
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatic alkyne insertion into six-membered cyclometalated pyridine complexes of iridium: different insertion modes and structurally novel products†
Xiaodan Chu,
Shaowei Zhang,
Zhuo Wang,
Tongyu Li and
Bolin Zhu *
*
Tianjin Key Laboratory of Structure and Performance for Functional Molecules, Key Laboratory of Inorganic–Organic Hybrid Functional Material Chemistry, Ministry of Education, College of Chemistry, Tianjin Normal University, Tianjin 300387, People's Republic of China. E-mail: hxxyzbl@gmail.com; Fax: +86 22 23766532; Tel: +86 22 23766515
First published on 14th February 2018
Abstract
Reactions of 2-benzoylpyridine or 2-benzylpyridine with [Cp*MCl2]2 (M = Ir, Rh) have been carried out in the presence of NaOAc in refluxing methanol, which form the corresponding six-membered cyclometalated products (1–3) except for the reaction of 2-benzylpyridine with [Cp*RhCl2]2. Insertion reactions of two six-membered cyclometalated pyridine iridium complexes (1 and 2) with terminal or internal aromatic alkynes were studied. Terminal alkynes p-XC6H4C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH (X = H, MeO, and F) with 1 give the corresponding five- and seven-membered doubly cycloiridated complexes 4a–c, internal alkynes p-XC6H4CCC6H4X-p (X = H, MeO, and Br) form the similar five- and seven-membered doubly cycloiridated complexes (5a,b) and/or di-insertion products (6a,c), whereas the acyl alkyne PhCCCOPh affords the novel spiro-metalated complex 7. For complex 2, internal alkynes p-XC6H4CCC6H4X-p (X = H, MeO, and Br) form similar five- and seven-membered doubly cycloiridated complexes (8a–c). However, in the case of PhCCCOPh, the reaction gives the novel four-membered cyclometalated complex 9. These results suggest that the products formed by alkyne insertion reactions of the six-membered cycloiridated pyridine complexes are very diverse. Plausible pathways for the formation of these novel insertion products were proposed. Molecular structures of seven cyclometalated complexes were determined by X-ray diffraction.
CH (X = H, MeO, and F) with 1 give the corresponding five- and seven-membered doubly cycloiridated complexes 4a–c, internal alkynes p-XC6H4CCC6H4X-p (X = H, MeO, and Br) form the similar five- and seven-membered doubly cycloiridated complexes (5a,b) and/or di-insertion products (6a,c), whereas the acyl alkyne PhCCCOPh affords the novel spiro-metalated complex 7. For complex 2, internal alkynes p-XC6H4CCC6H4X-p (X = H, MeO, and Br) form similar five- and seven-membered doubly cycloiridated complexes (8a–c). However, in the case of PhCCCOPh, the reaction gives the novel four-membered cyclometalated complex 9. These results suggest that the products formed by alkyne insertion reactions of the six-membered cycloiridated pyridine complexes are very diverse. Plausible pathways for the formation of these novel insertion products were proposed. Molecular structures of seven cyclometalated complexes were determined by X-ray diffraction.
Introduction
In the last decade, the reactions of N-containing substrates with alkynes catalyzed by transition metal complexes (Rh and Ir) have been widely studied, which is mainly due to the reactions providing an economic and convenient method to generate organic heterocyclic compounds.1 Up to now, the generally accepted catalytic cycle mainly includes: (i) initial coordination of the transition metal center (Rh and Ir) via a donor atom (N), (ii) a geometrically accessible C–H bond is activated by the metal atom to give a five-membered ring, (iii) alkyne insertion into the cyclometalated M–C bond (M = Rh, Ir) to form a seven-membered intermediate, and (iv) reductive elimination to give the organic heterocyclic product. In order to improve understanding selectivity and reactivity issues in the catalytic processes, synthesis of five-membered cyclometalated complexes2–9 and their stoichiometric reactions with alkynes have been well studied.10 For example, cyclometalation of [Cp*MCl2]2 (M = Ir, Rh) with 2-phenylpyridine has been widely reported, the resulting five-membered cyclometalated products (M = Ir, Rh) were formed either in stoichiometric reactions or in catalytic ones.2a,c,3,4c,10f The insertion reactions of alkynes were summarized in Scheme 1, the most common products were seven-membered cyclometalated compounds (I) which were formed by 1,2-alkyne insertion into M–C bond of five-membered ring.10c,d,f In the case of phenylacetylene, the reactions afforded the double-insertion products (II).10d When the reactions were carried out in the presence of NaBArF4, the competition of 1,2-alkyne insertion and vinylidene rearrangement/1,1-insertion was observed to generate the corresponding ion pair compounds (III and IV).3a However, synthesis of the similar six-membered cyclometalated complexes (Rh and Ir)2b,3b,11 and their stoichiometric reactions with alkynes were less reported. For comparison of reactivity and understanding the possibility of existence in the catalytic process, we now report the synthesis and structures of six-membered cyclometalated pyridine complexes (M = Ir, Rh) and their insertion reactions with aromatic alkynes. | ||
| Scheme 1 Reactions of five-membered cyclometalated complexes with alkynes. | ||
Results and discussion
Reaction of [Cp*MCl2]2 (M = Ir, Rh) with 2-benzoylpyridine or 2-benzylpyridine
By the modified methods reported by Davies2b and Jones,3b we reinvestigated the reaction of [Cp*MCl2]2 (M = Ir, Rh) with 2-benzoylpyridine or 2-benzylpyridine in the presence of NaOAc in the refluxing methanol, which was slightly different from the reported reaction conditions, CH2Cl2 as solvent at room temperature. Cyclometalation similarly occurred to yield the corresponding six-membered ring products 1–3 via an intramolecular C(sp2)–H bond activation (Scheme 2). All spectroscopic data we obtained for 1 and 2 are in complete agreement with those published by Davies and Jones, as well as the high yields (85% and 92%). It is worth mentioning that the similar cyclometalated rhodium complex 3 was successfully formed and isolated in our current conditions, although the yield is relatively low (41%). This is different from the product obtained from the same reaction but in the conditions of CH2Cl2 as solvent at room temperature, which gave a Cp* C–H bond activated complex.2b The reaction of 2-benzylpyridine with [Cp*RhCl2]2 and NaOAc was also attempted in the refluxing methanol, unfortunately it failed to give any products except the starting materials. The 1H NMR spectrum of 3 showed the expected four signals for an orthometalated phenyl group and four for the pyridine group, one singlet at δ = 1.34 ppm for Cp* group. The 13C NMR spectrum showed the metalated carbon atom at δ = 172.9 ppm (1JRh–C = 30.0), and the carbonyl group at δ = 191.2 ppm. The product 3 was recrystallized from dichloromethane/hexane and the structure was determined by X-ray crystallography.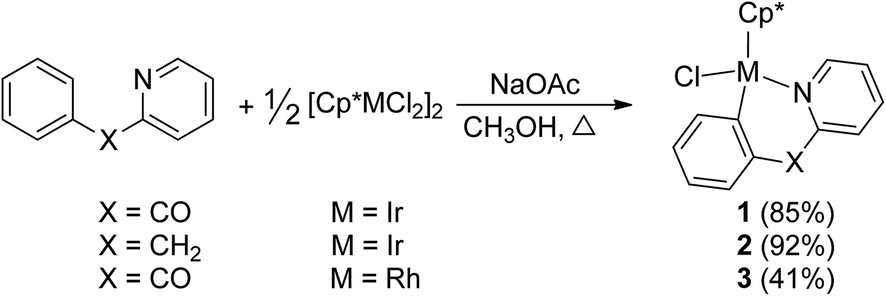 | ||
| Scheme 2 Reaction of [Cp*MCl2]2 (M = Ir, Rh) with 2-benzoylpyridine or 2-benzylpyridine. | ||
The structure of 3 is shown in Fig. 1 with selected bond lengths and angles, which reveals a classic three-legged piano stool geometry. The six-membered ring in the structure confirms the activation of a C(sp2)–H bond on α position of phenyl ring. The Rh(1)–N(1) bond distance is 2.079(4) Å, which is slightly shorter than those (2.099(3) and 2.1000(15) Å) in five-membered ring analogues Cp*RhCl(L) (HL = 2-phenylpyridine2c and 2-(4-methoxyphenyl)pyridine).3a
 | ||
| Fig. 1 Thermal ellipsoid drawing of 3 showing the labeling scheme and 50% probability ellipsoids; hydrogens are omitted for clarity. Selected bond lengths [Å] and angles [°] are Rh(1)–Cl(1) 2.4125(15), Rh(1)–N(1) 2.079(4), Rh(1)–C(8) 2.017(5), Rh(1)–Cp(centroid) 1.826, ∠Cl(1)–Rh(1)–N(1) 89.35(15), ∠Cl(1)–Rh(1)–C(8) 89.55(16), ∠C(8)–Rh(1)–N(1) 87.33(19). | ||
Reactions of 1 with terminal alkynes
Reactions of 1 with arylacetylene p-XC6H4CCH (X = H, MeO, and F) in methanol at room temperature resulted in the five-membered and seven-membered doubly cyclometalated complexes 4a–c in moderate yields (Scheme 3). Obviously, the products were different with those double-insertion compounds (II in Scheme 1) generated from the similar reactions of five-membered cyclometalated complexes (M = Ir, Rh) with phenylacetylene.10d It was observed that the reactions with arylacetylene containing an electron-donating substituent (p-OMe) is faster than the reaction with that containing an electron-withdrawing substituent (p-F). The 1H NMR spectrum of 4a showed eleven multiplets, integrating to 12H, assigned to protons of three aromatic rings, one singlet at δ = 6.81 ppm corresponding to the only vinyl proton, and one singlet at δ = 1.45 ppm for Cp* group.
 | ||
| Scheme 3 Reactions of 1 with arylacetylenes. | ||
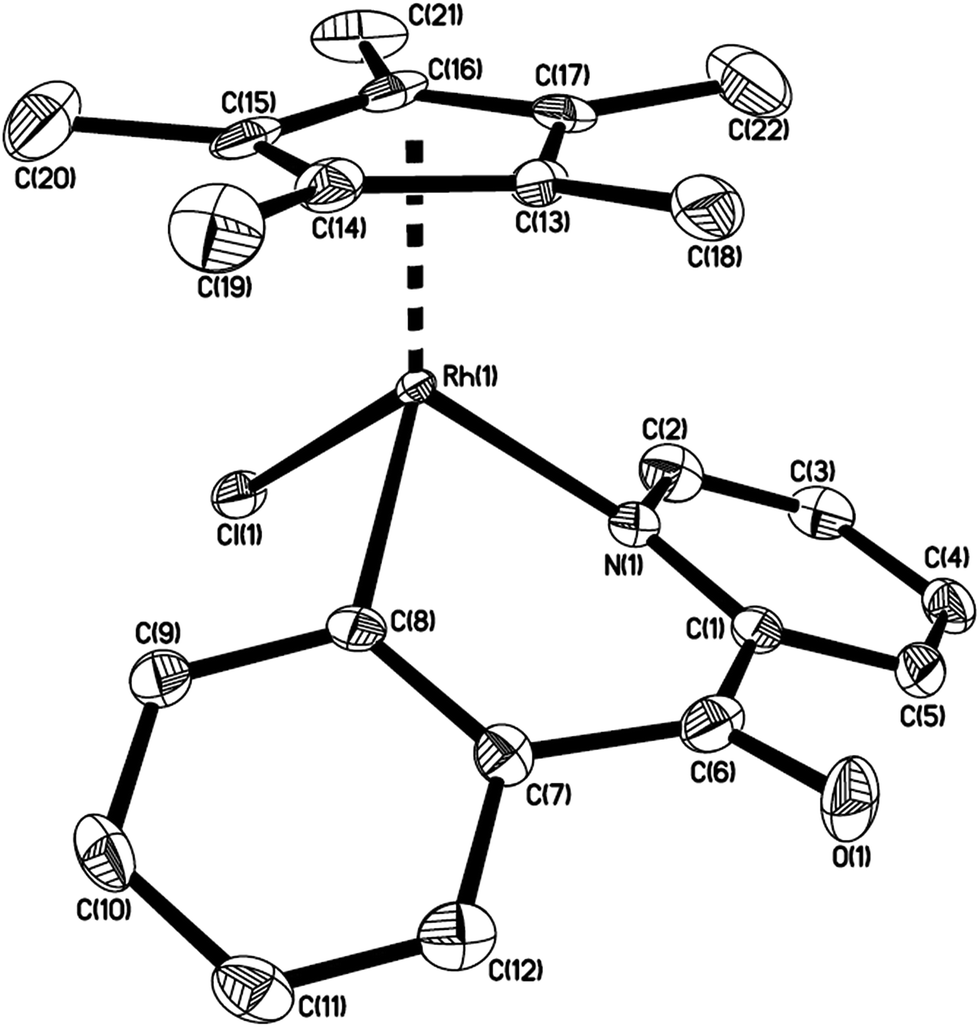
The structure of 4c is shown in Fig. 2 with selected bond lengths and angles. The iridium(III) center is coordinated by a η5 Cp*, one nitrogen atom N(1), and two metalated carbon atoms C(13) and C(16), which come from p-FC6H4CCH. Two fused rings constitute the molecular framework, one is a five-membered ring, and the other is a seven-membered one, they share two adjacent atoms Ir(1) and C(13). For the five-membered ring, all five atoms in the ring are almost in the same plane. The Ir(1)–N(1) bond distance (2.065(12) Å) is slightly shorter than that (2.085(3) Å) in the analogue which is generated from the reaction of 2-(2,3,4,5-tetrafluorophenyl)pyridine cycloiridated complex with diphenylacetylene.10a
 | ||
| Fig. 2 Thermal ellipsoid drawing of 4c showing the labeling scheme and 30% probability ellipsoids; hydrogens are omitted for clarity. Selected bond lengths [Å] and angles [°] are Ir(1)–N(1) 2.065(12), Ir(1)–C(13) 2.062(12), Ir(1)–C(16) 2.090(13), C(13)–C(14) 1.378(19), C(8)–C(13) 1.455(19), Ir(1)–Cp(centroid) 1.862, ∠N(1)–Ir(1)–C(13) 85.0(5), ∠N(1)–Ir(1)–C(16) 88.6(5), ∠C(13)–Ir(1)–C(16) 78.3(5), ∠Ir(1)–C(13)–C(14) 117.2(11), ∠Ir(1)–C(13)–C(8) 121.1(9). | ||
Reactions of 1 with internal alkynes
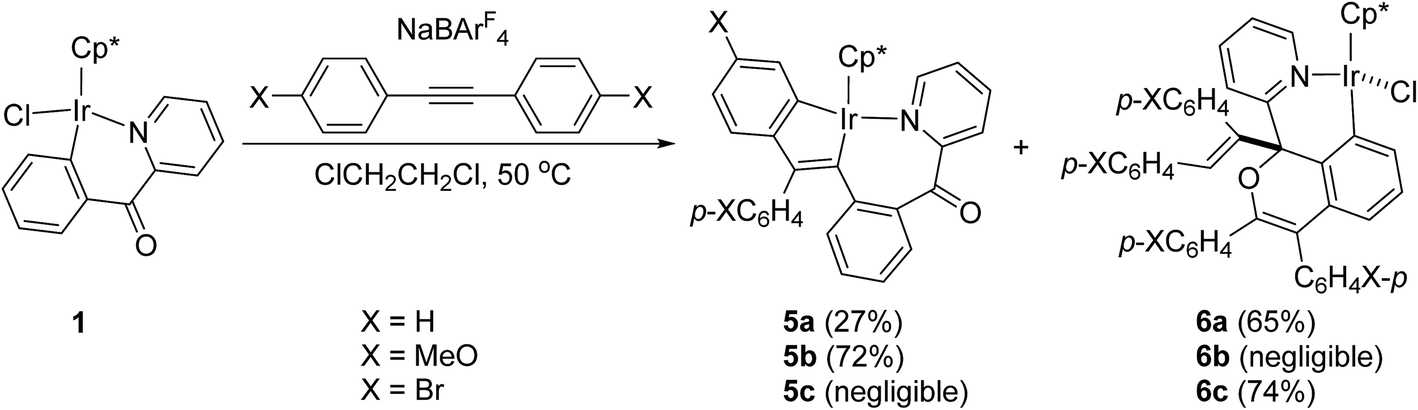
Since six-membered cyclometalated complex 1 showed different reactivity with arylacetylene from the five-membered one, the reaction of 1 with diphenylacetylene was carried out under the same conditions. Unfortunately, neither the expected 1,2-alkyne insertion product nor the double-insertion product was obtained except the starting materials. However, when the reaction was performed in the presence of NaBArF4 in dichloroethane at 50 °C, the five-membered and seven-membered doubly cyclometalated complex 5a was formed, which showed the similar structure to complexes 4a–c (Scheme 4). Besides, the reaction also produced the other product 6a in which 2 equiv. of diphenylacetylene has involved, but different from the double-insertion product mentioned in Scheme 1.10d We considered that NaBArF4 was required in this reaction to help to remove the chloride in 1, then the bulky diphenylacetylene ligand could readily approaching metal center for the subsequent coordination; while in the reaction of 1 with phenylacetylene, the phenylacetylene ligand is relatively less bulky which could attack the metal center directly when the chloride is leaving, so the presence of NaBArF4 was not necessary. Similarly, para-substituted diphenylacetylene derivatives p-XC6H4CCC6H4X-p were applied to the above reaction. When X was electron-donating substituent (MeO), the reaction mainly afforded the corresponding product 5b and trace amounts of 6b, when X was electron-withdrawing substituent (Br), the reaction mainly afforded the corresponding product 6c and trace amounts of 5c.
 | ||
| Scheme 4 Reactions of 1 with diarylacetylenes. | ||
The molecular structure of 5a was confirmed by X-ray single crystal diffraction study, which exhibited the structure similar to 4c. The crystal structure and crystallographic data for 5a were provided in the ESI.†
By careful analysis of the crystal structures of 4a and 5a, we found that one carbon atom is inserted into Ir–C(phenyl) bond of 1, and the two phenyl groups in 5a are bound to the same carbon atom, which indicates that the migration of one phenyl group of diphenylacetylene occurs during the reaction. Besides, we also noticed that an ortho C–H bond of phenyl group from alkyne is activated by the iridium center. Based on these results described above, we proposed a plausible pathway for the formation of 4 or 5 in Scheme 5. Firstly, the chloride of 1 dissociates to form a 16-electron cation, and the aromatic alkyne η2-coordinates to metal center to give complex A, which undergoes vinylidene rearrangement to give the phenylvinylidene intermediate B. This is followed by 1,1-insertion of the vinylidene ligand into the Ir–C(phenyl) bond to form C. In the last step, intramolecular activation of an ortho C–H bond of phenyl group occurs to build the second metallacycle, which comes with an elimination of HX to afford the final product 4 or 5. Ishii group reported similar reactions of five-membered cycloiridated pyridine complex with diphenylacetylene derivatives in the presence of NaBArF4,10a which afforded a nine-membered iridacycle complex with a Ir–(vinyl CH) agnostic interaction (IV in Scheme 1), and then followed by a further stirring in MeOH to give the doubly cyclometalated analogues of 4 or 5. Besides, recently we reported similar reactions of five-membered cycloiridated phosphine complex with aromatic alkynes in the absence of NaBArF4, which also produced the corresponding five-membered and six-membered doubly cyclometalated products.12 Based on our results and reported examples of reactions of cycloiridated complexes with aromatic alkynes, we may deduce that this type of doubly cyclometalated complexes are another general products which formed through vinylidene rearrangement of alkynes/1,1-insertion except for the normal seven-membered cyclometalated products generated from 1,2-alkyne insertion, sometimes there exists competition between above two pathways.
 | ||
| Scheme 5 A plausible pathway for formation of 4 or 5. | ||
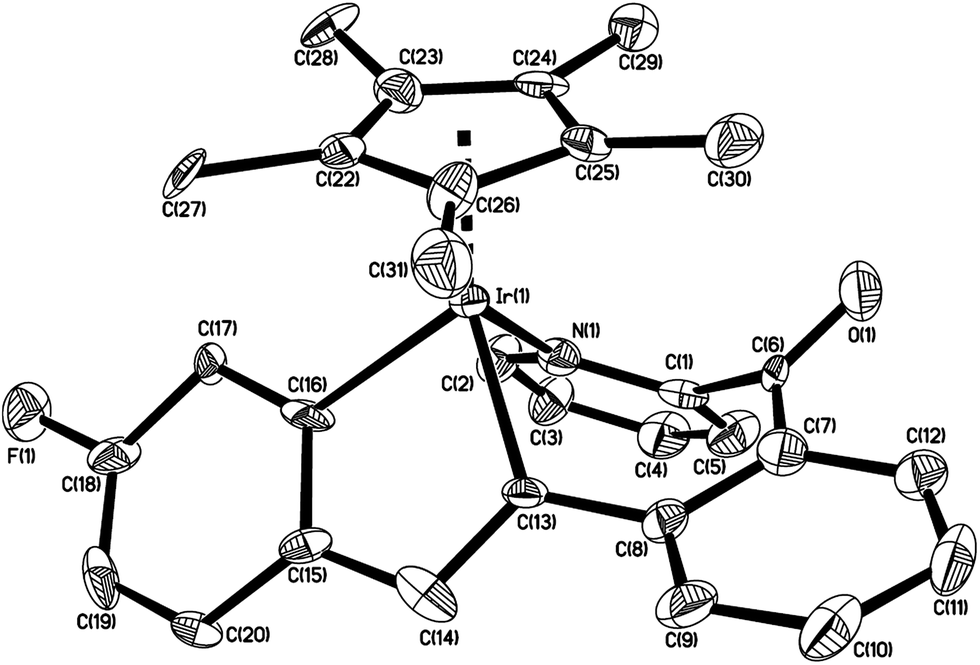
The structure of 6c is shown in Fig. 3 with selected bond lengths and angles, which confirms that the two molecules of p-BrC6H4CCC6H4Br-p are involved in building the molecular framework. The Ir(1)–C(8) bond distance (2.049(5) Å) is slightly longer than that (2.030(5) Å) in the parent 2-benzoylpyridine cycloiridated complex;2b and the Ir(1)–N(1) bond distance (2.106(5) Å) is also slightly longer than that (2.083(4) Å); however the N–Ir–C angles are almost same (86.39(19) and 86.75(16)°, respectively). The angle between the pyridine and phenyl planes (originally from 2-benzoylpyridine ligand) in 6c is 47.2°, which compares with 34.9° in the parent 2-benzoylpyridine cycloiridated complex.2b To the best of our knowledge, the similar structure of 6c has never been reported. Unfortunately, no plausible pathway has yet been proposed to account for the formation of 6c.
 | ||
| Fig. 3 Thermal ellipsoid drawing of 6c showing the labeling scheme and 30% probability ellipsoids; hydrogens are omitted for clarity. Selected bond lengths [Å] and angles [°] are Ir(1)–N(1) 2.106(5), Ir(1)–C(8) 2.049(5), Ir(1)–Cl(1) 2.4122(12), C(1)–N(1) 1.348(7), C(1)–C(6) 1.536(7), C(6)–C(7) 1.518(7), C(7)–C(8) 1.401(7), C(6)–C(33) 1.550(7), C(6)–O(1) 1.459(6), C(33)–C(34) 1.322(8), O(1)–C(20) 1.381(6), C(13)–C(20) 1.347(8), C(12)–C(13) 1.483(7), Ir(1)–Cp(centroid) 1.843, ∠N(1)–Ir(1)–C(8) 86.39(19), ∠C(1)–C(6)–C(7) 114.1(4), ∠C(6)–O(1)–C(20) 117.5(4). | ||
Reaction of 1 with acyl alkyne PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CCOPh
CCOPh
Apparently, in the reactions of six-membered cyclometalated complex 1 with terminal or internal aromatic alkynes, vinylidene arrangement of alkynes was more favored than 1,2-insertion into the Ir–C bond, while the latter was the main insertion mode in the reactions of five-membered cyclometalated complexes with alkynes (Scheme 1). Here we further investigated the reactivity of 1 with acyl alkyne PhCCCOPh, which prefers to undergo a vinylidene arrangement as a reactive substrate.13 When a mixture of 1 with PhCCCOPh and NaBArF4 in C2H4Cl2 was stirred at room temperature for 30 min, an orange crystal was obtained after recrystallization. X-ray diffraction study disclosed that the orange crystal was the novel spiro-metalated complex 7 (Scheme 6).
 | ||
| Scheme 6 Reaction of 1 with PhCCCOPh. | ||
The structure of 7 was shown in Fig. 4. The iridium(III) center is coordinated by a η5 Cp*, one pyridine ligand, one chlorine atom Cl(1), and one metalated oxygen atom O(1), which comes from the bridging carbonyl group. Two spiro five-membered rings constitute the molecular framework, one is a five-membered ring (C(16)–C(17)–C(18)–C(23)–C(30)), and the other is a metalated five-membered one (Ir(1)–N(1)–C(11)–C(16)–O(1)), they share one carbon atom C(16). All five atoms in each ring are perfectly in the same plane, and the dihedral angle between these two planes is 87.5°, which means that they are almost perpendicular. The Ir(1)–N(1) bond distance (2.098(4) Å) is also slightly longer than that (2.074(5) Å) in the 2-phenylpyridine cycloiridated complex.2c
 | ||
| Fig. 4 Thermal ellipsoid drawing of 7 showing the labeling scheme and 30% probability ellipsoids; hydrogens are omitted for clarity. Selected bond lengths [Å] and angles [°] are Ir(1)–N(1) 2.098(4), Ir(1)–O(1) 2.080(3), Ir(1)–Cl(1) 2.4207(11), C(16)–O(1) 1.392(6), C(16)–C(11) 1.534(6), C(16)–C(17) 1.525(6), C(16)–C(30) 1.567(6), C(23)–C(30) 1.347(7), C(18)–C(23) 1.491(7), Ir(1)–Cp(centroid) 1.766, ∠N(1)–Ir(1)–O(1) 77.27(13), ∠C(11)–C(16)–O(1) 111.3(4), ∠C(17)–C(16)–C(30) 99.7(4). | ||
By careful analysis of the crystal structure of 7, we found that the bridging carbonyl group was involved in the reaction. A plausible pathway for the formation of 7 was proposed in Scheme 7. Firstly, the chloride of 1 dissociates to free up a coordinate space for aromatic alkyne, and then generate a η2-alkyne complex D, which undergoes 1,2-alkyne insertion to give the eight-membered metallacycle E. The most noteworthy is the regioselectivity of the alkyne insertion in this system, the benzoyl group is found on the carbon atom adjacent to the metal, while the phenyl group is on the carbon atom adjacent to the phenyl pyridine. This regioselectivity is the same as that found by Ishii for the reaction of this alkyne with the similar five-membered cycloiridated pyridine complex,10a also the same as that reported by Davies in the reaction of ester alkyne PhCCCO2Et with the five-membered cycloiridated oxazoline complex.10e Subsequently, this is followed by an intramolecular nucleophilic addition of metal organic reagent to the bridging carbonyl group to form the spiro-metalated intermediate F. In the last step, recoordination of chlorine to metal center in F forms the final neutral product 7. Apparently, 1,2-insertion into the Ir–Cphenyl bond was more favored in this case than vinylidene rearrangement of alkyne.
 | ||
| Scheme 7 A plausible pathway for formation of 7. | ||
Reactions of 2 with internal alkynes
For comparison, we also studied the reactions of the other six-membered cycloiridated pyridine complex 2 with diarylacetylenes. When a mixture of 2 with p-XC6H4CCC6H4X-p (X = H, MeO, or Br) and NaBArF4 in C2H4Cl2 was stirred at room temperature for 0.5–1.5 h, the five-membered and seven-membered doubly cyclometalated complex 8a–c was formed, which showed the similar structure to complex 5a–c (Scheme 8). In this case, there was no other products obtained, which was different from the case of 1, the latter also formed the other product 6a, the two diarylacetylene involved product. For the effect of substituent on the reactivity, it was observed that the reaction with arylacetylene containing an electron-donating substituent (p-OMe) is faster than the reaction with that containing an electron-withdrawing substituent (p-F), which is consistent with the case of 1.
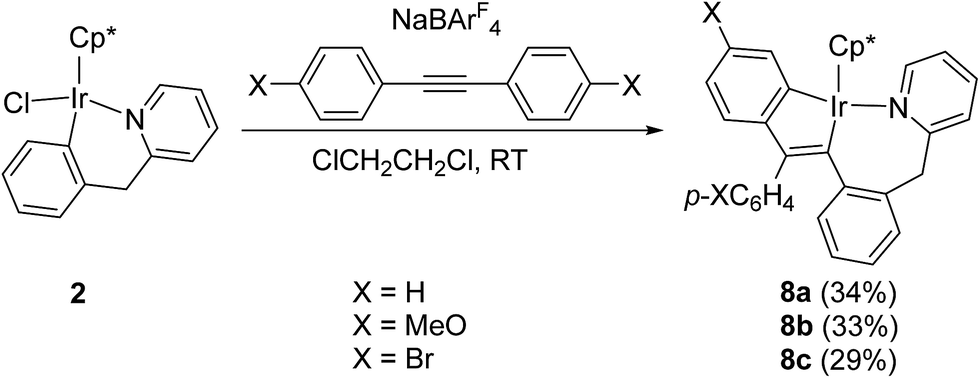 | ||
| Scheme 8 Reactions of 1 with diarylacetylenes. | ||
The structure of 8b is shown in Fig. 5. The iridium(III) center is coordinated by a η5 Cp*, one nitrogen atom N(1), and two metalated carbon atoms C(23) and C(26), which come from p-MeOC6H4CCC6H4OMe-p. Two fused rings constitute the molecular framework, one is a five-membered ring, and the other is a seven-membered one, they share two adjacent atoms Ir(1) and C(23). For the five-membered ring, all five atoms in this ring are almost in the same plane. The Ir(1)–N(1) bond distance (2.103(4) Å) is slightly shorter than that (2.065(12) Å) in 4c, and almost the same as that (2.088(6) Å) in 5a.
 | ||
| Fig. 5 Thermal ellipsoid drawing of 8b showing the labeling scheme and 30% probability ellipsoids; hydrogens are omitted for clarity. Selected bond lengths [Å] and angles [°] are Ir(1)–N(1) 2.103(4), Ir(1)–C(23) 2.085(4), Ir(1)–C(26) 2.035(4), C(23)–C(24) 1.364(5), C(18)–C(23) 1.485(6), Ir(1)–Cp(centroid) 1.878, ∠N(1)–Ir(1)–C(23) 81.31(15), ∠N(1)–Ir(1)–C(26) 88.06(15), ∠C(23)–Ir(1)–C(26) 78.34(17), ∠Ir(1)–C(23)–C(24) 116.4(3), ∠Ir(1)–C(23)–C(18) 122.7(3). | ||
Reaction of 2 with acyl alkyne PhCCCOPh
For comparison with 1, reaction of six-membered cyclometalated complex 2 was similarly investigated. When a mixture of 2 with PhCCCOPh and NaBArF4 in C2H4Cl2 was stirred at room temperature for 25 min, a yellow crystal was obtained after recrystallization. X-ray diffraction study disclosed that the yellow crystal was the novel four-membered cyclometalated complex 9 (Scheme 9).
 | ||
| Scheme 9 Reaction of 2 with PhCCCOPh. | ||
The structure of 9 was shown in Fig. 6. For clarity, the anion BArF4− part is omitted. The iridium cationic center is coordinated by a η5 Cp*, a η2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, one nitrogen atom N(1), and one metalated carbon atoms C(16), which comes from the bridging methylene group. The four-membered ring (Ir(1)–N(1)–C(15)–C(16)) adopts a nonplanar “puckered” conformation. The Ir(1)–C(16) bond distance is 2.125(4) Å, which is shorter than those (2.235(4) and 2.193(4) Å) in Ir(1)(η2–(C(23)C(30))) unit, but longer than that (2.062(12) Å) in five-membered metallacycle analogue 4c, which might be ascribed to the strain of four-membered ring.
C double bond, one nitrogen atom N(1), and one metalated carbon atoms C(16), which comes from the bridging methylene group. The four-membered ring (Ir(1)–N(1)–C(15)–C(16)) adopts a nonplanar “puckered” conformation. The Ir(1)–C(16) bond distance is 2.125(4) Å, which is shorter than those (2.235(4) and 2.193(4) Å) in Ir(1)(η2–(C(23)C(30))) unit, but longer than that (2.062(12) Å) in five-membered metallacycle analogue 4c, which might be ascribed to the strain of four-membered ring.
 | ||
| Fig. 6 Thermal ellipsoid drawing of 9 showing the labeling scheme and 30% probability ellipsoids; anion and hydrogens are omitted for clarity. Selected bond lengths [Å] and angles [°] are Ir(1)–N(1) 2.084(3), Ir(1)–C(16) 2.125(4), Ir(1)–C(23) 2.235(4), Ir(1)–C(30) 2.193(4), C(23)–C(30) 1.505(6), Ir(1)–Cp(centroid) 1.891, ∠N(1)–Ir(1)–C(16) 64.44(15), ∠C(16)–Ir(1)–C(23) 80.96(15), ∠C(23)–Ir(1)–C(30) 37.80(14), ∠C(24)–C(23)–C(30) 126.5(4), ∠C(23)–C(30)–C(31) 130.9(4), ∠C(16)–C(17)–C(22)–C(23) −5.1(5), ∠Ir(1)–N(1)–C(15)–C(16) 17.3(3). | ||
By careful analysis of the crystal structure of 9, we found that the bridging methylene group was involved in the reaction. We proposed a plausible pathway for the formation of 9 in Scheme 10. The first two steps are similar to that for the formation of 7, which afford the corresponding η2-alkyne intermediate G and 16e− unsaturated iridium species H. This is followed by oxidative addition of a C–H bond of methylene group to form I. In the final step, reductive elimination of vinyl and hydro groups on the metal to give an alkene, which subsequently coordinates to metal center in η2-mode to give the final product 9. Also, 1,2-insertion into the Ir–Cphenyl bond was more favored in this case than vinylidene rearrangement of alkyne.
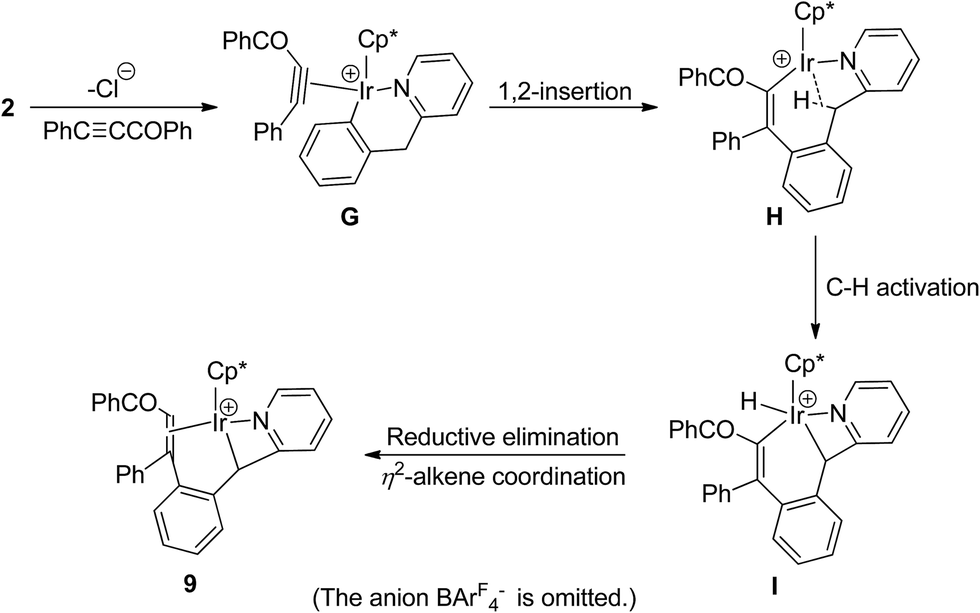 | ||
| Scheme 10 A plausible pathway for formation of 9. | ||
By comparing the above two pathways for formation of 7 and 9 (Schemes 8 and 10), we observed something in common between them: both of two eight-membered metallacycle intermediates (E and H) underwent an intramolecular reaction. The former is a nucleophilic addition to the bridging carbonyl group, and the latter is oxidative addition of a C–H bond on methylene group. The driving force for the above two reactions may come from the flexibility of eight-membered rings and the stability of the resulting products. While the similar reaction of five-membered cyclometalated iridium complex [Cp*IrCl(ppy-F4)] (ppy-F4 = 2,3,4,5-tetrafluoro-6-(2-pyridyl)phenyl) with PhCCCOPh afforded the ten-membered iridacycle complex and the vinyliridium complex by the corresponding 1,2-alkyne insertion and vinylidene rearrangement/1,1-insertion.10a
In summary, insertion reactions of the six-membered cyclometalated pyridine iridium complexes with terminal or internal aromatic alkynes were firstly reported, which showed different reactivity from those of the five-membered cyclometalated iridium analogues. The resulting stable products presented the wide diversity, which were formed from either 1,2-alkyne insertion or vinylidene rearrangement/1,1-alkyne insertion mode and then further intramolecular transformation. These results may also explain why [Cp*IrCl2]2 is catalytically inactive in the catalytic reaction of 2-benzoylpyridine or 2-benzylpyridine with aromatic alkynes.
Experimental
General considerations
All reactions were carried out under nitrogen using standard Schlenk and vacuum line techniques; however, the workup was carried out in air unless stated otherwise. All solvents were distilled from appropriate drying agents under nitrogen prior to use. 1H (400 MHz), and 13C (100 MHz) NMR spectra were recorded on a Bruker AV400 instrument at room temperature with CDCl3 as solvent. Chemical shifts were recorded in ppm, referenced to residual 1H and 13C signals of the non-deuterated CDCl3 (δ 7.26 and 77.16) as internal standards. Elemental analyses were performed on a Perkin-Elmer 240C analyzer. [Cp*MCl2]2 (M = Ir, Rh) were purchased from Strem, NaBArF4 was purchased from Alfa Aesar, 2-benzoylpyridine, 2-benzylpyridine, p-XC6H4CCH (X = H, MeO, and F), and p-XC6H4CCC6H4X-p (X = H and Br) were purchased from Sigma-Aldrich and used as received. p-MeOC6H4CCC6H4MeO-p14 and PhCCCOPh15 were synthesized according to the literature.
General procedures for the reactions of [Cp*MCl2]2 (M = Ir, Rh) with 2-benzoylpyridine or 2-benzylpyridine
A mixture of [Cp*MCl2]2 (M = Ir, Rh) (0.05 mmol), phosphine or 2-benzoylpyridine or 2-benzylpyridine (2.5 equiv.), and sodium acetate (4.0 equiv.) in methanol (10 mL) was stirred for 2–6 h at 60 °C. The solvent was then removed under vacuum and the residue was chromatographed on a silica gel column with a mixture of petrol ether/ethyl acetate (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent. The products were recrystallized from n-hexane/CH2Cl2 (1:1) at −10 °C to afford 1–3 as orange, yellow, or red crystals.
:1) as the eluent. The products were recrystallized from n-hexane/CH2Cl2 (1:1) at −10 °C to afford 1–3 as orange, yellow, or red crystals.
1, reaction time 6 h, orange crystals (85% yield). The NMR data for 1 are consistent with reported data.2b
2, reaction time 2 h, yellow crystals (92% yield). The NMR data for 2 are consistent with reported data.3b
3, reaction time 5 h, red crystals (41% yield). 1H NMR δ 9.27 (d, J = 5.3 Hz, 1H), 8.18 (d, J = 7.5 Hz, 1H), 7.94 (t, J = 7.3 Hz, 1H), 7.87 (dd, J = 11.2, 7.7 Hz, 2H), 7.50 (dd, J = 11.9, 5.7 Hz, 1H), 7.31 (d, J = 6.0 Hz, 1H), 7.09 (dd, J = 13.6, 6.0 Hz, 1H), 1.34 (s, 15H, Cp*). 13C{1H} NMR: δ 191.2, 172.9 (d, J = 30 Hz), 156.1, 155.3, 139.8, 138.2, 137.1, 132.2, 129.4, 127.1, 125.6, 123.6, 96.8 (d, J = 6.3 Hz), 8.7. Anal. calcd for C22H23ClNORh: C, 57.97; H, 5.09. Found: C, 57.74; H, 5.06.
General procedures for the insertion reactions of alkynes into Ir–C bond of 1
A mixture of 1 (15 mg, 0.03 mmol) and terminal alkynes (5.0 equiv.) in methanol (4 mL) was stirred for 1 h at room temperature. The solvent was then removed under vacuum and the residue was chromatographed on a silica gel column with a mixture of petrol ether/ethyl acetate (6:1) as the eluent. The products were recrystallized from n-hexane/CH2Cl2 at −10 °C to afford 4a–c as red or deep red crystals.
4a, red crystals (48% yield). 1H NMR (400 MHz, CDCl3) δ 8.50 (d, J = 5.1 Hz, 1H), 7.83 (dd, J = 7.8, 1.1 Hz, 1H), 7.70 (dd, J = 5.8, 2.7 Hz, 1H), 7.54 (d, J = 7.8, 1.5 Hz, 1H), 7.41 (d, J = 7.5, 1.3 Hz, 1H), 7.30 (d, J = 6.8 Hz, 1H), 7.18–7.13 (m, 1H), 7.09 (dd, J = 7.9, 0.9 Hz, 1H), 7.04 (dd, J = 5.9, 2.8 Hz, 1H), 6.89–6.79 (m, 4H), 1.45 (s, 15H, Cp*). 13C{1H} NMR: δ 194.1, 168.4, 161.1, 160.6, 156.3, 156.2, 152.6, 141.9, 136.9, 135.0, 133.2, 131.9, 130.5, 125.8, 125.1, 124.8 (two peaks), 124.5, 122.3, 121.4, 90.1, 8.8. Anal. calcd for C30H28IrNO: C, 59.00; H, 4.62. Found: C, 58.85; H, 4.71.
4b, deep red crystals (52% yield). 1H NMR (400 MHz, CDCl3) δ 8.53 (d, J = 5.6 Hz, 1H), 7.81 (d, J = 7.7 Hz, 1H), 7.54 (t, J = 7.6 Hz, 1H), 7.38 (d, J = 7.4 Hz, 1H), 7.33 (d, J = 2.3 Hz, 1H), 7.28 (d, J = 7.4 Hz, 1H), 7.15–7.07 (m, 2H), 6.98 (d, J = 8.1 Hz, 1H), 6.84 (t, J = 6.8 Hz, 1H), 6.76 (s, 1H), 6.44 (d, J = 5.6 Hz, 1H), 3.89 (s, 3H, CH3), 1.45 (s, 15H, Cp*). 13C{1H} NMR: δ 194.2, 168.4, 163.0, 156.6, 156.4, 154.2, 152.5, 152.3, 141.1, 136.9, 133.2, 131.7, 130.5, 125.9, 124.8, 122.2, 121.5, 105.5, 90.0, 55.4, 8.8. Anal. calcd for C31H30IrNO2: C, 58.11; H, 4.72. Found: C, 58.06; H, 4.57.
4c, deep red crystals (42% yield). 1H NMR (400 MHz, CDCl3) δ 8.45 (d, J = 5.9 Hz, 1H), 7.84–7.81 (m, 1H), 7.57 (td, J = 7.8, 1.5 Hz, 1H), 7.44–7.38 (m, 2H), 7.29 (s, 1H), 7.16 (t, J = 7.5 Hz, 1H), 7.10 (d, J = 6.9 Hz, 1H), 7.00–6.95 (m, 1H), 6.87 (m, 1H), 6.76 (s, 1H), 6.58–6.52 (m, 1H), 1.44 (s, 15H, Cp*). 13C{1H} NMR: δ 194.1, 168.4, 159.1, 156.5, 156.1, 154.3, 152.6, 140.6, 137.1, 133.3, 131.8, 130.5, 125.8, 125.1, 124.9, 121.7, 121.2, 121.1, 108.0, 107.8, 90.1, 8.7. Anal. calcd for C30H27FIrNO: C, 57.31; H, 4.33. Found: C, 57.43; H, 4.46.
General procedures for the insertion reactions of alkynes into M–C bond of 1 or 2 in the presence of NaBArF4
A mixture of 1 or 2 (0.02 mmol), aromatic alkynes (1.2 equiv.) and NaBArF4 (1.2 equiv.) in 1,2-dichloroethane (5 mL) was stirred for 25 min to 1.5 h at room temperature or at 50 °C. The solvent was then removed under vacuum and the residue was chromatographed on a silica gel column with a mixture of petrol ether/ethyl acetate as the eluent. The products were recrystallized from n-hexane/CH2Cl2 at −10 °C to afford 5–9.5a, reaction time 1 h, 50 °C, orange crystals (27% yield). 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 5.8 Hz, 1H), 7.90 (dd, J = 7.8, 1.3 Hz, 1H), 7.75 (d, J = 7.2 Hz, 1H), 7.60 (td, J = 7.7, 1.5 Hz, 1H), 7.31 (m, 1H), 7.22 (d, J = 6.8 Hz, 1H), 7.13 (m, 1H), 7.06–6.98 (m, 3H), 6.95–6.89 (m, 3H), 6.79 (m, 2H), 6.58 (m, 2H), 1.46 (s, 15H, Cp*). Anal. calcd for C36H32IrNO: C, 62.95; H, 4.70. Found: C, 63.06; H, 4.73.
6a, reaction time 1 h, 50 °C, orange crystals (65% yield). 1H NMR (400 MHz, CDCl3) δ 9.10 (dd, J = 5.9, 1.4 Hz, 1H), 7.64 (dd, J = 7.6, 1.2 Hz, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.50–7.44 (m, 1H), 7.38–7.31 (m, 3H), 7.24 (d, J = 7.3 Hz, 2H), 7.16–6.91 (m, 14H), 6.66 (dd, J = 9.2, 7.6 Hz, 3H), 6.40 (dd, J = 7.6, 1.2 Hz, 2H), 1.54 (s, 15H, Cp*). 13C{1H} NMR: δ 161.9, 159.1, 146.0, 143.6, 141.6, 139.5, 138.3, 137.0, 136.5, 136.3, 136.2, 134.8, 132.8, 131.9, 129.7, 128.7 (two peaks), 128.4, 128.3, 128.2, 128.1, 127.5, 127.2 (two peaks), 127.1, 127.0, 124.8, 120.7, 120.5, 89.6, 89.3, 9.6. Anal. calcd for C50H43ClIrNO: C, 66.61; H, 4.81. Found: C, 66.39; H, 4.88.
5b, reaction time 40 min, room temperature, green crystals (72% yield). 1H NMR (400 MHz, CDCl3) δ 8.39 (d, J = 5.0 Hz, 1H), 7.88 (dd, J = 7.8, 1.1 Hz, 1H), 7.59 (td, J = 7.8, 1.4 Hz, 1H), 7.40 (d, J = 2.5 Hz, 1H), 7.21 (d, J = 6.9 Hz, 1H), 7.03 (d, J = 7.5, 1.4 Hz, 1H), 6.92 (t, J = 7.1 Hz, 1H), 6.80 (m, 1H), 6.67 (m, 4H), 6.58 (d, J = 7.6 Hz, 1H), 6.52 (d, J = 8.2 Hz, 1H), 6.37 (dd, J = 8.3, 2.5 Hz, 1H), 3.88 (s, 3H, CH3), 3.72 (s, 3H, CH3), 1.46 (s, 15H, Cp*). 13C{1H} NMR: δ 194.1, 168.6, 162.9, 157.3, 157.2, 154.5, 152.6, 150.1, 148.7, 137.1, 134.0, 132.3, 132.1, 131.7, 130.3, 129.2, 124.4, 124.2, 123.7, 123.1, 122.0, 113.1, 105.0, 90.3, 55.4, 55.2, 8.8. Anal. calcd for C38H36IrNO3: C, 61.11; H, 4.86. Found: C, 60.95; H, 4.92.
6c, reaction time 1.5 h, 50 °C, orange crystals (74% yield). 1H NMR (400 MHz, CDCl3) δ 9.08 (d, J = 5.8 Hz, 1H), 7.64 (m, 1H), 7.55 (d, J = 4.1 Hz, 2H), 7.49 (d, J = 8.4 Hz, 2H), 7.24 (m, 2H), 7.20 (d, J = 8.4 Hz, 2H), 7.17 (m, 1H), 7.14 (m, 1H), 7.03 (d, J = 7.6 Hz, 2H), 6.99 (t, J = 7.6 Hz, 2H), 6.92 (d, J = 8.4 Hz, 2H), 6.53–6.48 (m, 3H), 6.34 (m, 1H), 6.21 (m, 1H), 5.88 (m, 1H), 1.51 (s, 15H, Cp*). Anal. calcd for C50H39Br4ClIrNO: C, 49.34; H, 3.23. Found: C, 49.55; H, 3.12.
7, reaction time 30 min, room temperature, orange crystals (61% yield). 1H NMR (400 MHz, CDCl3) δ 8.64 (d, J = 5.4 Hz, 1H), 7.70 (s, 3H), 7.52 (dd, J = 11.0, 4.3 Hz, 2H), 7.28 (d, J = 6.9 Hz, 1H), 7.25–7.20 (m, 5H), 7.16–7.07 (m, 5H), 6.78 (d, J = 8.0 Hz, 1H), 1.49 (s, 15H, Cp*). Anal. calcd for C37H33ClIrNO2: C, 59.15; H, 4.43. Found: C, 59.13; H, 4.61.
8a, reaction time 40 min, room temperature, brown crystals (34% yield). 1H NMR (400 MHz, CDCl3) δ 7.68 (t, J = 7.3 Hz, 2H), 7.45–7.42 (m, 1H), 7.15 (m, 3H), 7.11 (dd, J = 8.3, 7.1 Hz, 1H), 6.84 (td, J = 7.4, 1.5 Hz, 2H), 6.80 (m, 4H), 6.76–6.68 (m, 2H), 6.62 (d, J = 7.1 Hz, 1H), 6.41 (dd, J = 7.4, 1.3 Hz, 1H), 4.14 (d, J = 19.2 Hz, 1H), 3.91 (d, J = 19.2 Hz, 1H), 1.67 (d, J = 3.3 Hz, 15H, Cp*). Anal. calcd for C36H34IrN: C, 64.26; H, 5.09. Found: C, 64.11; H, 5.22.
8b, reaction time 25 min, room temperature, brown crystals (33% yield). 1H NMR (400 MHz, CDCl3) δ 8.53 (d, J = 4.4 Hz, 1H), 7.51 (d, J = 2.6 Hz, 1H), 7.38 (m, 1H), 7.27 (m, 5H), 7.14 (d, J = 7.0 Hz, 1H), 6.88 (m, 1H), 6.83 (m, 1H), 6.57 (d, J = 8.3 Hz, 1H), 6.50 (t, J = 6.0 Hz, 1H), 6.43 (d, J = 8.1 Hz, 1H), 6.34 (m, 1H), 4.29 (d, J = 13.9 Hz, 1H), 4.08 (d, J = 14.3 Hz, 1H), 3.89 (s, 3H, CH3), 3.69 (s, 3H, CH3), 1.57 (s, 15H, Cp*). Anal. calcd for C38H38IrNO2: C, 62.27; H, 5.23. Found: C, 62.22; H, 5.39.
8c, reaction time 1.5 h, room temperature, brown crystals (29% yield). 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 1.9 Hz, 1H), 7.67 (d, J = 8.3 Hz, 1H), 7.35 (m, 1H), 7.26 (m, 3H), 7.20–7.17 (m, 3H), 7.15 (t, J = 5.3 Hz, 1H), 6.92 (m, 2H), 6.84 (m, 1H), 6.67 (d, J = 7.2 Hz, 1H), 6.26 (d, J = 7.9 Hz, 1H), 4.13 (d, J = 19.9 Hz, 1H), 3.94 (d, J = 19.3 Hz, 1H), 1.66 (s, 15H, Cp*). 13C{1H} NMR: δ 182.1, 154.1, 150.3, 147.0, 146.9, 138.0, 136.9, 135.1, 132.4, 131.6, 128.3, 127.5, 127.1, 126.2, 123.7, 122.7, 119.8, 117.4, 114.7, 93.9, 91.3, 48.6, 9.0. Anal. calcd for C36H32Br2IrN: C, 52.05; H, 3.88. Found: C, 51.79; H, 4.03.
9, reaction time 25 min, room temperature, brown crystals (28% yield). 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 7.5 Hz, 2H), 7.72–7.65 (m, 11H), 7.60 (t, J = 7.4 Hz, 1H), 7.49 (s, 4H), 7.44 (t, J = 8.0 Hz, 3H), 7.30 (d, J = 7.8 Hz, 1H), 7.24–7.16 (m, 4H), 7.10–6.97 (m, 4H), 6.82 (d, J = 8.0 Hz, 1H), 4.27 (s, 1H), 1.16 (s, 15H, Cp*). Anal. calcd for C69H47BF24IrNO: C, 52.95; H, 3.03. Found: C, 52.76; H, 2.92.
Crystallography
Single crystals of complexes 3, 4c, 5a, 6c, 7, 8b, and 9 (CCDC reference numbers 1589484–1589490†) suitable for X-ray diffraction were obtained by crystallization from n-hexane/CH2Cl2 (1:1). Data collection was performed on an Oxford Diffraction SuperNova diffractometer, processed with CrysalisPro, and processed with Olex2, using ShelXT as the solution program and refined with SheLX-2014/7, or on a Bruker SMART 1000, using graphite-monochromated Mo Kα radiation (ω–2θ scans, λ = 0.71073 Å). The crystal data and summary of X-ray data collection are presented in Tables S1 and S2 in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (21572160), and the Program for Innovative Research Team in University of Tianjin (TD12-5038).Notes and references
- (a) J. M. Villar, J. Suárez, J. A. Varela and C. Saá, Org. Lett., 2017, 21, 1702–1705 CrossRef PubMed; (b) D. Ghorai, C. Dutta and J. Choudhury, ACS Catal., 2016, 6, 709–713 CrossRef CAS; (c) W. Zhang, C.-Q. Wang, H. Lin, L. Dong and Y.-J. Xu, Chem.–Eur. J., 2016, 22, 907–910 CrossRef CAS PubMed; (d) Y.-X. Lao, S.-S. Zhang, X.-G. Liu, C.-Y. Jiang, J.-Q. Wu, Q. Li, Z.-S. Huang and H. Wang, Adv. Synth. Catal., 2016, 358, 2186–2191 CrossRef CAS; (e) C.-Q. Wang, W. Zhang, H. Lin, F. Su and L. Dong, Heterocycles, 2016, 92, 1674–1686 CrossRef CAS; (f) D. L. Davies, C. E. Ellul, S. A. Macgregor, C. L. McMullin and K. Singh, J. Am. Chem. Soc., 2015, 137, 9659–9669 CrossRef CAS PubMed; (g) R. Morioka, K. Nobushige, T. Satoh, K. Hirano and M. Miura, Org. Lett., 2015, 17, 3130–3133 CrossRef CAS PubMed; (h) Z. Qi, S. Yu and X. Li, J. Org. Chem., 2015, 80, 3471–3479 CrossRef CAS PubMed; (i) D. Ghorai and J. Choudhury, Chem. Commun., 2014, 50, 15159–15162 RSC; (j) W. Han, G. Zhang, G. Li and H. Huang, Org. Lett., 2014, 16, 3532–3535 CrossRef CAS PubMed; (k) A. G. Algarra, W. B. Cross, D. L. Davies, Q. Khamker, S. A. Macgregor, C. L. McMullin and K. Singh, J. Org. Chem., 2014, 79, 1954–1970 CrossRef CAS PubMed; (l) X.-C. Huang, X.-H. Yang, R.-J. Song and J.-H. Li, J. Org. Chem., 2014, 79, 1025–1031 CrossRef CAS PubMed; (m) C. Ma, C. Ai, Z. Li, B. Li, H. Song, S. Xu and B. Wang, Organometallics, 2014, 33, 5164–5172 CrossRef CAS; (n) G. Zhang, L. Yang, Y. Wang, Y. Xie and H. Huang, J. Am. Chem. Soc., 2013, 135, 8850–8853 CrossRef CAS PubMed; (o) C.-Z. Luo, P. Gandeepan, J. Jayakumar, K. Parthasarathy, Y.-W. Chang and C.-H. Cheng, Chem.–Eur. J., 2013, 19, 14181–14186 CrossRef CAS PubMed; (p) T. Fukutani, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Commun., 2009, 5141–5143 RSC.
- (a) K. J. T. Car, D. L. Davies, S. A. Macgregor, K. Singh and B. Villa-Marcos, Chem. Sci., 2014, 5, 2340–2346 RSC; (b) Y. Boutadla, D. L. Davies, R. C. Jones and K. Singh, Chem.–Eur. J., 2011, 17, 3438–3448 CrossRef CAS PubMed; (c) Y. Boutadla, O. Al-Duaij, D. L. Davies, G. A. Griffith and K. Singh, Organometallics, 2009, 28, 433–440 CrossRef CAS; (d) D. L. Davies, S. M. A. Donald, O. Al-Duaij, J. Fawcett, C. Little and S. A. Macgregor, Organometallics, 2006, 25, 5976–5978 CrossRef CAS; (e) D. L. Davies, O. Al-Duaij, J. Fawcett, M. Giardiello, S. T. Hilton and D. R. Russell, Dalton Trans., 2003, 4132–4138 RSC.
- (a) A. P. Walsh and W. D. Jones, Organometallics, 2015, 34, 3400–3407 CrossRef CAS; (b) L. Li, W. W. Brennessel and W. D. Jones, Organometallics, 2009, 28, 3492–3500 CrossRef CAS.
- (a) L. Barloy, J.-I. Issenhuth, M. G. Weaver, N. Pannetier, C. Sirlin and M. Pfeffer, Organometallics, 2011, 30, 1168–1174 CrossRef CAS; (b) R. M. Haak, F. Berthiol, T. Jerphagnon, A. J. A. Gayet, C. Tarabiono, C. P. Postema, V. Ritleng, M. Pfeffer, D. B. Janssen, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Am. Chem. Soc., 2008, 130, 13508–13509 CrossRef CAS PubMed; (c) C. Scheeren, F. Maasarani, A. Hijazi, J.-P. Djukic, M. Pfeffer, S. D. Zarić, X.-F. Le Goff and L. Ricard, Organometallics, 2007, 26, 3336–3345 CrossRef CAS.
- (a) Y. Kashiwame, S. Kuwata and T. Ikariya, Chem.–Eur. J., 2010, 16, 766–770 CrossRef CAS PubMed; (b) S. Arita, T. Koike, Y. Kayaki and T. Ikariya, Organometallics, 2008, 27, 2795–2802 CrossRef CAS.
- M. E. Tauchert, C. D. Incarvito, A. L. Rheingold, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2012, 134, 1482–1485 CrossRef CAS PubMed.
- (a) B.-B. Guo, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2017, 46, 8190–8197 RSC; (b) L. Lin, Y.-Y. Zhang, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2016, 45, 7014–7021 RSC; (c) Y.-F. Han and G.-X. Jin, Chem. Soc. Rev., 2014, 43, 2799–2823 RSC; (d) Y.-F. Han, H. Li, P. Hu and G.-X. Jin, Organometallics, 2011, 30, 905–911 CrossRef CAS; (e) Y.-F. Han, H. Li, L.-H. Weng and G.-X. Jin, Chem. Commun., 2010, 46, 3556–3558 RSC.
- L. S. Park-Gehrke, J. Freudenthal, W. Kaminsky, A. G. DiPasquale and J. M. Mayer, Dalton Trans., 2009, 1972–1983 RSC.
- W. Bauer, M. Prem, K. Polborn, K. Sünkel, W. Steglich and W. Beck, Eur. J. Inorg. Chem., 1998, 485–493 CrossRef CAS.
- (a) Y. Ikeda, S. Kodama, N. Tsuchida and Y. Ishii, Dalton Trans., 2015, 44, 17448–17452 RSC; (b) M. F. Espada, M. L. Poveda and E. Carmona, Organometallics, 2014, 33, 7164–7175 CrossRef CAS; (c) Y. Boutadla, D. L. Davies, O. Al-Duaij, J. Fawcett, R. C. Jones and K. Singh, Dalton Trans., 2010, 39, 10447–10457 RSC; (d) L. Li, Y. Jiao, W. W. Brennessel and W. D. Jones, Organometallics, 2010, 29, 4593–4605 CrossRef CAS; (e) D. L. Davies, O. Al-Duaij, J. Fawcett and K. Singh, Organometallics, 2010, 29, 1413–1420 CrossRef CAS; (f) L. Li, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2008, 130, 12414–12419 CrossRef CAS PubMed.
- A. Savini, G. Bellachioma, G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Chem. Commun., 2010, 46, 9218–9219 RSC.
- R. Sun, S. Zhang, X. Chu and B. Zhu, Organometallics, 2017, 36, 1133–1141 CrossRef CAS.
- (a) F. E. Fernández, M. C. Puerta and P. Valerga, Inorg. Chem., 2013, 52, 6502–6509 CrossRef PubMed; (b) Y. Mutoh, K. Imai, Y. Kimura, Y. Ikeda and Y. Ishii, Organometallics, 2011, 30, 204–207 CrossRef CAS; (c) V. K. Singh, E. Bustelo, I. de los Ríos, I. Macías-Arce, M. C. Puerta, P. Valerga, M. A. Ortuño, G. Ujaque and A. Lledós, Organometallics, 2011, 30, 4014–4031 CrossRef CAS; (d) E. Bustelo, I. de los Ríos, M. C. Puerta and P. Valerga, Organometallics, 2010, 29, 1740–1749 CrossRef; (e) M. J. Shaw, S. W. Bryant and N. Rath, Eur. J. Inorg. Chem., 2007, 3943–3946 CrossRef CAS.
- Á. M. Martínez, J. Echavarren, I. Alonso, N. Rodríguez, R. G. Arrayás and J. C. Carretero, Chem. Sci., 2015, 6, 5802–5814 RSC.
- R. L. Sahani and R.-S. Liu, Angew. Chem., Int. Ed., 2017, 56, 12736–12740 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1589484–1589490. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra13547e |
| This journal is © The Royal Society of Chemistry 2018 |