66
Generation of iminoxyl radical IV (stage C) can occur via oxidation of oxime III with Cu2+,8 as well as with radicals I and II formed from t-BuOOH.47,67 At stage D, β-dicarbonyl compound V forms a Cu2+ chelate complex VI,22,68 which reacts with iminoxyl radical IV to form the final coupling product VII (stage E).
Formation of peroxidation byproduct VIII takes place in stage F by the interaction of complex VI with tert-butylperoxyl radical I.22 In the absence of oxime, peroxidation products 4a,b were obtained with moderate yields (Scheme 4).
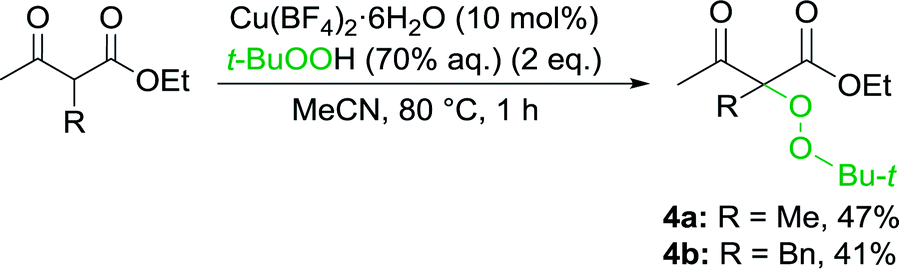 | ||
| Scheme 4 Reaction of β-keto esters with Cu(II)/t-BuOOH (in the absence of oxime) with the formation of peroxidation products 4a,b. | ||
Redox properties of oxime 2a in the presence of Cu(BF4)2 were studied by cyclic voltammetry (Fig. 1). In the absence of copper, only a slight reversible peak of oxidation of oxime into the radical was observed at 0.32 V. When copper salt was added, oxime oxidation peak increased significantly, and the peak of Cu+/Cu2+ oxidation appeared in the more anodic region (0.97 V).
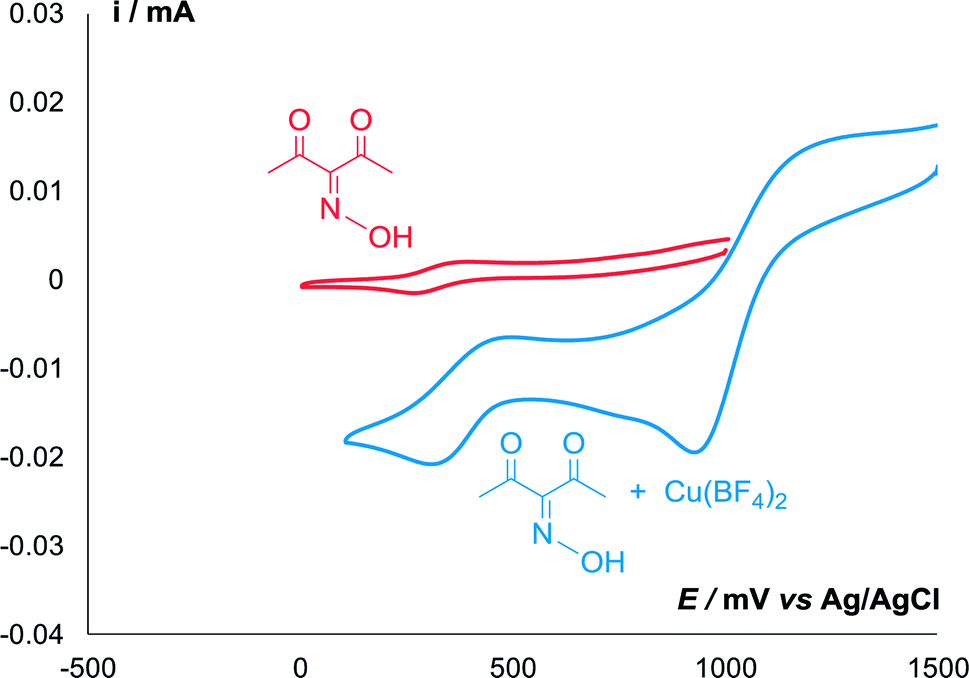 | ||
| Fig. 1 CV curves of 5 mmol L−1 of oxime 2a in the presence (blue) and absence (red) of equimolar amount of Cu(BF4)2 in 0.1 M n-Bu4NBF4/MeCN at a scan rate of 0.1 V s−1 at 298 K. | ||
Thus, Cu(BF4)2 is a strong oxidizing agent for oxidation of oxime 2a to iminoxyl radical IV, and it also can serve as an effective mediator for the electrochemical generation of iminoxyl radicals.
Conversion of oxime 2a into radical IV under the action of Cu(BF4)2 and t-BuOOH separately or in combination was studied by EPR spectroscopy (Fig. 2). The concentrations of reagents were the same as in the general conditions of the C–O coupling (Table 3). The highest amount of iminoxyl radical was formed in the presence of Cu(BF4)2 (Run 1). Mixing the reagents at room temperature led to a rapid change in color from almost colorless to the red which is typical of radical IV.39 The concentration of IV was estimated using a 0.002 M solution of 4-benzoyloxy-2,2,6,6-tetramethylpiperidine 1-oxyl in MeCN as external standard. The concentration of IV according to EPR did not varied in time interval of 5–60 min after the beginning of the reaction. The estimated amount of iminoxyl radical IV was 0.05 mmol which corresponds to a 25% conversion of Cu(BF4)2 (reduction of Cu2+ to Cu+).
 | ||
| Fig. 2 EPR monitoring of formation of iminoxyl radical IV from oxime 2a under action of Cu(BF4)2 and t-BuOOH. | ||
Surprisingly, the amount of iminoxyl radical formed in the presence of both Cu(BF4)2 and t-BuOOH (Run 2, ca. 0.015 mmol of IV) was lower than in the presence of Cu(BF4)2 alone (Run 1, ca. 0.05 mmol of IV). Apparently, addition of t-BuOOH 70% aq. affected the coordination environment of Cu2+ ions, which caused a decrease in Cu2+ oxidative potential, which was confirmed by the CV method (Fig. 3). Addition of t-BuOOH 70% aq. to a solution of the oxime 2a and Cu(BF4)2 leads to a negative shift of the copper reduction peak by 170 mV.
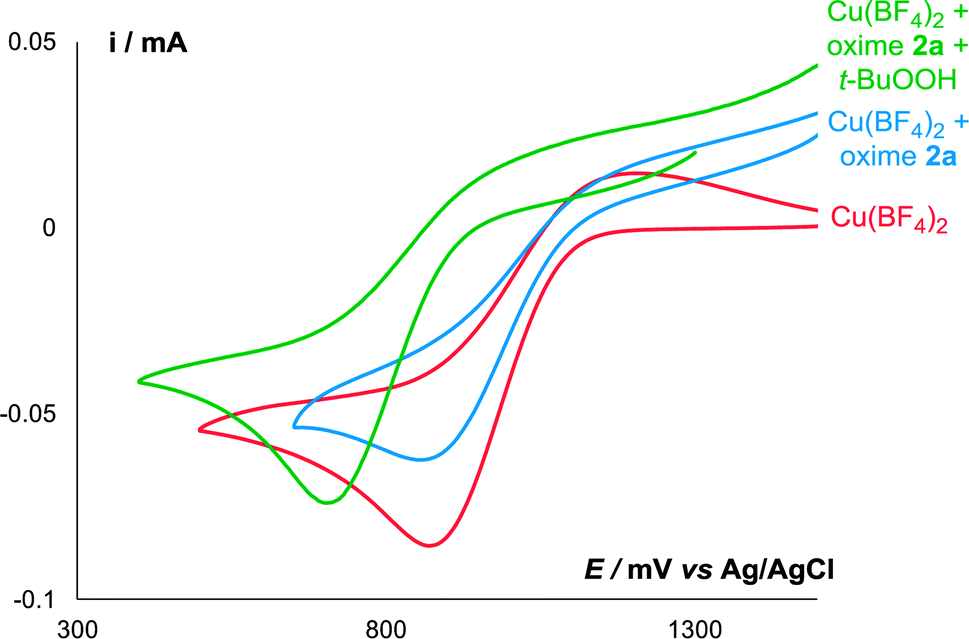 | ||
| Fig. 3 CV curves of Cu(BF4)2 (red), Cu(BF4)2 in the presence of oxime 2a (blue), and Cu(BF4)2 in the presence of oxime 2a and t-BuOOH (70% aq.) (green) in 0.1 M n-Bu4NBF4/MeCN at a scan rate of 0.1 V s−1 at 298 K. | ||
Reoxidation of Cu+ to Cu2+ by t-BuOOH is supposed to be rather slow at room temperature, and therefore EPR experiment with Cu(BF4)2/t-BuOOH system at 80 °C was performed (Fig. 2, Run 3). In this case the concentration of IV decreased significantly, presumably, due to decomposition.69–74 In the presence of t-BuOOH without Cu(BF4)2, iminoxyl radicals IV formed very slowly at 80 °C; maximum concentration was observed after 60 min (Run 4, ca. 0.003 mmol of IV). The formation of IV in this run can be explained by the interaction of oxime 2a with radicals formed by a homolytic cleavage of O–O bond in t-BuOOH.
The results of EPR monitoring and CV experiments indicate a complex character of the interaction between oxime, Cu(BF4)2 and t-BuOOH, but iminoxyl radical IV is the dominating O-centered radical in all cases.
Conclusions
In the reaction system containing β-dicarbonyl compounds, iminoxyl and peroxyl radicals, the reaction conditions were found which allow to synthesize C–O coupling products with iminoxyl radicals preferably over peroxyl radicals. It was shown that the system consisting of oxime, Cu(II) and t-BuOOH is a convenient source of iminoxyl radicals. The first cross-dehydrogenative C–O coupling of β-dicarbonyl compounds with α-keto oximes was accomplished using the proposed system with catalytic amounts of metal-containing oxidant. Kharasch peroxidation of β-dicarbonyl compounds with tert-butylperoxyl radicals was observed in the absence of oxime. Apparently, oximes are effective scavengers of peroxyl radicals in the discovered process, which leads to a change of the process route.It appears that the ability of oximes to scavenge O-centered radicals found in this work opens the new way for their application as reagents for C–O coupling, inhibitors of radical processes and test reagents for confirmation of the radical mechanism of various reactions.
Experimental
Experimental for Table 1
2-Methylacetoacetate 1a (144 mg, 1 mmol), 3-(hydroxyimino)pentan-2,4-dione 2a (129 mg, 1 mmol), MeCN (5 mL), and metal salt (10–55 mg, 0.1 mmol) were successively loaded into a round bottom flask. The mixture was heated on oil bath (80 °C) with stirring by a magnetic bar and an oxidant (200–615 mg, 2–3 mmol) was added for 10 seconds; stirring was continued for 1 h at 80 °C. In the entries 1 and 7 oxygen gas was bubbled through the reaction mixture (0.3 mL s−1) until the end of the synthesis; entries 2–6, 8–23 were carried out in air atmosphere. In the entry 20 mixture of β-keto ester 1a (144 mg, 1 mmol), oxime 2a (194 mg, 1.5 mmol), Mn(OAc)3·2H2O 95% (28 mg, 0.1 mmol, 10 mol%), t-BuOOH 70% aq. (386 mg, 3 mmol) and MeCN (2.5 mL) was stirred for 48 h at 20–25 °C.Reaction mixture was cooled to the room temperature, diluted with CH2Cl2 (10 mL) and water (30 mL) and shaken. Organic layer was separated and aqueous layer was extracted with CH2Cl2 (3 × 10 mL). All the organic extracts were combined, washed with aqueous solution of Na2S2O4 (200 mg in 20 mL of water), then with water (20 mL), dried over Na2SO4, rotary evaporated at 40–60 °C under water-jet vacuum (20–30 mmHg). C–O coupling product 3a was isolated by column chromatography on silica gel using CH2Cl2/EtOAc eluent with volume part of EtOAc 2.5%.
Experimental for Table 2
2-Methylacetoacetate 1a (144–288 mg, 1–2 mmol), 3-(hydroxyimino)pentan-2,4-dione 2a (129–387 mg, 1–3 mmol), MeCN (5 mL), and Cu(BF4)2·6H2O (35 mg, 0.1 mmol) were successively loaded into a round bottom flask. The mixture was heated on oil bath (80 °C) with stirring by a magnetic bar and t-BuOOH (70% aqueous solution, 257–386 mg, 2–3 mmol) was added for 10 seconds; stirring was continued for 1 h at 80 °C.Reaction mixture was cooled to the room temperature, diluted with CH2Cl2 (10 mL) and water (30 mL) and shaken. Organic layer was separated and aqueous layer was extracted with CH2Cl2 (3 × 10 mL). All the organic extracts were combined, washed with aqueous solution of Na2S2O4 (200 mg in 20 mL of water), then with water (20 mL), dried over Na2SO4, rotary evaporated at 40–60 °C under water-jet vacuum (20–30 mm Hg). Yields of 3a and 4a were determined by 1H NMR using p-methoxyacetophenone as an internal standard. In the entries 2 and 3 product 3a was isolated by column chromatography on silica gel using CH2Cl2/EtOAc eluent with volume part of EtOAc 2.5% (isolated yields are given in parenthesis).
Experimental for Table 3
β-Dicarbonyl compound 1a–h (114–254 mg, 1 mmol), oxime 2a–f (194–380 mg, 1.5 mmol), MeCN (5 mL), and Cu(BF4)2·6H2O (35 mg, 0.1 mmol) were successively loaded into a round bottom flask. The mixture was heated on oil bath (80 °C) with stirring by a magnetic bar and t-BuOOH (70% aqueous solution, 257 mg, 2 mmol) was added for 10 seconds; stirring was continued for 1 h at 80 °C.Reaction mixture was cooled to the room temperature, diluted with CH2Cl2 (10 mL) and water (30 mL) and shaken. Organic layer was separated and aqueous layer was extracted with CH2Cl2 (3 × 10 mL). All the organic extracts were combined, washed with aqueous solution of Na2S2O4 (200 mg in 20 mL of water), then with water (20 mL), dried over Na2SO4, rotary evaporated at 40–60 °C under water-jet vacuum (20–30 mmHg). C–O coupling product 3a–o was isolated by column chromatography on silica gel using CH2Cl2/EtOAc eluent.
Experimental for Scheme 4
To a stirred at 80 °C mixture of β-keto ester 1a,f (144–220 mg, 1 mmol), Cu(BF4)2·6H2O (35 mg, 0.1 mmol) and MeCN (5 mL) t-BuOOH (70% aqueous solution, 257 mg, 2 mmol) was added for 10 seconds; stirring was continued at 80 °C for 1 h.Reaction mixture was cooled to the room temperature, diluted with CH2Cl2 (10 mL) and water (30 mL) and shaken. Organic layer was separated and aqueous layer was extracted with CH2Cl2 (3 × 10 mL). All the organic extracts were combined, washed with aqueous solution of Na2S2O4 (200 mg in 20 mL of water), then with water (20 mL), dried over Na2SO4, rotary evaporated at 40–60 °C under water-jet vacuum (20–30 mmHg). Peroxidation products 4a,b were isolated by column chromatography on silica gel using CH2Cl2/EtOAc eluent with volume part of EtOAc 3%.
Experimental for Fig. 1
Oxime 2a (3.22 mg, 0.025 mmol) and 0.1 M n-Bu4NBF4/MeCN (2.5 mL) were placed into electrochemical cell, the resultant solution was stirred by bubbling with argon at 25 °C. Then a solution of Cu(BF4)2·6H2O (8.62 mg, 0.025 mmol) in 0.1 M n-Bu4NBF4/MeCN (2.5 mL) was added. The bubbling was stopped after 15 min and voltammetry curve was recorded.Experimental for Fig. 2
EPR spectra were recorded at 18–22 °C using following parameters: microwave frequency — ≈9.6 GHz, central field — 3340 G, hf (100 kHz) field modulation amplitude — 1.0 G, microwave power — 31 mW, scan range — 300 G, receiver gain — 1.25 × 104. Sample solutions in 100 μL glass capillaries (inner diameter 1.2 mm) were placed into the EPR cavity. In all runs, EPR spectra were recorded after 5, 30 and 60 min of reaction. In runs 3 and 4 conducted at 80 °C, samples were taken from the reaction mixture at given times (5, 30 and 60 min), allowed to cool to 18–22 °C in air (about 5 min) and then EPR spectra were recorded. In all cases, EPR signal of diacetyliminoxyl radical IV (g = 2.0044, aN = 28.1 G)8 was observed. The amount of radical IV formed in the reaction was estimated by double integration of its EPR spectrum. A 0.002 M solution of 4-benzoyloxy-2,2,6,6-tetramethylpiperidine 1-oxyl (4-BzO-TEMPO) in MeCN was used as external concentration standard. Detailed procedures for runs 1–4 are given below. Run 1: to a stirred at 18–22 °C solution of oxime 2a (387.3 mg, 3 mmol) in MeCN (5 mL) a solution of Cu(BF4)2·6H2O (69.0 mg, 0.2 mmol) in MeCN (5 mL) was added. Run 2: to a stirred at 18–22 °C solution of oxime 2a (387.3 mg, 3 mmol) in MeCN (5 mL) a solution of Cu(BF4)2·6H2O (69.0 mg, 0.2 mmol) in MeCN (5 mL) was added, then t-BuOOH (70% aq.) (515 mg, 4 mmol) was added. Run 3: to a stirred at 18–22 °C solution of oxime 2a (387.3 mg, 3 mmol) in MeCN (5 mL) a solution of Cu(BF4)2·6H2O (69.0 mg, 0.2 mmol) in MeCN (5 mL) was added, then t-BuOOH (70% aq.) (515 mg, 4 mmol) was added. Then the obtained solution was stirred on the oil bath (80 °C). Run 4: to a stirred at 18–22 °C solution of oxime 2a (387.3 mg, 3 mmol) in MeCN (5 mL) a solution of t-BuOOH (70% aq.) (515 mg, 4 mmol) in MeCN (5 mL) was added. Then the obtained solution was stirred on the oil bath (80 °C).Experimental for Fig. 3
Three solutions were studied by CV with scan rate 100 mV s−1. Solution of Cu(BF4)2: Cu(BF4)2·6H2O (34.5 mg, 0.1 mmol) was dissolved in 0.1 M n-Bu4NBF4/MeCN (5 mL). Solution of Cu(BF4)2 + oxime 2a: Cu(BF4)2·6H2O (34.5 mg, 0.1 mmol) was dissolved in 0.1 M n-Bu4NBF4/MeCN (5 mL), then oxime 2a (193.7 mg, 1.5 mmol) was added. Solution of Cu(BF4)2 + oxime 2a + t-BuOOH: Cu(BF4)2·6H2O (34.5 mg, 0.1 mmol) was dissolved in 0.1 M n-Bu4NBF4/MeCN (5 mL), then oxime 2a (194 mg, 1.5 mmol) and t-BuOOH 70% aq. (257 mg, 2 mmol) were added. Solutions were deaerated by bubbling argon for 15 min before recording a cyclic voltammogram.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (Grant 17-73-10405).Notes and references
- H. M. Davies and D. Morton, J. Org. Chem., 2016, 81, 343–350 CrossRef CAS PubMed.
- H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed.
- C. J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- I. B. Krylov, V. A. Vil and A. O. Terent'ev, Beilstein J. Org. Chem., 2015, 11, 92–146 CrossRef PubMed.
- C.-J. Li, From C–H to C–C Bonds: Cross-Dehydrogenative-Coupling, Royal Society of Chemistry, 2014 Search PubMed.
- C.-J. Li and Z. Li, Pure Appl. Chem., 2006, 78, 935–945 CAS.
- I. B. Krylov, A. O. Terent'ev, V. P. Timofeev, B. N. Shelimov, R. A. Novikov, V. M. Merkulova and G. I. Nikishin, Adv. Synth. Catal., 2014, 356, 2266–2280 CrossRef CAS.
- I. B. Krylov and A. O. Terent'ev, Russ. J. Org. Chem., 2015, 51, 10–13 CrossRef CAS.
- A. O. Terent'ev, I. B. Krylov, V. P. Timofeev, Z. A. Starikova, V. M. Merkulova, A. I. Ilovaisky and G. I. Nikishin, Adv. Synth. Catal., 2013, 355, 2375–2390 CrossRef.
- M. Baidya, K. A. Griffin and H. Yamamoto, J. Am. Chem. Soc., 2012, 134, 18566–18569 CrossRef CAS PubMed.
- A. O. Terent'ev, I. B. Krylov, M. Y. Sharipov, Z. M. Kazanskaya and G. I. Nikishin, Tetrahedron, 2012, 68, 10263–10271 CrossRef.
- M. Mahesh, V. Panduranga, G. Prabhu, R. Kumar L, P. V. Ramana and V. V. Sureshbabu, Synth. Commun., 2017, 47, 716–721 CrossRef CAS.
- T. Dohi and Y. Kita, Top. Curr. Chem., 2016, 373, 1–23 CrossRef CAS PubMed.
- Y. Kita and T. Dohi, Chem. Rec., 2015, 15, 886–906 CrossRef CAS PubMed.
- T. C. Turner, K. Shibayama and D. L. Boger, Org. Lett., 2013, 15, 1100–1103 CrossRef CAS PubMed.
- H. Tohma, H. Morioka, S. Takizawa, M. Arisawa and Y. Kita, Tetrahedron, 2001, 57, 345–352 CrossRef CAS.
- L. Lv and Z. Li, Top. Curr. Chem., 2016, 374, 38 CrossRef PubMed.
- Z. Li, D. S. Bohle and C. J. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8928–8933 CrossRef CAS PubMed.
- H.-F. He, K. Wang, B. Xing, G. Sheng, T. Ma and W. Bao, Synlett, 2012, 24, 211–214 Search PubMed.
- J. Jin, Y. Li, Z.-j. Wang, W.-x. Qian and W.-l. Bao, Eur. J. Org. Chem., 2010, 2010, 1235–1238 CrossRef.
- A. O. Terent'ev, D. A. Borisov, I. A. Yaremenko, V. V. Chernyshev and G. I. Nikishin, J. Org. Chem., 2010, 75, 5065–5071 CrossRef PubMed.
- G. Nikishin, A. Terent'ev, D. Borisov, V. Semenov, V. Chernyshev and V. Dembitsky, Synthesis, 2011, 2011, 2091–2100 CrossRef.
- X. Zhu, Y. F. Wang, W. Ren, F. L. Zhang and S. Chiba, Org. Lett., 2013, 15, 3214–3217 CrossRef CAS PubMed.
- Y. Y. Liu, X. H. Yang, J. Yang, R. J. Song and J. H. Li, Chem. Commun., 2014, 50, 6906–6908 RSC.
- D. Shi, H.-T. Qin, C. Zhu and F. Liu, Eur. J. Org. Chem., 2015, 2015, 5084–5088 CrossRef CAS.
- B. C. Lemercier and J. G. Pierce, Org. Lett., 2015, 17, 4542–4545 CrossRef CAS PubMed.
- B. Han, X. L. Yang, R. Fang, W. Yu, C. Wang, X. Y. Duan and S. Liu, Angew. Chem., Int. Ed. Engl., 2012, 51, 8816–8820 CrossRef CAS PubMed.
- W. Li, P. Jia, B. Han, D. Li and W. Yu, Tetrahedron, 2013, 69, 3274–3280 CrossRef CAS.
- X. L. Yang, F. Chen, N. N. Zhou, W. Yu and B. Han, Org. Lett., 2014, 16, 6476–6479 CrossRef CAS PubMed.
- X. X. Peng, Y. J. Deng, X. L. Yang, L. Zhang, W. Yu and B. Han, Org. Lett., 2014, 16, 4650–4653 CrossRef CAS PubMed.
- L. Zhu, G. Wang, Q. Guo, Z. Xu, D. Zhang and R. Wang, Org. Lett., 2014, 16, 5390–5393 CrossRef CAS PubMed.
- L. Zhu, H. Yu, Z. Xu, X. Jiang, L. Lin and R. Wang, Org. Lett., 2014, 16, 1562–1565 CrossRef CAS PubMed.
- R.-H. Liu, D. Wei, B. Han and W. Yu, ACS Catal., 2016, 6, 6525–6530 CrossRef CAS.
- X.-W. Zhang, Z.-F. Xiao, Y.-J. Zhuang, M.-M. Wang and Y.-B. Kang, Adv. Synth. Catal., 2016, 358, 1942–1945 CrossRef CAS.
- F. Chen, X. L. Yang, Z. W. Wu and B. Han, J. Org. Chem., 2016, 81, 3042–3050 CrossRef CAS PubMed.
- X.-L. Yang, Y. Long, F. Chen and B. Han, Org. Chem. Front., 2016, 3, 184–189 RSC.
- W. Zhang, Y. Su, K. H. Wang, L. Wu, B. Chang, Y. Shi, D. Huang and Y. Hu, Org. Lett., 2017, 19, 376–379 CrossRef CAS PubMed.
- I. B. Krylov, S. A. Paveliev, B. N. Shelimov, B. V. Lokshin, I. A. Garbuzova, V. A. Tafeenko, V. V. Chernyshev, A. S. Budnikov, G. I. Nikishin and A. O. Terent'ev, Org. Chem. Front., 2017, 4, 1947–1957 RSC.
- E. G. Bagryanskaya and S. R. Marque, Chem. Rev., 2014, 114, 5011–5056 CrossRef CAS PubMed.
- S. Wertz and A. Studer, Green Chem., 2013, 15, 3116–3134 RSC.
- F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800–3842 CrossRef PubMed.
- G. V. Korolev and A. P. Marchenko, Russ. Chem. Rev., 2000, 69, 409–434 CrossRef CAS.
- E. G. Bagryanskaya, O. A. Krumkacheva, M. V. Fedin and S. R. Marque, Methods Enzymol., 2015, 563, 365–396 Search PubMed.
- E. S. Babaylova, A. A. Malygin, A. A. Lomzov, D. V. Pyshnyi, M. Yulikov, G. Jeschke, O. A. Krumkacheva, M. V. Fedin, G. G. Karpova and E. G. Bagryanskaya, Nucleic Acids Res., 2016, 44, 7935–7943 CrossRef CAS PubMed.
- E. V. Tretyakov and V. I. Ovcharenko, Russ. Chem. Rev., 2009, 78, 971–1012 CrossRef CAS.
- J. L. Brokenshire, J. R. Roberts and K. U. Ingold, J. Am. Chem. Soc., 1972, 94, 7040–7049 CrossRef CAS.
- A. Corsaro, U. Chiacchio and V. Pistarà, Synthesis, 2001, 13, 1903–1931 CrossRef.
- Z.-H. Ren, M.-N. Zhao and Z.-H. Guan, RSC Adv., 2016, 6, 16516–16519 RSC.
- Z. Mirjafary, M. Abdoli, H. Saeidian, S. Boroon and A. Kakanejadifard, RSC Adv., 2015, 5, 79361–79384 RSC.
- E. Vessally, J. Iran. Chem. Soc., 2016, 13, 2325 CrossRef CAS.
- V. P. Ananikov, D. B. Eremin, S. A. Yakukhnov, A. D. Dilman, V. V. Levin, M. P. Egorov, S. S. Karlov, L. M. Kustov, A. L. Tarasov, A. A. Greish, A. A. Shesterkina, A. M. Sakharov, Z. N. Nysenko, A. B. Sheremetev, A. Y. Stakheev, I. S. Mashkovsky, A. Y. Sukhorukov, S. L. Ioffe, A. O. Terent'ev, V. A. Vil', Y. V. Tomilov, R. A. Novikov, S. G. Zlotin, A. S. Kucherenko, N. E. Ustyuzhanina, V. B. Krylov, Y. E. Tsvetkov, M. L. Gening and N. E. Nifantiev, Mendeleev Commun., 2017, 27, 425–438 CrossRef CAS.
- J. Cossy, D. Belotti, V. Bellosta and D. Brocca, Tetrahedron Lett., 1994, 35, 6089–6092 CrossRef CAS.
- K. M. Steward and J. S. Johnson, Org. Lett., 2011, 13, 2426–2429 CrossRef CAS PubMed.
- P. J. Zhou, C. K. Li, S. F. Zhou, A. Shoberu and J. P. Zou, Org. Biomol. Chem., 2017, 15, 2629–2637 CAS.
- Y. Yuan, X. Ji and D. Zhao, Eur. J. Org. Chem., 2010, 2010, 5274–5278 CrossRef.
- S. W. Ashford and K. C. Grega, J. Org. Chem., 2001, 66, 1523–1524 CrossRef CAS PubMed.
- J. Yu, J. Cui and C. Zhang, Eur. J. Org. Chem., 2010, 2010, 7020–7026 CrossRef.
- H. Sajiki, Y. Monguchi, T. Takahashi, Y. Iida, Y. Fujiwara, Y. Inagaki and T. Maegawa, Synlett, 2008, 2008, 2291–2294 CrossRef.
- J. Christoffers, T. Werner, S. Unger and W. Frey, Eur. J. Org. Chem., 2003, 2003, 425–431 CrossRef.
- A. O. Terent'ev, M. Y. Sharipov, I. B. Krylov, D. V. Gaidarenko and G. I. Nikishin, Org. Biomol. Chem., 2015, 13, 1439–1445 Search PubMed.
- J. K. Kochi, Tetrahedron, 1962, 18, 483–497 CrossRef CAS.
- F. Minisci, F. Fontana, S. Araneo, F. Recupero, S. Banfi and S. Quici, J. Am. Chem. Soc., 1995, 117, 226–232 CrossRef CAS.
- G. B. Shul'pin, J. Gradinaru and Y. N. Kozlov, Org. Biomol. Chem., 2003, 1, 3611 Search PubMed.
- M. S. Kharasch and G. Sosnovsky, Tetrahedron, 1958, 3, 105–112 CrossRef CAS.
- C. M. Jones and M. J. Burkitt, J. Am. Chem. Soc., 2003, 125, 6946–6954 CrossRef CAS PubMed.
- E. M. Pliss, M. E. Soloviev, I. V. Tikhonov, D. V. Loshadkin and A. L. Buchachenko, Russ. J. Phys. Chem. B, 2016, 10, 417–420 CrossRef CAS.
- J. P. Fackler, Metal β-Ketoenolate Complexes, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1966 Search PubMed.
- D. H. R. Barton, V. N. Le Gloahec and J. Smith, Tetrahedron Lett., 1998, 39, 7483–7486 CrossRef CAS.
- S. Goldstein and A. Samuni, J. Phys. Chem. A, 2007, 111, 1066–1072 CrossRef CAS PubMed.
- E. M. Pliss, I. V. Tikhonov and A. I. Rusakov, Russ. J. Phys. Chem. B, 2012, 6, 376–383 CrossRef CAS.
- I. V. Tikhonov, E. M. Pliss, L. I. Borodin, V. D. Sen' and T. S. Kuznetsova, Russ. Chem. Bull., 2016, 64, 2438–2443 CrossRef.
- I. V. Tikhonov, E. M. Pliss, D. A. Bogoyavlenskii, M. P. Berezin and V. D. Sen', Russ. Chem. Bull., 2016, 64, 2433–2437 CrossRef.
- S. Hoffman, A. Jezierski and B. Jezowska-Trezebiatowska, Bull. Pol. Acad. Sci., Chem., 1985, 34, 251–255 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization of new compounds. See DOI: 10.1039/c7ra13587d |
| This journal is © The Royal Society of Chemistry 2018 |