
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA self-assembled silicon/phenolic resin-based carbon core–shell nanocomposite as an anode material for lithium-ion batteries
Zhiyao Luab,
Bing Li *ab,
Daijun Yangab,
Hong Lvab,
Mingzhe Xueab and
Cunman Zhang*ab
*ab,
Daijun Yangab,
Hong Lvab,
Mingzhe Xueab and
Cunman Zhang*ab
aSchool of Automotive Studies, Tongji University, Shanghai 201804, P. R. China. E-mail: libing210@tongji.edu.cn; zhangcunman@tongji.edu.cn
bClean Energy Automotive Engineering Center, Tongji University, Shanghai 201804, P. R. China
First published on 17th January 2018
Abstract
Silicon, with advantages such as high theoretical capacity and relatively low working potential, has been regarded as promising when it is used for lithium-ion battery anodes. However, its practical application is impeded by the intrinsic low electrical conductivity and the dramatic volume change during the lithiation/delithiation process, which leads to a rapid capacity fading of the electrode. In this regard, we design silicon nanoparticles homogeneously coated with a phenolic resin-based carbon layer as a core–shell nanocomposite via a facile self-assembly method followed by carbonization. The surrounding carbon shell, confirmed by transmission electron microscopy and Raman spectroscopy, is not only beneficial to the formation of a stable solid electrolyte interface film, but the electrical conductivity of the electrode is also enhanced. A high and stable specific capacity of nearly 1000 mA h g−1 is achieved at C/3 after 200 cycles with a coulombic efficiency of >99.6%. The entire synthesis process is quite simple and easy to scale up, thus having great potential for commercial applications.
1 Introduction
Lithium-ion batteries (LIBs) with high energy density and long cycle life have been widely used in the past few decades as efficient electrochemical energy storage devices in the fields of portable electronic products and electric vehicles.1,2 However, with the increasing demands on LIBs such as higher energy density, higher power density and longer cycling life, the performance of the current LIBs, especially those applied in electric vehicles, is no longer satisfactory.2–5 For this purpose, silicon (Si), which is well-known for its significantly high theoretical capacity of 4200 mA h g−1 and a relatively low working potential of less than 0.5 V (vs. Li/Li+), has been considered as the most promising anode material for next-generation LIBs.6–9 Nevertheless, the practical application of silicon-based materials is restricted by two main problems: (1) the intrinsic low electrical conductivity of silicon; (2) the dramatic volume change (∼300%) of silicon during the Li-ion insertion/extraction process.7,10–12 Such volume change seriously causes structural pulverization, and the electrical contact is lost as well. The anode materials are constantly exposed to the electrolyte during cycling, leading to the continuous formation of the solid electrolyte interface (SEI) layer.13–15 As a result, the cycling performance of the battery degrades fast.To overcome the aforementioned problems, numerous approaches have been explored. Among various solutions, the utilization of nanostructured silicon materials, including nanoparticles,16,17 nanospheres,18,19 nanotubes,20 nanowires21 and nanosheets,22 has proven to be effective, as the nanoscale materials could alleviate the mechanical stresses due to the volume change and offer a fast electronic and ionic conduction pathway. Furthermore, covering silicon materials with a protective layer such as carbon materials23–27 or metal oxides28–31 is also considered to be a useful method. Such coating can protect silicon particles from being directly exposed to the electrolyte and limit the side reaction. For example, Sourice et al.32 synthesized carbon-covered silicon nanoparticles through a one-step laser pyrolysis process with two stages. The electrode based on such material exhibited a high capacity of 2400 mA h g−1 at C/10. However, the gaseous silicon source (SiH4) was highly toxic and inflammable, which would substantially increase the manufacturing costs. Bai et al.30 coated silicon nanospheres with titanium dioxide (TiO2) by atomic layer deposition (ALD). A high specific charge capacity of 1580.3 mA h g−1 was achieved at C/10 after 50 cycles. Although the electrode exhibited a superior electrochemical performance, the manufacturing process using ALD technique was expensive, and the electrical conductivity was still relatively low caused by the TiO2 layer. Recently, many researchers have laid great emphasis on silicon nanostructure with void spaces, including porous silicon,33,34 hollow silicon31 and yolk–shell structures.35–37 Such constructions do improve the electrode properties, as the void spaces can effectively buffer the volume expansion of silicon during lithiation process. Nonetheless, the introduction of void space is usually realized by using a hazardous hydrofluoric acid (HF), and the fabricating process becomes more complex as well. Meanwhile, the compacted density of the electrode will also be negatively affected.
Herein, we report a facile solvent evaporation induced self-assembly method followed by carbonization process for preparing a carbon-coated silicon core–shell nanocomposite as a kind of anode material for LIBs. The phenolic resin-based carbon shell can not only accommodate the large volume change of silicon but also isolate the inner silicon core from electrolyte. By having formed a stable SEI layer, the cycling stability of the electrode is improved. At the same time, the electrical conductivity is strengthened by the carbon coating. As a consequence, the well-designed Si@C core–shell nanocomposite exhibits an outstanding specific capacity of 1107.5 mA h g−1 at C/3 over 50 cycles and nearly 1000 mA h g−1 after 200 cycles with a high coulombic efficiency of >99.6%, as well as a good rate capability. Moreover, the synthesis process of such material is quite simple and facile for large-scale production with low cost, which has an advantage in the commercial applications of next-generation LIBs.
2 Experimental
2.1 Synthesis of Si@C core–shell nanocomposite
The commercial silicon nanoparticles (Si NPs), which have a diameter of around 100 nm on average, were purchased from MTI Corporation. An ethanol-soluble phenolic resin with a low molecular weight, which was synthesized with phenol and formaldehyde via a simple polymerization method, was prepared for the subsequent coating process.38 For a typical preparation of carbon-coated silicon material (named as Si@C-2), 2 g commercial silicon nanoparticles were added into 100 mL ethanol and treated by ultrasonic wave for 1 h to be fully dispersed. 0.5 g resin, which had been dissolved in 50 mL ethanol in advance, was then added into the silicon suspension, followed by continuous stirring at room temperature for 4 h. Subsequently, the ethanol as the solvent in the suspension was evaporated by using a vacuum rotary evaporator at 100 °C for 2 h. The as-obtained Si@resin core–shell nanoparticles were finally calcinated in a tube furnace under nitrogen atmosphere at 700 °C for 2 h. The heating rate was set at 5 °C min−1. For comparison, different amount of phenolic resin was added (e.g., 0.35 g and 1 g resin) under the same condition as described above to coat silicon nanoparticles with different thickness (named as Si@C-1 and Si@C-3, respectively).2.2 Physical characterization
Several instruments were utilized to examine the morphology of the samples, as well as their structure. Transmission electron microscopy (TEM) analysis was carried out by a transmission electron microscope (FEI Tecnai T20, USA). To prepare the samples for TEM measurement, the powdered nanocomposite was fully dispersed in ethanol via ultrasonic wave and then dried on a copper grid. X-ray diffraction (XRD) patterns were obtained by a diffractometer (Bruker D8 Advance, Germany) using Cu Kα radiation. Raman spectra were tested through a Raman spectrometer (Bruker Optics Senterra R200-L, Germany) with a He–Ne laser wavelength of 632.8 nm. The thermal properties of the samples were characterized by using a thermogravimetric analyzer (Netzsch STA449C, Germany) from room temperature to 900 °C in air with a heating rate of 10 °C min−1.2.3 Electrochemical characterization
For the fabrication of working electrodes, 70 wt% of active material was mixed with 10 wt% of Super P (Kejing Star), 5 wt% of graphite (Shanshan Technology) and 15 wt% of sodium alginate (Sinopharm Chemical) in deionized water. The well-mixed slurry was then pasted onto a copper foil by using a doctor blade, which was 200 μm thick. After drying the obtained film in an oven at 70 °C for 3 h, it was cut into discs with a diameter of 12 mm, followed by being thoroughly dried in a vacuum oven at 80 °C overnight. The mass loading of active material was about 1.7 mg cm−2. CR2032-type coin cells were assembled in MBraun glove box under argon atmosphere, using lithium foil as both the counter and reference electrode and Celgard 2325 separator film. In preparation for the electrolyte, 1.0 M LiPF6 (Tinci Materials) was dissolved in dimethyl carbonate (DMC) and fluoroethylene carbonate (FEC), which had a volume ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The galvanostatic charge/discharge cycling test was conducted at 0.01–1.0 V (vs. Li/Li+) at room temperature with Land test system. Cyclic voltammetry (CV) measurements were performed at 0.01–1.0 V (vs. Li/Li+) with scanning rate of 0.1 mV s−1, using an electrochemical station (CHI 660E, Shanghai). Electrochemical impedance spectroscopy (EIS) was tested from 100 kHz to 0.01 Hz with AC amplitude of 5 mV.
:1. The galvanostatic charge/discharge cycling test was conducted at 0.01–1.0 V (vs. Li/Li+) at room temperature with Land test system. Cyclic voltammetry (CV) measurements were performed at 0.01–1.0 V (vs. Li/Li+) with scanning rate of 0.1 mV s−1, using an electrochemical station (CHI 660E, Shanghai). Electrochemical impedance spectroscopy (EIS) was tested from 100 kHz to 0.01 Hz with AC amplitude of 5 mV.
3 Results and discussion
The schematic illustration of the synthesis process for fabricating Si@C core–shell nanocomposite is presented in Fig. 1. In the first step, commercially available silicon nanoparticles are coated with a low-molecular-weight phenolic resin via a solvent evaporation induced self-assembly method. In this process, the native oxide layer on the surface of the silicon nanoparticles could strongly interacts with the resin via the formation of hydrogen bonds, since such phenolic resin has plenty of hydroxyl groups (–OH).39,40 For this reason, the resin could be homogeneously coated on the silicon surface during the evaporation of ethanol. Secondly, the Si@resin core–shell nanoparticles are carbonized under nitrogen atmosphere to form a carbon shell outside the silicon core. Therefore, the whole two synthesis procedures are quite facile and easy to scale up, which has great potential for low-cost commercial applications.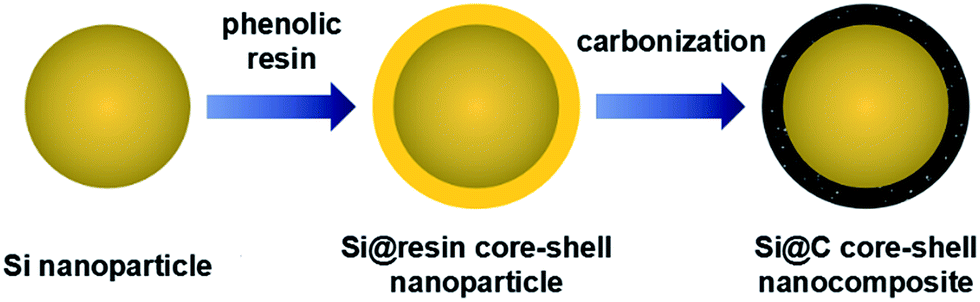 | ||
| Fig. 1 Schematic illustration of Si@C core–shell nanocomposite. | ||
As can be seen in the TEM images in Fig. 2, the microstructure of Si@C core–shell nanocomposite can be clearly observed. The average diameter of the pristine commercial silicon nanoparticles is around 100 nm, while a uniform carbon layer, which is less than 10 nm thick, is homogeneously formed surrounding the silicon core. With an increasing adding amount of the phenolic resin during the fabricating process (0.35, 0.5 and 1 g resin for Si@C-1, Si@C-2 and Si@C-3, respectively), the thickness of the carbon coating layer increases accordingly.
 | ||
| Fig. 2 TEM images of (a) Si@C-1, (b) Si@C-2 and (c) Si@C-3 materials. (d) HRTEM image of the Si@C-2 material. | ||
Fig. 3a shows the XRD patterns of Si@C core–shell nanocomposite and the silicon nanoparticles. For all the carbon-coated silicon samples, five distinct diffraction peaks can be observed at 28.4, 47.3, 56.1, 69.1 and 76.4°, which are matched well with the pristine silicon corresponding to the (111), (220), (311), (400) and (331) plane, respectively (JCPDS no. 65-1060). Additionally, no other peaks are visible, indicating that the carbon coating layer is very thin and has little influence on the silicon structure. Fig. 3b displays the Raman spectra of different samples, revealing the characteristics of the carbon layer. Two carbon peaks located at about 1340 and 1590 cm−1 are respectively related to the disorder-induced D band and graphitic G band of carbon. The intensity ratio of ID/IG is around 0.9, suggesting an amorphous carbon layer with a poor graphitization degree in such core–shell structure. In addition, the intensity of the silicon characteristic peaks at around 515 cm−1 decreases dramatically, which demonstrates that the silicon nanoparticles are well encapsulated in the carbon frameworks.41,42 Moreover, the silicon contents in the three samples are measured by thermogravimetric analysis (TGA). As shown in Fig. 3c, the main loss of weight occurring at 400–650 °C is attributed to the carbon combustion. When the temperature continues climbing, there occurs a slight weight increase due to the oxidation of silicon, which can be confirmed by the curve of the pure silicon sample. According to the remaining weight in the thermogravimetric curves, the mass percentages of silicon are determined to be 90.1, 86.3 and 83.9 wt% for Si@C-1, Si@C-2 and Si@C-3, respectively, which corresponds to the increasing adding amount of the phenolic resin.
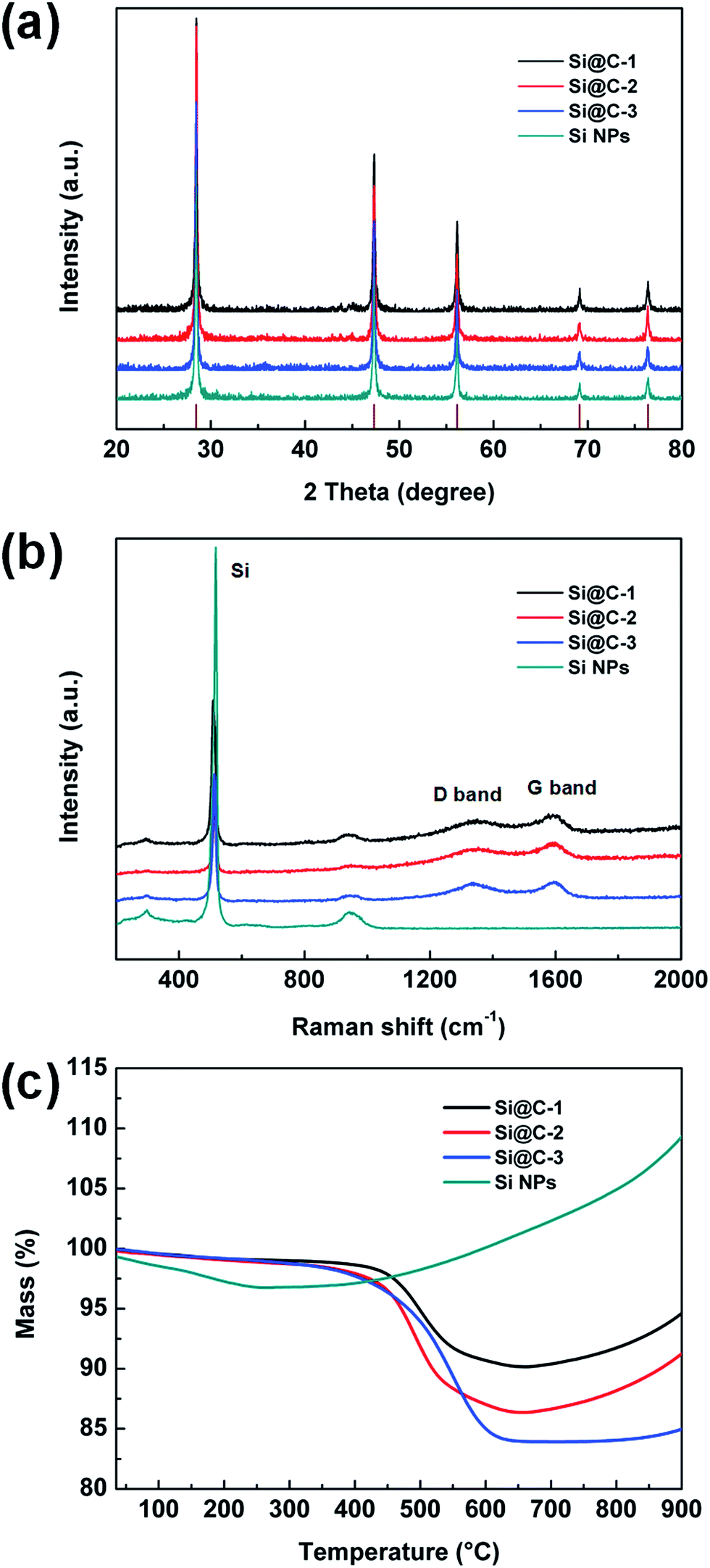 | ||
| Fig. 3 (a) XRD patterns, (b) Raman spectra and (c) TGA curves of various samples. | ||
To characterize the electrochemical properties of the Si@C core–shell nanocomposite electrodes, cyclic voltammetry (CV) measurements are first obtained at 0.01–1.0 V (vs. Li/Li+), as exhibited in Fig. 4a. The cathodic peak at about 0.2 V during lithiation process, which does not occur in the first cycle, indicates the evolution of silicon from crystalline structure to amorphous one. The two anodic peaks at around 0.36 and 0.53 V could be ascribed to the delithiation of the lithium–silicon alloy.43 Meanwhile, it can be seen that the intensity of the two peaks increases in the second cycle, which is due to the activation process in the initial cycles. Fig. 4b displays voltage profiles of the as-prepared electrode at typical cycles at C/20 rate for the first cycle and C/3 for the rest of the cycles (1C = 1.4 A g−1). A distinct and long plateau profile occurs at about 0.1 V in the first lithiation process, which is in accordance with the alloying behavior of crystalline silicon.44–47 The initial discharge and charge specific capacity achieve 2282.8 and 1523.2 mA h g−1 respectively, meaning an initial CE of 66.7% due to the formation of SEI film as well as lithium ions irreversibly intercalating into silicon and amorphous carbon. It should be noted that no obvious change can be observed in later cycles, which suggests a stable performance of the electrode.
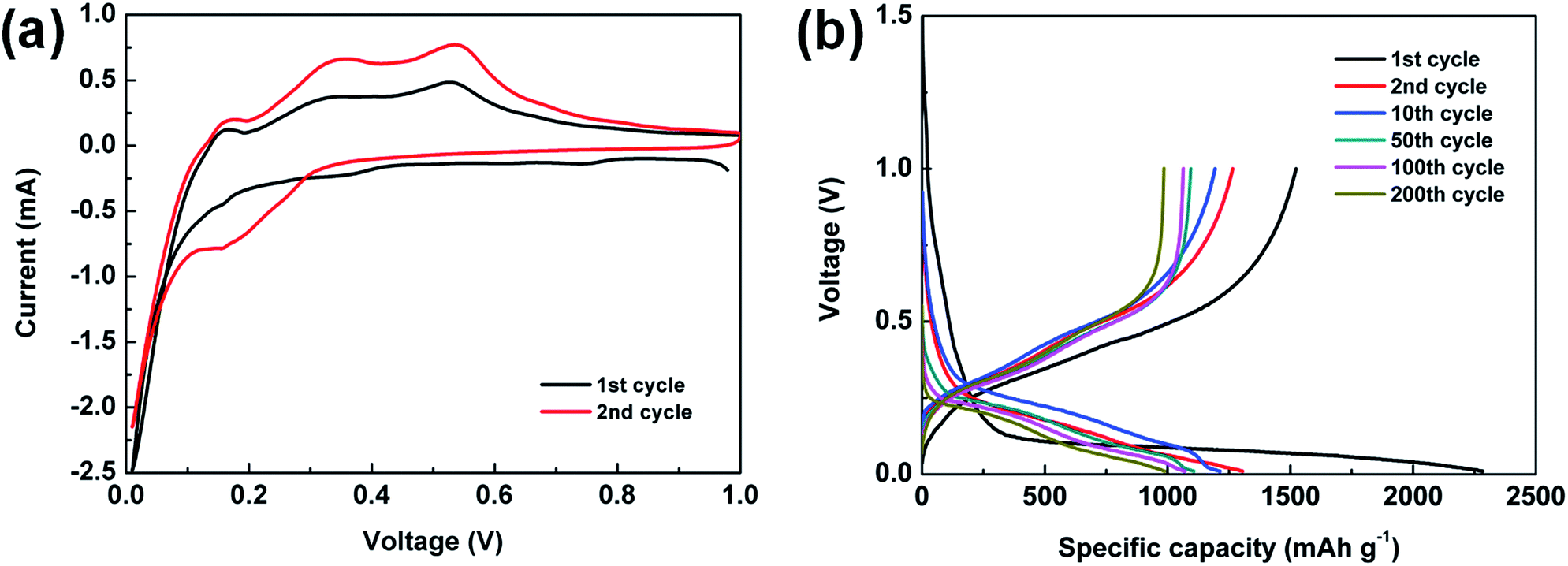 | ||
| Fig. 4 (a) CV curves of the Si@C-2 electrode. (b) Voltage profiles of the Si@C-2 electrode at a rate of C/20 for the first cycle and C/3 for the 2nd, 10th, 50th, 100th and 200th cycles. | ||
The galvanostatic charge/discharge cycling test is also performed between 0.01 and 1.0 V at C/20 for the first cycle and C/3 for the rest. The cycling performances of various electrodes based on the silicon nanoparticles and Si@C core–shell nanocomposite with different silicon contents are under consideration. As displayed in Fig. 5a, the pure silicon electrode delivers a high initial discharge capacity of 2823.6 mA h g−1, but experiences a fast capacity fading (<200 mA h g−1 after 20 cycles), which is ascribed to the large volume change of silicon during lithiation/delithiation process. For the Si@C-2 material with a silicon content of 86.3 wt%, a superior reversible specific capacity of 1107.5 mA h g−1 is achieved over 50 cycles. The results indicate that the carbon coating layer, which is derived from phenolic resin, could effectively buffer the volume expansion of silicon and relax the internal stress, thus tremendously improving the cycling stability of the electrode. However, compared with Si@C-2, both the Si@C-1 and Si@C-3 electrodes obtain a relatively lower capacity. On one hand, if the carbon shell is not thick enough, it will fail to bear the mechanical stresses and start to break up during cycling, exposing the active material to the electrolyte. On the other hand, an excessive amount of carbon content could negatively affect the capacity of the nanocomposite electrode due to the low lithium storage capacity of hard carbon. Meanwhile, with an increasing thickness of carbon coating layer, the toughness of the carbon shell decreases, which may lead to the destruction of the electrode structure. Further, the cycling performance of Si@C-2 electrode in the long term is exhibited in Fig. 5b. After experiencing a slight increase in capacity at early cycles due to the electrochemical activation process, the carbon-coated silicon anode delivers a high reversible capacity of nearly 1000 mA h g−1 over 200 cycles, which is almost three times the theoretical specific capacity of graphite (372 mA h g−1). In addition, the CE rises to 96.9% after the first cycle and stabilizes at more than 99.6% during the long-term cycles, indicating that such carbon frameworks also benefit the formation of a stable SEI layer and the restraint of the side reaction from electrolyte.
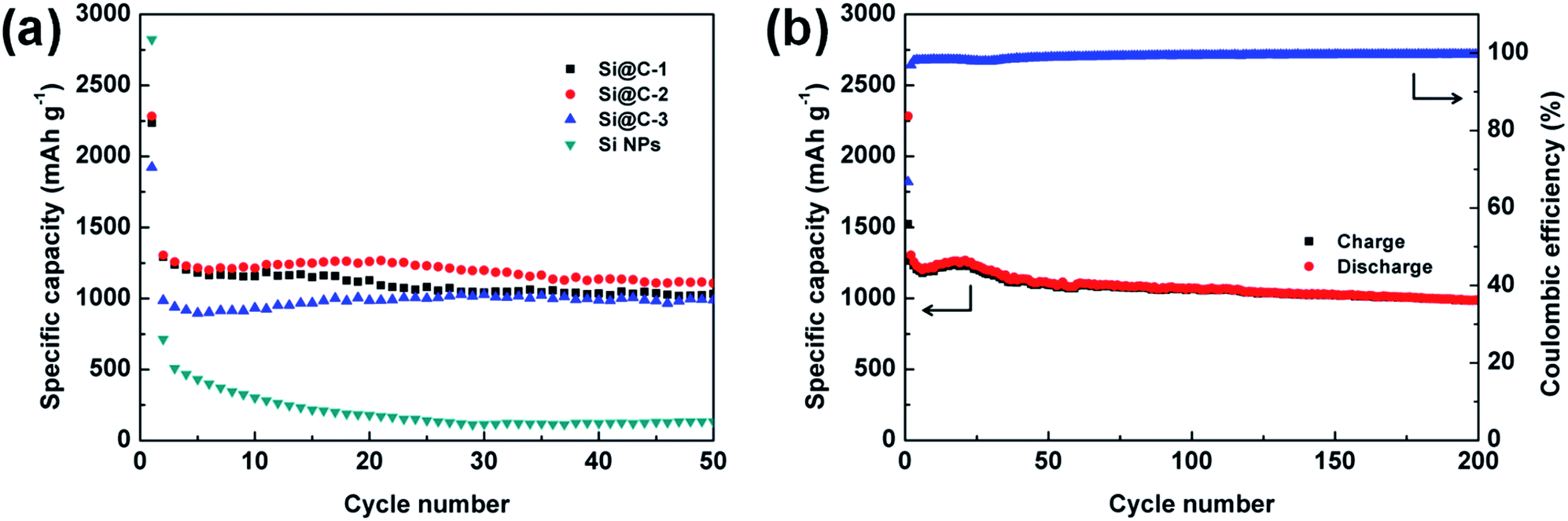 | ||
| Fig. 5 (a) Discharge capacities of the electrodes based on Si@C-1, Si@C-2, Si@C-3 materials and silicon nanoparticles for 50 cycles. (b) Charge/discharge profiles and coulombic efficiency of the Si@C-2 electrode at a rate of C/20 for the first cycle and C/3 for later cycles. | ||
To further investigate the rate performance of Si@C core–shell nanocomposite electrode, the rate capability of the three samples is measured with different current densities, which can be seen in Fig. 6. The discharge specific capacity of Si@C-2 electrode varies from 1507.8 to 482.3 mA h g−1 with a rising rate from C/10 to 2C, respectively, demonstrating a descending trend. Remarkably, when the C-rate returns to C/10, a capacity of 1513 mA h g−1 could still be achieved, revealing a good recovery of the electrode after charging/discharging at high current densities. This could be attributed to the stable SEI layer outside the core–shell nanocomposite and the enhancement of electrical conductivity caused by the carbon frameworks.
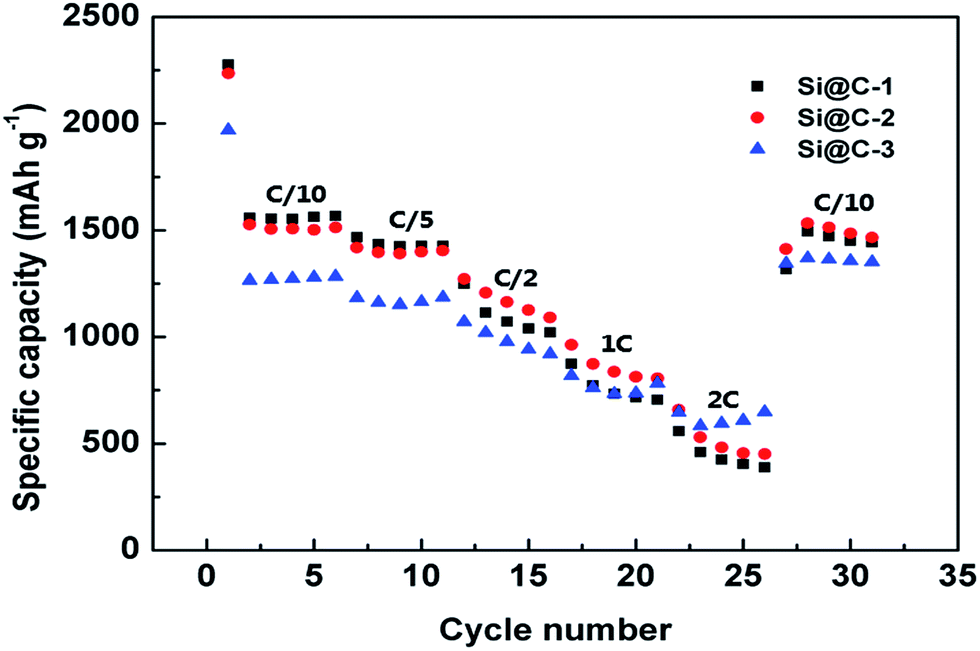 | ||
| Fig. 6 Rate capabilities of Si@C-1, Si@C-2 and Si@C-3 electrodes at various C-rates. | ||
Fig. 7 shows the electrochemical impedance spectroscopy (EIS) illustrated via Nyquist plots within the frequency range between 100 kHz and 0.01 Hz. An appropriate equivalent circuit is also used for the impedance analysis through some kinetic parameters. According to the fitting results, the ohmic resistance (RS) from the cell components, separator and electrolyte is around 3.0 Ω among the three samples, which indicates a good condition of the cells. More importantly, with an increasing carbon content of the three electrodes, the charge transfer resistance (Rct) decreases correspondingly, respectively with 42.9, 8.01 and 4.81 Ω. The improvement of the electron transfer kinetics is undoubted, as the surrounding carbon shell does play a critical role in raising the electronic conductivity.
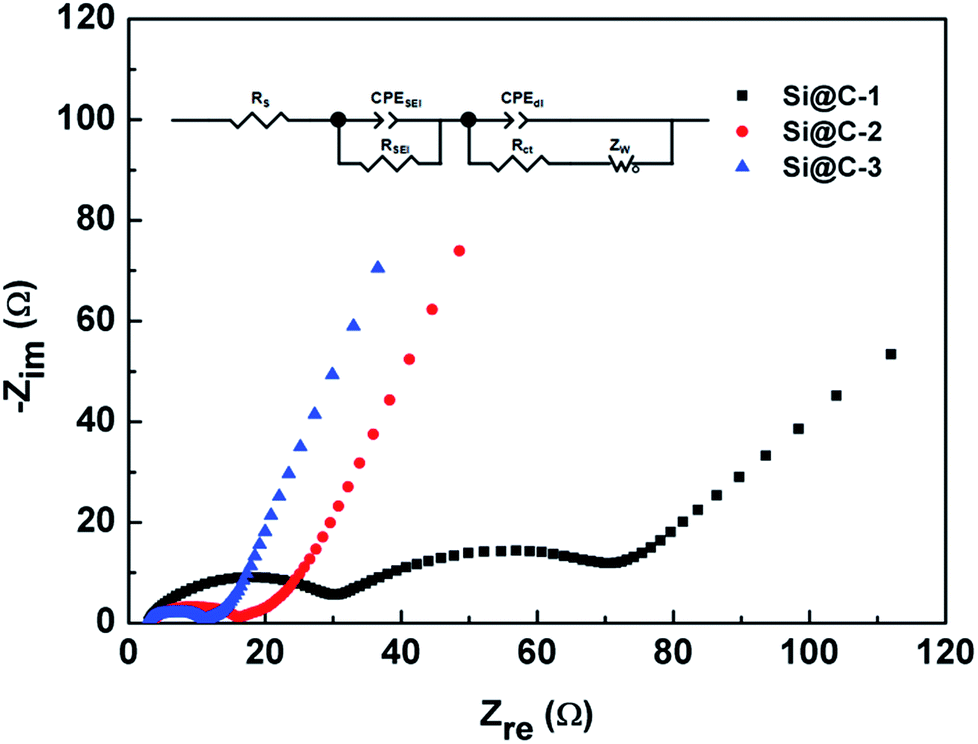 | ||
| Fig. 7 Nyquist plots of the different electrodes after 10 cycles and the equivalent circuit. | ||
4 Conclusions
In summary, a carbon-coated silicon core–shell nanocomposite has been developed through a facile self-assembly method and a subsequent carbonization process. By effectively solving the main problems of the silicon materials, the Si@C core–shell nanocomposite electrode exhibits a much better cycling performance than the pure silicon electrode. With the original adding amount of silicon nanoparticles and phenolic resin at the mass ratio of 4:1 and a silicon content of about 86 wt% after carbonization, the Si@C-2 electrode delivers the optimal electrochemical performance of 1107.5 mA h g−1 at C/3 over 50 cycles and nearly 1000 mA h g−1 over 200 cycles with CE of more than 99.6%. Furthermore, our synthesis process is quite simple and scalable, presenting a promising route to fabricate the silicon-based anode materials for LIBs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21676204) and Young Talents “Climbing” Program of Tongji University, China.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- H. Li, Z. X. Wang, L. Q. Chen and X. J. Huang, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- T. Song, J. L. Xia, J. H. Lee, D. H. Lee, M. S. Kwon, J. M. Choi, J. Wu, S. K. Doo, H. Chang, W. Il Park, D. S. Zang, H. Kim, Y. G. Huang, K. C. Hwang, J. A. Rogers and U. Paik, Nano Lett., 2010, 10, 1710–1716 CrossRef CAS PubMed.
- Z. L. Zhang, Y. H. Wang, W. F. Ren, Q. Q. Tan, Y. F. Chen, H. Li, Z. Y. Zhong and F. B. Su, Angew. Chem., Int. Ed., 2014, 53, 5165–5169 CAS.
- H. J. Tian, X. J. Tan, F. X. Xin, C. S. Wang and W. Q. Han, Nano Energy, 2015, 11, 490–499 CrossRef CAS.
- N. Lin, Y. Han, L. B. Wang, J. B. Zhou, J. Zhou, Y. C. Zhu and Y. T. Qian, Angew. Chem., Int. Ed., 2015, 54, 3822–3825 CrossRef CAS PubMed.
- X. F. Gao, J. Y. Li, Y. Y. Xie, D. S. Guan and C. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 7855–7862 CAS.
- Y. He, X. Q. Yu, Y. H. Wang, H. Li and X. J. Huang, Adv. Mater., 2011, 23, 4938–4941 CrossRef CAS PubMed.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966–4984 CrossRef CAS PubMed.
- M. H. Park, M. G. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Nano Lett., 2009, 9, 3844–3847 CrossRef CAS PubMed.
- B. Li, F. Yao, J. J. Bae, J. Chang, M. R. Zamfir, D. T. Le, D. T. Pham, H. Yue and Y. H. Lee, Sci. Rep., 2015, 5, 7659 CrossRef CAS PubMed.
- X. H. Liu, J. Zhang, W. P. Si, L. X. Xi, B. Eichler, C. L. Yan and O. G. Schmidt, ACS Nano, 2015, 9, 1198–1205 CrossRef CAS PubMed.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522–1531 CrossRef CAS PubMed.
- T. H. Hwang, Y. M. Lee, B. S. Kong, J. S. Seo and J. W. Choi, Nano Lett., 2012, 12, 802–807 CrossRef CAS PubMed.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. A. Liu, L. B. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949–2954 CrossRef CAS PubMed.
- H. Ma, F. Y. Cheng, J. Chen, J. Z. Zhao, C. S. Li, Z. L. Tao and J. Liang, Adv. Mater., 2007, 19, 4067–4070 CrossRef CAS.
- J. K. Yoo, J. Kim, Y. S. Jung and K. Kang, Adv. Mater., 2012, 24, 5452–5456 CrossRef CAS PubMed.
- C. K. Chan, H. L. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- Z. Y. Lu, J. X. Zhu, D. H. Sim, W. W. Zhou, W. H. Ship, H. H. Hng and Q. Y. Yan, Chem. Mater., 2011, 23, 5293–5295 CrossRef CAS.
- N. Liu, Z. D. Lu, J. Zhao, M. T. McDowell, H. W. Lee, W. T. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- W. Sun, R. Z. Hu, M. Zhang, J. W. Liu and M. Zhu, J. Power Sources, 2016, 318, 113–120 CrossRef CAS.
- D. Q. Chen, W. J. Liao, Y. Yang and J. B. Zhao, J. Power Sources, 2016, 315, 236–241 CrossRef CAS.
- S. Fang, L. F. Shen, Z. K. Tong, H. Zheng, F. Zhang and X. G. Zhang, Nanoscale, 2015, 7, 7409–7414 RSC.
- L. Shi, W. K. Wang, A. B. Wang, K. G. Yuan, Z. Q. Jin and Y. S. Yang, J. Mater. Chem. A, 2015, 3, 18190–18197 CAS.
- X. J. Bai, Y. Y. Yu, H. H. Kung, B. Wang and J. M. Jiang, J. Power Sources, 2016, 306, 42–48 CrossRef CAS.
- G. Jeong, J. G. Kim, M. S. Park, M. Seo, S. M. Hwang, Y. U. Kim, Y. J. Kim, J. H. Kim and S. X. Dou, ACS Nano, 2014, 8, 2977–2985 CrossRef CAS PubMed.
- Y. Bai, D. Yan, C. Y. Yu, L. N. Cao, C. L. Wang, J. S. Zhang, H. Y. Zhu, Y. S. Hu, S. Dai, J. L. Lu and W. F. Zhang, J. Power Sources, 2016, 308, 75–82 CrossRef CAS.
- Z. W. Zhou, Y. T. Liu, X. M. Xie and X. Y. Ye, ACS Appl. Mater. Interfaces, 2016, 8, 7092–7100 CAS.
- J. Sourice, A. Quinsac, Y. Leconte, O. Sublemontier, W. Porcher, C. Haon, A. Bordes, E. De Vito, A. Boulineau, S. J. S. Larbi, N. Herlin-Boime and C. Reynaud, ACS Appl. Mater. Interfaces, 2015, 7, 6637–6644 CAS.
- Z. D. Lu, N. Liu, H. W. Lee, J. Zhao, W. Y. Li, Y. Z. Li and Y. Cui, ACS Nano, 2015, 9, 2540–2547 CrossRef CAS PubMed.
- L. S. Jiao, J. Y. Liu, H. Y. Li, T. S. Wu, F. H. Li, H. Y. Wang and L. Niu, J. Power Sources, 2016, 315, 9–15 CrossRef CAS.
- L. Y. Yang, H. Z. Li, J. Liu, Z. Q. Sun, S. S. Tang and M. Lei, Sci. Rep., 2015, 5, 10908 CrossRef CAS PubMed.
- J. P. Yang, Y. X. Wang, S. L. Chou, R. Y. Zhang, Y. F. Xu, J. W. Fan, W. X. Zhang, H. K. Liu, D. Y. Zhao and S. X. Dou, Nano Energy, 2015, 18, 133–142 CrossRef CAS.
- J. Xie, L. Tong, L. W. Su, Y. W. Xu, L. B. Wang and Y. H. Wang, J. Power Sources, 2017, 342, 529–536 CrossRef CAS.
- Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, H. F. Yang, Z. Li, C. Z. Yu, B. Tu and D. Y. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS PubMed.
- Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, L. Cheng, D. Feng, Z. X. Wu, Z. X. Chen, Y. Wan, A. Stein and D. Y. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- J. Park, G. P. Kim, I. Nam, S. Park and J. Yi, Nanotechnology, 2013, 24, 025602 CrossRef PubMed.
- W. Luo, Y. X. Wang, S. L. Chou, Y. F. Xu, W. Li, B. Kong, S. X. Dou, H. K. Liu and J. P. Yang, Nano Energy, 2016, 27, 255–264 CrossRef CAS.
- W. Luo, Y. X. Wang, L. J. Wang, W. Jiang, S. L. Chou, S. X. Dou, H. K. Liu and J. P. Yang, ACS Nano, 2016, 10, 10524–10532 CrossRef CAS PubMed.
- X. S. Zhou, L. J. Wan and Y. G. Guo, Small, 2013, 9, 2684–2688 CrossRef CAS PubMed.
- J. K. Feng, Z. Zhang, L. J. Ci, W. Zhai, Q. Ai and S. L. Xiong, J. Power Sources, 2015, 287, 177–183 CrossRef CAS.
- X. S. Zhou, Y. X. Yin, L. J. Wan and Y. G. Guo, Chem. Commun., 2012, 48, 2198–2200 RSC.
- B. Wang, X. L. Li, B. Luo, Y. Y. Jia and L. J. Zhi, Nanoscale, 2013, 5, 1470–1474 RSC.
- C. D. Wang, Y. S. Chui, R. G. Ma, T. L. Wong, J. G. Ren, Q. H. Wu, X. F. Chen and W. J. Zhang, J. Mater. Chem. A, 2013, 1, 10092–10098 CAS.
| This journal is © The Royal Society of Chemistry 2018 |