
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radical-initiated alkene hydroauration as a route to gold(III) alkyls: an experimental and computational study†
Anna Pintus‡
 and
Manfred Bochmann*
and
Manfred Bochmann*
School of Chemistry, University of East Anglia, Norwich Research Park, Norwich NR4 7TJ, UK. E-mail: m.bochmann@uea.ac.uk; Tel: +44(0)-1603-592044
First published on 12th January 2018
Abstract
The hydroauration of functionalised 1-alkenes by the gold(III) hydride (C^NOMe^C)AuH is initiated by organic radicals and proceeds via (C^N^C)Au(II) radical intermediates following a bimolecular outer-sphere mechanism. The outcome of these reactions is determined by the stability of the gold-substituted radicals, and chemoselectivity correlates with the degree of spin delocalisation in the alkylgold radical intermediates. The reaction is sensitive to steric as well as electronic factors; disubstituted alkenes and alkenes that form unstable radicals give product mixtures or are unreactive. As DFT calculations show, the reactions agree well with the calculated reaction enthalpies and the standard free energy change for the reaction of the gold(II) radical with the respective alkene.
Introduction
We recently reported the synthesis of the first example of a gold(III) hydride complex (C^N^C)AuH, based on the stabilisation provided by a C^N^C pincer ligand framework [(C^N^C) = 2,6-(C6H3But)2pyridine].1 Cyclometallated C^N^C pincer complexes of gold(III)2,3 have proved particularly useful for the stabilisation of otherwise non-isolable species, including gold(III) alkene,4 alkyne,5 CO6 and peroxide complexes.7 (C^N^C)AuH proved to be thermally stable and did not react with air, moisture or even acetic acid and was also unreactive to alkenes and alkynes. On the other hand, it did react with allenes to give gold vinyl complexes in high yield.1 This lack of reactivity is not entirely unsurprising: gold(III) adheres strictly to a square-planar coordination geometry and in (C^N^C)AuH all four coordination sites are occupied, so that these pincer compounds lack the ability to bind unsaturated substrates. However, we discovered that alternative reaction pathways become accessible in the presence of traces of organic radicals capable of abstracting the hydrogen ligand and thus generating (C^N^C)Au(II)˙ radical species. These Au(II) radicals can readily bind alkynes and lead to alkyne hydroauration in a bimolecular outer-sphere process (Scheme 1). Increasing the concentration of radicals greatly increases the rate of insertion reactions into Au–H bonds. This pathway allows the hydroauration of a range of substituted alkynes to give (Z)-vinylgold complexes (C^N^C)Au–C(R1)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(R2) with almost quantitative stereo- and regioselectivity. These reactions are tolerant of a large variety of functional groups including hydroxide and carboxylic acid functions.8 There is a growing realisation of the role that single-electron transfer steps and gold(II) intermediates may play in gold-mediated reactions.9
CH(R2) with almost quantitative stereo- and regioselectivity. These reactions are tolerant of a large variety of functional groups including hydroxide and carboxylic acid functions.8 There is a growing realisation of the role that single-electron transfer steps and gold(II) intermediates may play in gold-mediated reactions.9
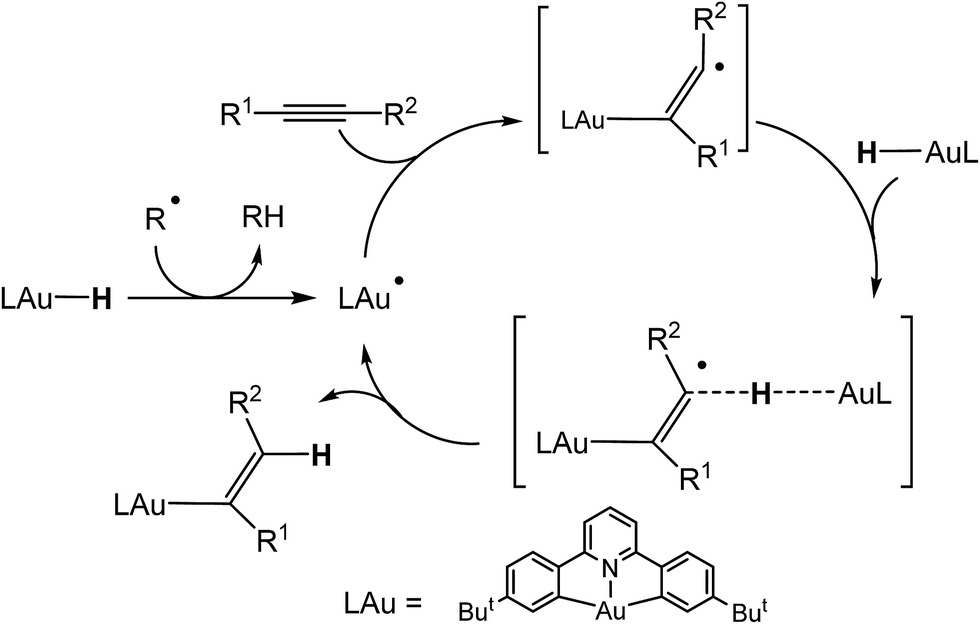 | ||
| Scheme 1 Mechanism of radical-mediated alkyne hydroauration with (C^N^C)AuH pincer complexes.8 | ||
We report here on the reactivity of in situ generated gold(II) radicals with 1-alkenes, which leads to the formation of gold(III) alkyl complexes. Alkyl complexes of C^N^C gold pincer complexes are accessible in a variety of ways: by alkylation with Grignard reagents or aluminium alkyls,10–12 by O-abstraction from (C^N^C)AuOMe with phosphines,13 or by the reaction of (C^N^C)AuOH with allylic alcohols,13 or by the reaction of (C^N^C)AuCl/base with C–H acidic alkanes CH2R1R2. This last method is very versatile and gives alkyls (C^N^C)AuCHR1R2 which carry functional groups in the α-position.12 Here we describe the hydroauration of alkenes to give gold(III) alkyls with functional groups in β-position. The experimental and computational results provide insights into the factors influencing radical-based hydroaurations of unsaturated substrates.
Results and discussion
For solubility reasons, from the library of differently substituted C^N^C gold(III) hydrides previously reported,1,8 we chose to carry out the reactions reported here using the p-OMe substituted gold hydride, (C^NOMe^C)AuH (1). This compound is accessible following literature procedures from (C^NOMe^C)AuCl and LiAlH4 in 85% yield.The reactivity of this complex towards different alkenes was investigated initially through scoping experiments carried out on a small scale, by mixing micromolar quantities of 1 and stoichiometric amounts of the alkene in toluene-d8 in a J-Young NMR tube (Scheme 2). Two molar equivalents of azobisisobutyronitrile (AIBN) were added, the mixture was shaken and heated in the dark to 50 °C to induce the decomposition of AIBN. The progress of the alkene hydroaurations was monitored by 1H NMR spectroscopy. At the end of the reaction the volatile components were removed in vacuo, the residue was washed with n-hexane followed by MeOH to remove any unreacted alkene and excess AIBN, and the residue was dissolved in CD2Cl2. The product was characterized spectroscopically. This method led to the formation of the alkyls 2–8 in high yields.
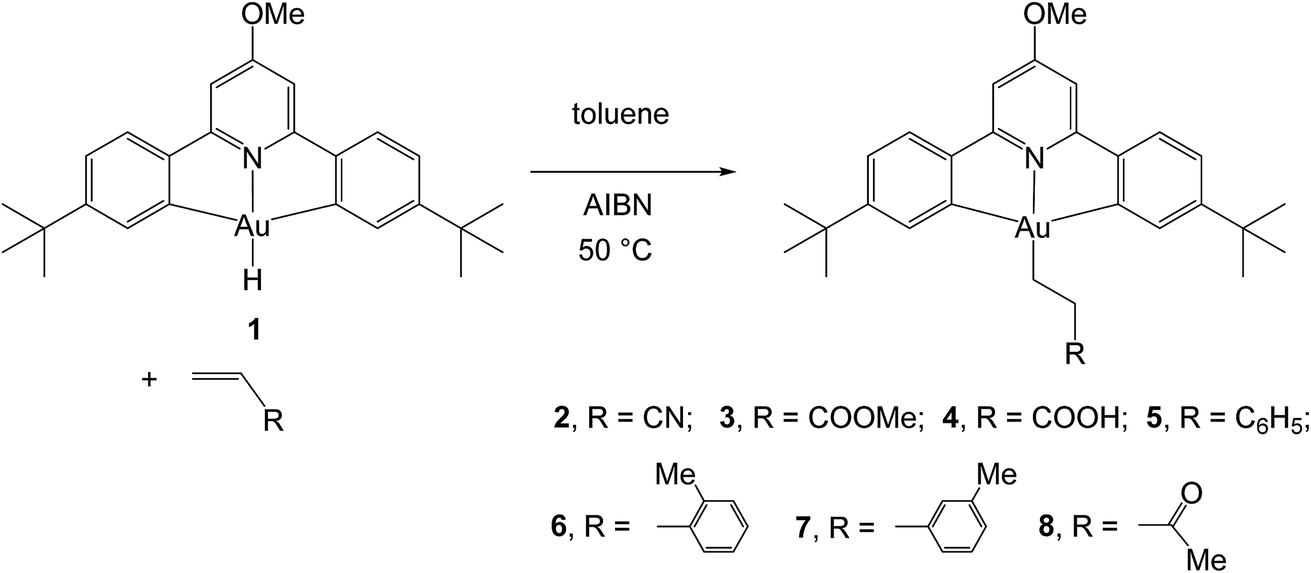 | ||
| Scheme 2 Synthesis of gold(III) alkyls 2–8 from the gold(III) hydride 1. | ||
For the alkenes CH2CHR [R = CN, COOMe, COOH, Ph, 2-MeC6H4, 3-MeC6H4, C(O)Me] this resulted in the clean and facile formation of the corresponding gold-alkyl products 2–8. These reactions were also conducted on a preparative scale and allowed the isolation of the gold alkyls 2–8 as microcrystalline powders in moderate yields, with losses being mainly due to the washing steps during purification. Attempts to obtain crystals suitable for X-ray diffraction were unfortunately not successful.
However, another series of alkenes gave slow reactions that led to mixtures of products which could not be purified. This behaviour was shown by unfunctionalised alkenes (1-pentene and 1-hexene), by allylic derivatives CH2CHR (R = CH2OH, CH2NH2, CH2COOH), and by more highly substituted and internal alkenes, notably 1,1-diphenylethene, α-methylstyrene, p-tert-butylstyrene, 2-methyl-3-butenol and 3-pentenoic acid. Finally, cis- and trans-stilbene, cis- and trans-2-pentene and cyclopentene proved entirely unreactive.
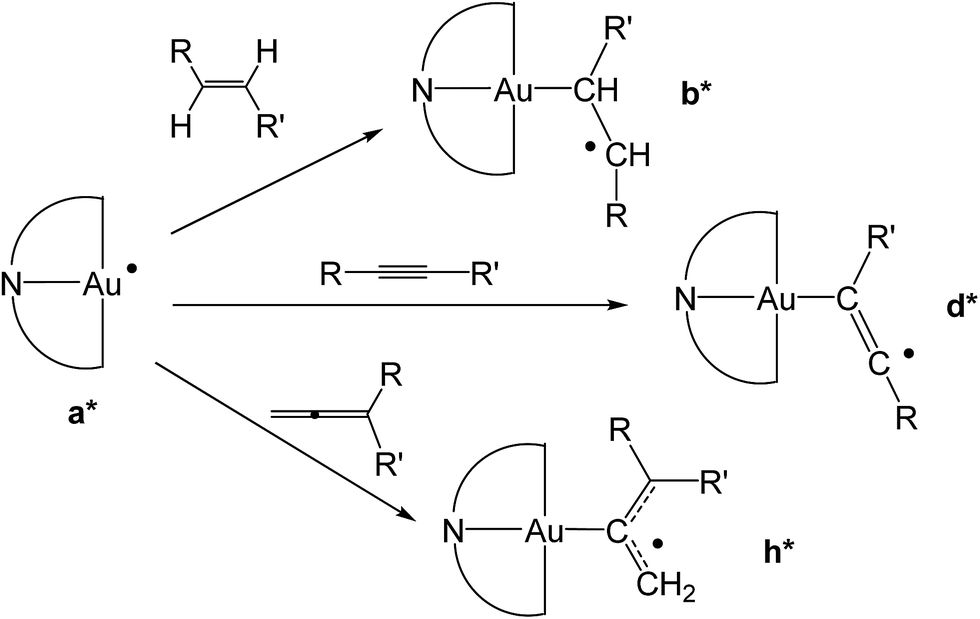
There were therefore two classes of alkenes: those that gave clean insertions into the Au–H bond, and those that showed borderline or no reactivity. In order to rationalize the reactivity differences observed for the various alkene substrates, a computational investigation was undertaken using density functional theory (DFT).14 It is proposed that the mechanism of alkene hydroauration follows the principles previously established for the correspondent alkyne reactions,8 as shown in Scheme 3, and involves various radical intermediates a, b and c.
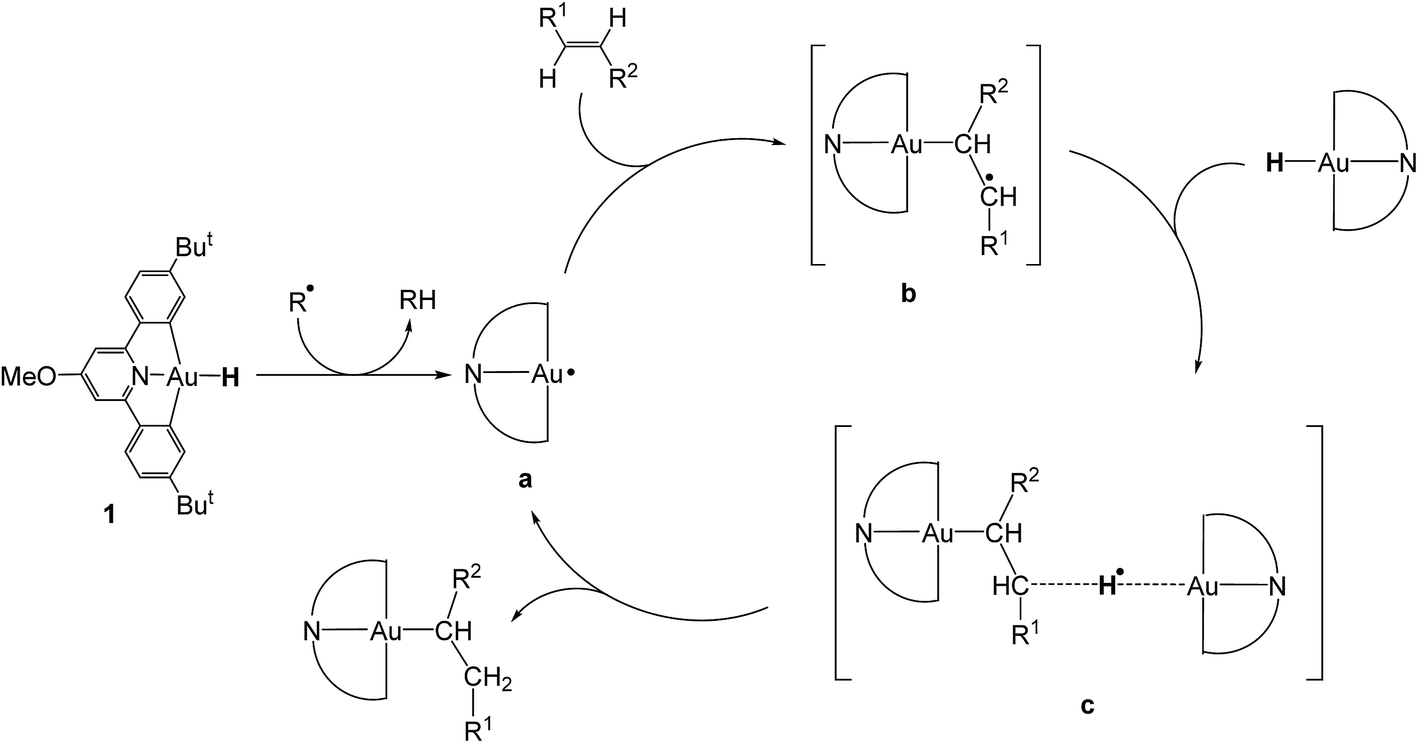 | ||
| Scheme 3 Proposed mechanism for the radical-initiated hydroauration of alkenes by 1. | ||
Our calculations focussed on the first step of the mechanism described above, the formation of the intermediate radical species b* from the gold(II) radical a* and the alkene substrate. The model for the pincer ligand was simplified by omitting the But and OMe substituents (denoted by *). The energetics of this reaction step were investigated by calculating the standard reaction enthalpies  , Gibbs free energy of reaction
, Gibbs free energy of reaction 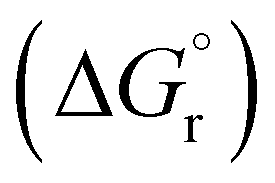 and the total electronic energy differences (ΔEtot) in the gas phase and under standard conditions.
and the total electronic energy differences (ΔEtot) in the gas phase and under standard conditions.
Very similar trends were calculated for the three parameters taken into consideration (see Table 1). In particular, the calculated values of  reflect the experimental observations quite accurately: the formation of alkyl radicals b1*–b5* is energetically favourable, as observed by the clean, near-quantitative formation of gold alkyls in these cases, while reactions leading to b6*–b15* are close to
reflect the experimental observations quite accurately: the formation of alkyl radicals b1*–b5* is energetically favourable, as observed by the clean, near-quantitative formation of gold alkyls in these cases, while reactions leading to b6*–b15* are close to 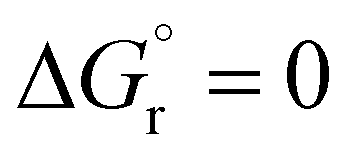 or positive and are therefore not predicted to proceed.
or positive and are therefore not predicted to proceed.
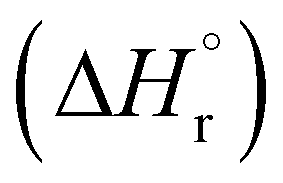 , Gibbs free energy of reaction
, Gibbs free energy of reaction 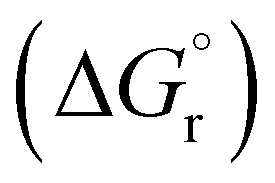 , and total electronic energy differences (ΔEtot) calculated in the gas phase at 298 K for the formation of gold(III) alkyl, vinyl and allenyl radicals from the corresponding unsaturated substrates and a* (Scheme 3)
, and total electronic energy differences (ΔEtot) calculated in the gas phase at 298 K for the formation of gold(III) alkyl, vinyl and allenyl radicals from the corresponding unsaturated substrates and a* (Scheme 3)
| Radical | R | R′ |
|
|
ΔEtot (kcal mol−1) |
|---|---|---|---|---|---|
| b1* | CN | H | −18.68 | −6.52 | −19.61 |
| b2* | COOMe | H | −16.84 | −4.25 | −17.86 |
| b3* | COOH | H | −17.03 | −4.47 | −18.04 |
| b4* | Ph | H | −18.50 | −5.79 | −19.34 |
| b5* | C(O)Me | H | −17.85 | −5.11 | −19.04 |
| b6* | CH2OH | H | −13.59 | −1.31 | −14.07 |
| b7* | CH2NH2 | H | −11.68 | 0.66 | −12.34 |
| b8* | CH2COOH | H | −13.39 | −0.49 | −14.00 |
| b9* | n-Pr | H | −11.32 | 0.92 | −11.83 |
| b10* | n-Bu | H | −11.26 | 0.87 | −11.81 |
| b11* | Ph | Ph | −11.70 | 1.94 | −12.30 |
| b12* | Me | Et | −7.33 | 4.89 | −7.95 |
| b13* | Et | Me | −7.56 | 4.36 | −8.06 |
| b14* | Me | CH2COOH | −8.32 | 4.78 | −8.84 |
| b15* | CH2COOH | Me | −8.24 | 4.28 | −9.12 |
| d1* | Ph | H | −23.20 | −12.48 | −23.94 |
| d2* | Ph | Ph | −17.47 | −6.60 | −17.98 |
| d3* | n-Bu | H | −16.67 | −5.17 | −17.65 |
| d4* | Ph | Me | −17.11 | −6.09 | −18.36 |
| d5* | SiMe3 | H | −17.46 | −6.36 | −18.48 |
| h1* | Me | Me | −29.18 | −16.22 | −29.23 |

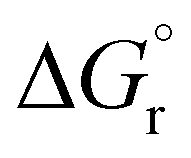
To provide a further insight into the observed trends, the calculations were extended to the previously investigated alkyne8 and allene1 substrates, and in particular to the formation of some of the corresponding vinyl and allyl radicals d* and h*, respectively, from the reaction with a* (Scheme 4). For all of these systems, the calculated values of  , ΔEtot and
, ΔEtot and 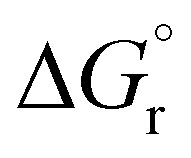 were more negative than in the case of the alkene substrates (Table 1 and Fig. 1). The reaction with allenes to give the allyl radical h1* proved energetically particularly favourable, in agreement with the experimentally observed facile hydroauration of allenes by (C^N^C)AuH.1 The formation of the vinyl radical d1* is also strongly exergonic, while there is little energy difference between the other mono- and disubstituted alkynes in this series.
were more negative than in the case of the alkene substrates (Table 1 and Fig. 1). The reaction with allenes to give the allyl radical h1* proved energetically particularly favourable, in agreement with the experimentally observed facile hydroauration of allenes by (C^N^C)AuH.1 The formation of the vinyl radical d1* is also strongly exergonic, while there is little energy difference between the other mono- and disubstituted alkynes in this series.
 | ||
| Scheme 4 Formation of differently substituted alkyl, vinyl and allyl radicals through the reaction of the gold(II) species a* with alkene, alkyne and allene substrates, respectively. | ||
 | ||
Fig. 1 Trend of  , (T = 298 K) calculated for the reactions depicted in Scheme 3 (kcal mol−1). The substituents for the radical species b1*–b15*, d1*–d5* and h1* are as listed in Table 1. , (T = 298 K) calculated for the reactions depicted in Scheme 3 (kcal mol−1). The substituents for the radical species b1*–b15*, d1*–d5* and h1* are as listed in Table 1. | ||
The chemoselectivity of the hydroauration was explored using the enynes 9 and 10 (Scheme 5), under analogous AIBN-initiated conditions. NMR spectroscopy showed that product mixtures are formed from attack on both the double and triple bonds, which in the case of 9 occurred with about equal probability, while 10 gave an approximately 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 mixture with predominant attack on CC. In agreement with this, calculations of the reaction of species a* with 2-methylbuten-3-yne showed essentially identical ΔEtot values for the formation of bb* (ΔEtot = −26.06 kcal mol−1) and dd* (ΔEtot = −26.11 kcal mol−1) (Scheme 5).
:20 mixture with predominant attack on CC. In agreement with this, calculations of the reaction of species a* with 2-methylbuten-3-yne showed essentially identical ΔEtot values for the formation of bb* (ΔEtot = −26.06 kcal mol−1) and dd* (ΔEtot = −26.11 kcal mol−1) (Scheme 5).
 | ||
| Scheme 5 Reactions with enynes. | ||
In order to rationalize observed alkene reactivity pattern, an investigation of the spin density in different b*, d* and h* radical intermediates was performed. As summarized in Fig. 2, in radicals b1*–b5*, d1*, and h1* a significant degree of spin delocalization is observed, while such delocalization does not arise in b6*–b10*. This suggests that the reason for the energy difference in the formation of radicals on reaction with the gold(II) species reflects these differences in spin delocalisation, the most stable radical intermediates being those stabilized by resonance. Accordingly, all of the alkyne and allene substrates previously explored,1,8 whose radicals can in all cases be stabilized by resonance, were observed to undergo facile hydroauration, while for the alkenes the reactivity depends on the nature of the substituent.
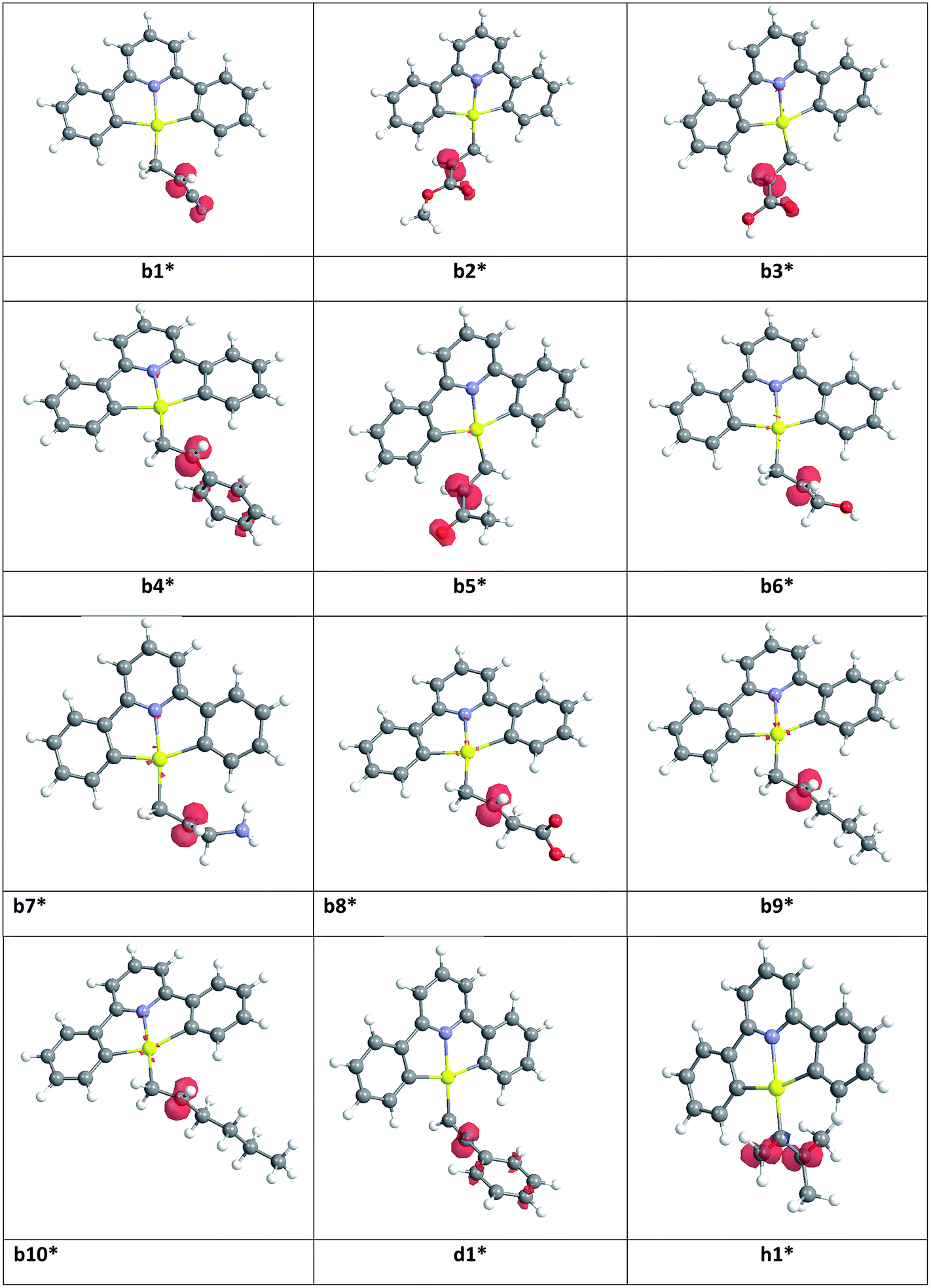 | ||
| Fig. 2 Spin density maps for radical species b1*–b10*, d1* and h1*. | ||
Conclusion
The hydroauration of 1-alkenes with the gold(III) hydride pincer complex (C^NOMe^C)AuH is initiated by radicals and appears to follow the same bimolecular outer-sphere mechanism that has previously been established for the regio- and stereoselective hydroauration of alkynes. The process involves the generation of a (C^N^C)Au(II) radical which reacts with alkenes to give a gold-substituted alkyl radical. According to DFT calculations, the determining factor for the reaction appears to be the energy change associated with the attack by a gold(II) radical species on the alkene. Alkenes leading to alkyl radicals with restricted spin delocalisation either reacted slowly to a mixture of products, or did not react at all. While this limits the scope of the method to some extent, the hydroauration of activated alkenes is a facile method for the metal alkyl-free generation of gold(III) alkyl complexes bearing a variety of functional groups in β-position, including cyano, keto, ester and carboxylic acid functions. Moreover, the present study suggests that the hydroauration by gold(III) hydrides can be extended to different classes of unsaturated species, and that the reactivity trend of different substrates can be rationalized and/or predicted based on the spin delocalization of the radical intermediates involved. The scope of such alkyls for C(sp2)–C(sp3) coupling reactions by reductive elimination using (aryl)(alkyl)gold(III) complexes is currently under investigation.Experimental
When required, manipulations were performed by using standard Schlenk techniques under dry nitrogen or a MBraun glove box. Nitrogen was purified by passing through columns of supported P2O5 with moisture indicator, and of activated 4 Å molecular sieves. Anhydrous solvents were freshly distilled from appropriate drying agents. Elemental analyses were carried out at London Metropolitan University. AIBN (BDH Chemicals) was degassed by evacuation and stored under N2 in the glovebox before use. The alkenes (Sigma Aldrich) were degassed by freeze–pump–thaw cycles and stored over activated 4 Å molecular sieves before use. Solvents, toluene-d8 and CD2Cl2 (Apollo Scientific) were degassed by three freeze–pump–thaw cycles and stored over activated 4 Å molecular sieves prior to use. 1H and 13C{1H} NMR spectra were recorded using a Bruker Avance DPX-300 spectrometer equipped with a 1H, BB smartprobe. 1H NMR spectra (300.13 MHz) were referenced to the residual protons of the deuterated solvent used. 13C{1H} NMR spectra (75.47 MHz) were referenced to the D-coupled 13C resonances of the NMR solvent.Preparation of (C^NOMe^C)AuH (1)
Complex 1 was prepared by a modification of a literature procedure.8 Under a N2 atmosphere, 0.40 g (0.66 mmol) of the chloro complex (C^NOMe^C)AuCl was charged in a dry Schlenk flask with 40 mL of dry toluene. The mixture was cooled to −78 °C and a solution of LiAlH4 in dry THF (0.05 M, 13 mL, 0.66 mmol) was added dropwise. The mixture was stirred at −78 °C in the dark for 15 min, yielding a dark suspension which was filtered under N2. The filtrate was evaporated to dryness to afford a brown powder. This was taken up in dichloromethane, and the resulting suspension was filtered over cotton in the dark, to give a pale-yellow filtrate. The solvent was removed under reduced pressure, and a white solid was obtained, yield 0.38 g (0.56 mmol, 85%).Synthesis and characterization of insertion products 2–8
The compound was synthesized from 1 (40.0 mg, 0.07 mmol), acrylonitrile (5 μL, 0.07 mmol) and 11.0 mg of AIBN (0.007 mmol), and the reaction resulted complete after 2 h at 50 °C. Yield: 20.0 mg (0.03 mmol), 45%. Anal. calcd for C29H33AuN2O: C, 55.95; H, 5.34; N, 4.50. Found: C, 55.41; H, 5.11; N, 5.04. 1H-NMR (300.13 MHz, CD2Cl2): 7.70 (d, 2H, 4J = 1.8 Hz, H8), 7.57 (d, 2H, 3J = 8.1 Hz, H5), 7.30 (dd, 2H, 3J = 8.1, 4J = 1.8 Hz, H6), 6.96 (s, 2H, H2), 4.01 (s, 3H, OMe), 2.89 (t, 2H, 3J = 8.0 Hz, H12), 2.08 (t, 2H, 3J = 8.0 Hz, H13), 1.38 ppm (s, 18H, H11). 13C{1H} NMR (75.47 MHz, CD2Cl2): δ 170.6 (s, C1), 166.7 (s, C9), 164.4 (s, C3), 154.4 (s, C7), 148.2 (s, C4), 130.1 (s, C8), 125.2 (s, C5), 123.7 (s, C6), 122.2 (s, CN), 102.4 (s, C2), 56.5 (s, OMe), 35.6 (s, C10), 31.4 (s, C11), 18.9 (s, C12), 16.7 ppm (s, C13).
This compound was synthesized from 1 (40.0 mg, 0.07 mmol), methyl acrylate (4 μL, 0.07 mmol) and 11.0 mg of AIBN (0.007 mmol). The reaction was complete after 70 min at 50 °C. Yield: 20.0 mg (0.03 mmol), 44%. Anal. calcd for C30H36AuNO3: C, 54.96; H, 5.54; N, 2.14. Found: C, 54.23; H, 5.78; N, 2.68. 1H-NMR (300.13 MHz, CD2Cl2): δ 7.75 (d, 2H, 4J = 1.6 Hz, H8), 7.55 (d, 2H, 3J = 8.1 Hz, H5), 7.27 (dd, 2H, 3J = 8.1, 4J = 1.6 Hz, H6), 6.95 (s, 2H, H2), 4.00 (s, 3H, OMe), 3.59 (s, 3H, COOMe), 2.84 (t, 2H, 3J = 8.0 Hz, H12), 2.10 (t, 2H, 3J = 8.0 Hz, H13), 1.38 ppm (s, 18H, H11). 13C{1H} NMR (75.47 MHz, CD2Cl2): δ 175.7 (s, COOMe), 170.6 (s, C1), 167.3 (s, C9), 164.3 (s, C3), 154.2 (s, C7), 148.2 (s, C4), 130.4 (s, C8), 125.0 (s, C5), 123.4 (s, C6), 102.4 (s, C2), 56.4 (s, OMe), 51.6 (s, COOMe), 35.2 (s, C10), 31.4 (s, C11), 25.4 (s, C12), 18.1 ppm (s, C13).
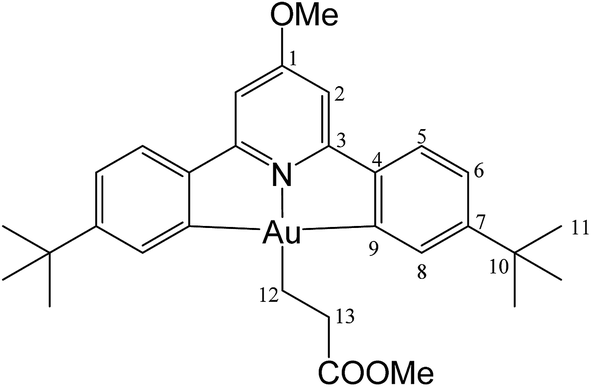
This compound was synthesized from 1 (40.0 mg, 0.07 mmol), acrylic acid (4 μL, 0.07 mmol) and 11.0 mg of AIBN (0.007 mmol), and the reaction resulted complete after 80 min at 50 °C. Yield: 20.5 mg (0.04 mmol), 56%. Anal. calcd for C29H34AuNO3: C, 54.29; H, 5.34; N, 2.18. Found: C, 53.73; H, 5.45; N, 2.76. 1H-NMR (300.13 MHz, CD2Cl2): δ 7.78 (d, 2H, 4J = 1.4 Hz, H8), 7.56 (d, 2H, 3J = 8.2 Hz, H5), 7.28 (dd, 2H, 3J = 8.2, 4J = 1.4 Hz, H6), 6.97 (s, 2H, H2), 4.01 (s, 3H, OMe), 2.89 (t, 2H, 3J = 8.0 Hz, H12), 2.13 (t, 2H, 3J = 8.0 Hz, H13), 1.38 ppm (s, 18H, H11). 13C{1H} NMR (75.47 MHz, CD2Cl2): δ 179.3 (s, COOH), 170.7 (s, C1), 167.2 (s, C9), 164.4 (s, C3), 154.4 (s, C7), 148.3 (s, C4), 130.4 (s, C8), 125.1 (s, C5), 123.5 (s, C6), 102.4 (s, C2), 56.5 (s, OMe), 35.6 (s, C10), 31.4 (s, C11), 25.4 (s, C12), 17.4 ppm (s, C13).

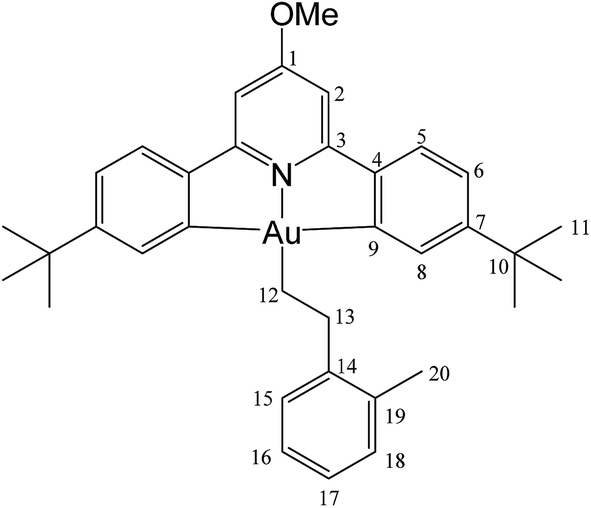
This compound was synthesized from 1 (40.0 mg, 0.07 mmol), styrene (4 μL, 0.07 mmol) and 11.0 mg of AIBN (0.007 mmol). The reaction was complete after 8 h at 50 °C. Yield: 6.0 g (0.009 mmol), 13%. Anal. calcd for C34H38AuNO: C, 60.62; H, 5.69; N, 2.08. Found: C, 59.92; H, 5.78; N, 2.48. 1H-NMR (300.13 MHz, CD2Cl2): δ 7.81 (d, 2H, 4J = 1.8 Hz, H8), 7.54 (d, 2H, 3J = 8.1 Hz, H5), 7.38 (d, 2H, 3J = 7.4 Hz, H15), 7.29 (m, 4H, H6 + H16), 7.16 (t, 1H, 3J = 7.4 Hz, H17), 6.94 (s, 2H, H2), 3.99 (s, 3H, OMe), 3.16 (t, 2H, 3J = 8.2 Hz, H12), 2.14 (t, 2H, 3J = 8.2 Hz, H13), 1.40 ppm (s, 18H, H11). 13C{1H} NMR (75.47 MHz, CD2Cl2): δ 170.6 (s, C1), 167.6 (s, C9), 164.2 (s, C3), 154.1 (s, C7), 148.4 (s, C4), 146.6 (s, C14), 130.4 (s, C8), 128.7 (s, C15 or C16), 128.7 (s, C15 or C16), 125.7 (s, C17), 125.0 (s, C5), 123.4 (s, C6), 102.4 (s, C2), 56.5 (s, OMe), 38.0 (s, C12), 35.6 (s, C10), 31.5 (s, C11), 27.6 ppm (s, C13).

This compound was synthesized from 1 (40.0 mg, 0.07 mmol), 2-methylstyrene (6 μL, 0.07 mmol) and 11.0 mg of AIBN (0.007 mmol), and the reaction resulted complete after 6 h at 50 °C. Anal. yield: 19.0 g (0.027 mmol), 39%. Calcd for C33H40AuNO: C, 61.13; H, 5.86; N, 2.04. Found: C, 60.65; H, 5.36; N, 2.38. 1H-NMR (300.13 MHz, CD2Cl2): δ 7.85 (d, 2H, 4J = 1.8 Hz, H8), 7.57 (d, 2H, 3J = 8.2 Hz, H5), 7.35 (d, 2H, 3J = 7.2 Hz, H15), 7.28 (dd, 2H, 3J = 8.2, 4J = 1.8 Hz, H6), 7.14 (m, 2H, H16 + H18), 7.08 (t, 1H, 3J = 7.2 Hz, H17), 6.96 (s, 2H, H2), 4.0 (s, 3H, OMe), 3.15 (t, 2H, 3J = 8.7 Hz, H12), 2.51 (s, 3H, Me), 2.07 (t, 2H, 3J = 8.7 Hz, H13), 1.39 ppm (s, 18H, H11). 13C{1H} NMR (75.47 MHz, CD2Cl2): δ 170.6 (s, C1), 167.6 (s, C9), 164.2 (s, C3), 154.1 (s, C7), 148.4 (s, C4), 144.5 (s, C14), 135.9 (s, C19), 130.5 (s, C15), 130.5 (s, C8), 129.5 (C16), 125.6 (s, C17 or C18), 125.9 (C17 or C18), 125.1 (s, C5), 123.4 (s, C6), 102.4 (s, C2), 56.4 (s, OMe), 35.6 (s, C10), 35.2 (s, C12), 31.6 (s, C11), 25.9 (s, C13), 19.8 ppm (s, Me).
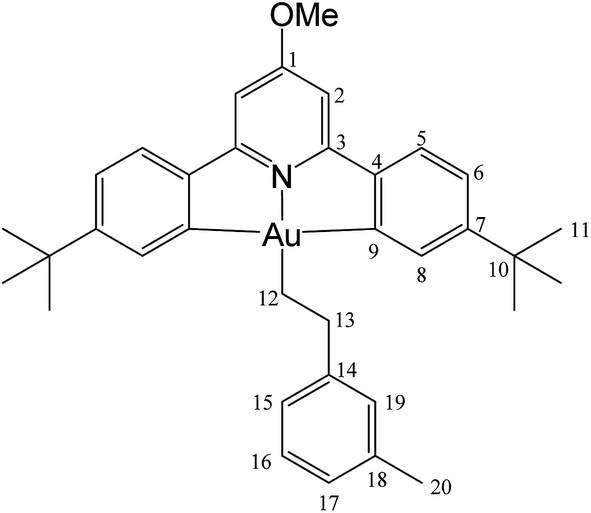
This compound was synthesized from 1 (40.0 mg, 0.07 mmol), 3-methylstyrene (9 μL, 0.07 mmol) and 11.0 mg of AIBN (0.007 mmol), and the reaction resulted complete after 8 h at 50 °C. Yield: 12.0 mg (0.02 mmol), 25%. Calcd for C33H40AuNO: C, 61.13; H, 5.86; N, 2.04. Found: C, 61.82; H, 5.97; N, 1.85. 1H-NMR (300.13 MHz, CD2Cl2): 7.83 (d, 2H, 4J = 1.9 Hz, H8), 7.58 (d, 2H, 3J = 8.1 Hz, H5), 7.29 (dd, 2H, 3J = 8.1, 4J = 1.9 Hz, H6), 7.20 (m, 3H, H15 + H17 + H19), 6.98 (m, 3H, H2 + H16), 4.0 (s, 3H, OMe), 3.11 (t, 2H, 3J = 8.1 Hz, H12), 2.33 (s, 3H, Me), 2.14 (t, 2H, 3J = 8.1 Hz, H13), 1.40 ppm (s, 18H, H11). 13C{1H} NMR (75.47 MHz, CD2Cl2): δ 170.6 (s, C1), 167.6 (s, C9), 164.2 (s, C3), 154.1 (s, C7), 148.4 (s, C4), 146.5 (s, C14), 138.2 (s, C19), 130.4 (s, C8), 129.6 (s, C15), 128.6 (C16), 126.5 (s, C17 or C18), 125.6 (C17 or C18), 125.1 (s, C5), 123.4 (s, C6), 102.4 (s, C2), 56.4 (s, OMe), 37.9 (s, C12), 35.6 (s, C10), 31.5 (s, C11), 27.7 (s, C13), 21.6 ppm (s, Me).
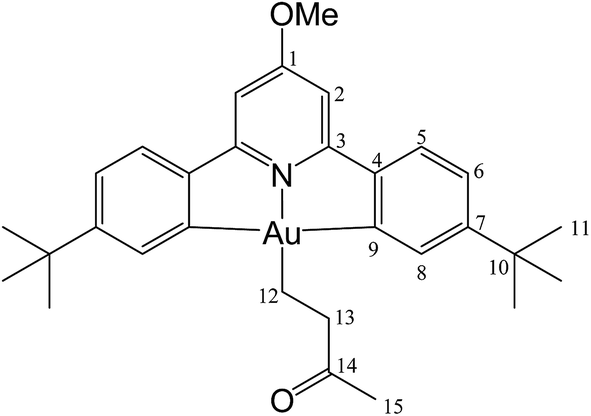
This compound was synthesized from 1 (40.0 mg, 0.07 mmol), 3-buten-2-one (6 μL, 0.07 mmol) and 11.0 mg of AIBN (0.007 mmol). The reaction was complete after 60 min at 50 °C. Yield: 35.0 mg (0.06 mmol), 78%. Anal. calcd for C30H36AuNO2: C, 56.34; H, 5.67; N, 2.19. Found: C, 56.21; H, 5.57; N, 2.30. 1H-NMR (300.13 MHz, CD2Cl2): δ 7.74 (d, 2H, 4J = 1.4 Hz, H8), 7.57 (d, 2H, 3J = 8.2 Hz, H5), 7.28 (dd, 2H, 3J = 8.2, 4J = 1.4 Hz, H6), 6.97 (s, 2H, H2), 4.01 (s, 3H, OMe), 2.93 (t, 2H, 3J = 8.1 Hz, H12), 2.93 (s, 3H, Me), 2.20 (t, 2H, 3J = 8.1 Hz, H13), 1.37 ppm (s, 18H, H11). 13C{1H} NMR (75.47 MHz, CD2Cl2): δ 210.6 (s, C(O)Me), 170.6 (s, C1), 167.3 (s, C9), 164.2 (s, C3), 154.2 (s, C7), 148.3 (s, C4), 130.3 (s, C8), 125.1 (s, C5), 123.5 (s, C6), 102.4 (s, C2), 56.4 (s, OMe), 45.4 (s, C(O)Me), 35.6 (s, C10), 31.4 (s, C11), 29.6 (s, C12), 17.4 ppm (s, C13).
DFT calculations
Theoretical calculations were performed at the Density Functional Theory (DFT) level14 using the Gaussian 09 package of programs.15 Geometry optimizations were carried out using the PBE0 (PBE1PBE) hybrid functional,16 along with the Schäfer, Horn, and Ahlrichs double-ζ plus polarization all-electron basis sets17 for all atomic species. The nature of the minima of each optimized structure was verified by harmonic frequency calculations. The software Chemissian18 was used for the preparation of spin density distribution figures.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the European Research Council. M. B. is an ERC Advanced Investigator Award holder (grant no. 338944-GOCAT).References
- D.-A. Roşca, D. A. Smith, D. L. Hughes and M. Bochmann, Angew. Chem., Int. Ed., 2012, 51, 10643 CrossRef PubMed.
- D.-A. Roşca, J. A. Wright and M. Bochmann, Dalton Trans., 2015, 44, 20785 RSC.
- R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 2 CrossRef , and references cited therein.
- N. Savjani, D.-A. Roşca, M. Schormann and M. Bochmann, Angew. Chem., Int. Ed., 2013, 52, 874 CrossRef CAS PubMed.
- L. Rocchigiani, J. Fernandez-Cestau, G. Agonigi, I. Chambrier, P. H. M. Budzelaar and M. Bochmann, Angew. Chem., Int. Ed., 2017, 56, 13861 CrossRef CAS PubMed.
- D.-A. Roşca, J. Fernandez-Cestau, J. Morris, J. A. Wright and M. Bochmann, Sci. Adv., 2015, 1, e1500761 Search PubMed.
- D. –A. Roşca, J. A. Wright, D. L. Hughes and M. Bochmann, Nat. Commun., 2013, 4, 2167 Search PubMed.
- A. Pintus, L. Rocchigiani, J. Fernandez-Cestau, P. H. M. Budzelaar and M. Bochmann, Angew. Chem., Int. Ed., 2016, 55, 12321 CrossRef CAS PubMed.
- (a) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505 CrossRef CAS PubMed; (b) A. Tlahuext-Aca, M. N. Hopkinson, B. Sahoo and F. Glorius, Chem. Sci., 2016, 7, 89 RSC; (c) M. S. Winston, W. J. Wolf and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 7777 CrossRef CAS PubMed; (d) X.-Z. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844 CrossRef CAS PubMed; (e) S. Kim, J. Rojas-Martin and F. D. Toste, Chem. Sci., 2016, 7, 85 RSC; (f) D. V. Patil, H. Yun and S. Shin, Adv. Synth. Catal., 2015, 357, 2622 CrossRef CAS; (g) L. Huang, F. Rominger, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2016, 52, 6435 RSC; (h) L. Huang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 4808 CrossRef CAS PubMed.
- (a) D. A. Smith, D.-A. Roşca and M. Bochmann, Organometallics, 2012, 31, 5988 CrossRef; (b) E. Langseth, C. H. Görlitz, R. H. Heyn and M. Tilset, Organometallics, 2012, 31, 6567 CrossRef CAS.
- W.-P. To, G. S. M. Tong, C.-W. Cheung, C. Yang, D. Zhoua and C.-M. Che, Inorg. Chem., 2017, 56, 5046 CrossRef CAS PubMed.
- J. Fernandez-Cestau, B. Bertrand, A. Pintus and M. Bochmann, Organometallics, 2017, 36, 3304 CrossRef CAS.
- I. Chambrier, D.-A. Roşca, J. Fernandez-Cestau, D. L. Hughes, P. H. M. Budzelaar and M. Bochmann, Organometallics, 2017, 36, 1358 CrossRef CAS PubMed.
- W. Koch and M. C. Holthausen, A Chemist's Guide to Density Functional Theory, Wiley-VCH, Weinheim, Germany, 2nd edn, 2002 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef.
- L. V. Skripnikov, Chemissian V. 4.43, Visualization Computer Program, http://www.chemissian.com, 2016 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13481a |
| ‡ Present address: Dipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, S.S. 554 Bivio per Sestu, 09042 Monserrato (CA), Italy. |
| This journal is © The Royal Society of Chemistry 2018 |