
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu-catalyzed mild and efficient oxidation of THβCs using air: application in practical total syntheses of perlolyrine and flazin†
Bo Zheng,
Tien Ha Trieu,
Tian-Zhuo Meng,
Xia Lu,
Jing Dong,
Qiang Zhang and
Xiao-Xin Shi *
*
Shanghai Key Laboratory of Chemical Biology, School of Pharmacy, East China University of Science and Technology, 130 Mei-Long Road, Shanghai 200237, P. R. China. E-mail: xxshi@ecust.edu.cn
First published on 12th February 2018
Abstract
A mild, efficient and environmentally benign method for synthesis of aromatic β-carbolines via Cu(II)-catalyzed oxidation of 1,2,3,4-tetrahydro-β-carbolines (THβCs) was developed, in which air (O2) was used as the clean oxidant. This method has advantages such as environmentally friendliness, mildness, very good tolerance of functional groups, high yielding and easy experiment operation. In addition, this new methodology was successfully applied in the efficient and practical total syntheses of β-carboline alkaloids perlolyrine and flazin.
Introduction
The aromatic β-carboline skeleton is ubiquitous in many alkaloids,1 bioactive congeners,2 agrochemicals3 and functional materials.4 In view of the significance of β-carbolines in drug discovery, agricultural chemistry and material science, development of practical, efficient and environmentally benign methods for syntheses of β-carbolines is of considerable interest and has attracted much attention from chemists.5Since 1,2,3,4-tetrahydro-β-carbolines (THβCs) can be readily and efficiently prepared via the Pictet–Spengler reaction,6 so the aromatization of THβCs via dehydrogenation or oxidation is an easy and good method to obtain β-carbolines. Dehydrogenation of THβCs usually employed precious metallic catalysts including Pd,7 Pt8 and complexes of Ru9 and Ir.10 Oxidation of THβCs normally needed stoichiometric strong oxidants such as KMnO4,11 MnO2,12 SeO2,13 DDQ,14 TCCA15 and IBX.16 However, uses of expensive metallic catalysts and hazardous strong oxidants are sometimes lack of cheapness, practicality and environmental friendliness, therefore novel practical, efficient and environmentally benign methods for the conversion of THβCs to β-carbolines are highly desirable.
Oxygen is an important component of air, which is one of the most abundant resources on the earth, so the air has been used as a clean and eco-friendly oxidant for various oxidation reactions in recent decades.17 On the other hand, copper is one of the most abundant metals on the earth's crust, thus copper salts are normally very cheap, and moreover copper is a low toxic transition metal, so an increasing amount of copper-catalyzed reactions have been developed recently.18 Herein, we describe a new practical and very mild Cu(II)-catalyzed oxidative conversion of THβCs to β-carbolines using air as the clean oxidant.
Results and discussion
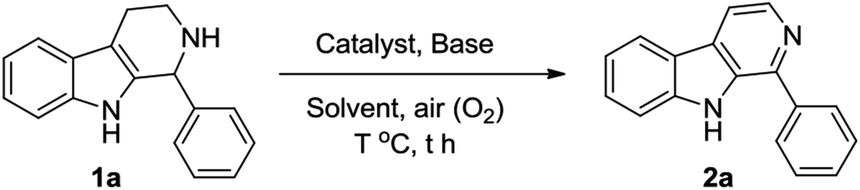
At first, we attempted to find out the optimized reaction conditions for the Cu-catalyzed oxidative conversion of THβCs 1 to β-carbolines 2. With the conversion 1-phenyl-THβC 1a to 1-phenyl-β-carboline 2a as the model reaction, we tried the reaction under various conditions, and results are summarized in Table 1. As can be seen from Table 1, various copper reagents have been tested as catalysts (Table 1, Entries 2–11), and it was found that cupric bromide is the best catalyst for the reaction. When 0.2 equivalents of CuBr2 was used as the catalyst, the aerobic oxidation of compound 1a took place smoothly at room temperature in DMSO in the presence of 2 equivalents of 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU) to afford the desired compound 2a in high yield (Entry 2). Several amines such as DBU, 1,5-diazabicyclo[4,3,0]non-5-ene (DBN), 4-dimethyl-aminopyridine (DMAP), pyridine and trimethylamine have been tested as bases (Entries 2 and 12–15), DBU was found to be the most appropriate base for the reaction. Amount of DBU has significant effect on the reaction (Entries 2 and 16–19), almost no desired product 2a could be obtained in the absence of DBU (Entry 16); and the reaction was hard to be complete even for a longer time, if less than 2.0 equivalents of DBU were used (Entries 17–19). Several other solvents such as N,N-dimethylformamide (DMF), acetonitrile, ethanol, tetrahydrofuran (THF), dichloromethane, ethyl acetate and acetone have also been examined (Entries 20–26); DMSO is obviously better than them. Additionally, when the reaction temperature was elevated, the reaction velocity increased, but the yield of desired product 2a obviously decreased (Entries 27–29).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalystb | Basec | Solvent | T (°C) | t (h) | Yieldd (%) |
| a All reactions were performed at T °C for t hours under an atmosphere of air.b 20% (mol%) of the catalyst was used.c 2.0 equivalents of the base were used unless otherwise indicated.d Isolated yields.e 1,8-Diazabicyclo[5,4,0]undec-7-ene.f Dimethyl sulfoxide.g 1,5-Diazabicyclo[4,3,0]non-5-ene.h 4-Dimethylaminopyridine.i 0.5 equivalent of DBU was used.j 1.0 equivalent of DBU was used.k 1.5 equivalents of DBU was used.l N,N-Dimethylformamide.m Tetrahydrofuran. | ||||||
| 1 | None | DBUe | DMSOf | 25 | 18 | 0 |
| 2 | CuBr2 | DBU | DMSO | 25 | 18 | 95 |
| 3 | CuCl2 | DBU | DMSO | 25 | 18 | 90 |
| 4 | Cu(OAc)2 | DBU | DMSO | 25 | 18 | 86 |
| 5 | CuBr | DBU | DMSO | 25 | 18 | 85 |
| 6 | CuCl | DBU | DMSO | 25 | 18 | 84 |
| 7 | CuI | DBU | DMSO | 25 | 18 | 82 |
| 8 | CuSO4 | DBU | DMSO | 25 | 24 | <5 |
| 9 | CuCO3 | DBU | DMSO | 25 | 24 | 11 |
| 10 | CuO | DBU | DMSO | 25 | 24 | <5 |
| 11 | Cu | DBU | DMSO | 25 | 24 | <5 |
| 12 | CuBr2 | DBNg | DMSO | 25 | 20 | 88 |
| 13 | CuBr2 | DMAPh | DMSO | 25 | 20 | 19 |
| 14 | CuBr2 | Py | DMSO | 25 | 20 | 12 |
| 15 | CuBr2 | Et3N | DMSO | 25 | 20 | 10 |
| 16 | CuBr2 | None | DMSO | 25 | 18 | 0 |
| 17 | CuBr2 | DBU (0.5)i | DMSO | 25 | 60 | 75 |
| 18 | CuBr2 | DBU (1.0)j | DMSO | 25 | 30 | 83 |
| 19 | CuBr2 | DBU (1.5)k | DMSO | 25 | 25 | 92 |
| 20 | CuBr2 | DBU | DMFl | 25 | 24 | 82 |
| 21 | CuBr2 | DBU | CH3CN | 25 | 24 | 65 |
| 22 | CuBr2 | DBU | EtOH | 25 | 24 | 23 |
| 23 | CuBr2 | DBU | THFm | 25 | 24 | 30 |
| 24 | CuBr2 | DBU | CH2Cl2 | 25 | 24 | 27 |
| 25 | CuBr2 | DBU | EtOAc | 25 | 24 | 12 |
| 26 | CuBr2 | DBU | Me2CO | 25 | 24 | 14 |
| 27 | CuBr2 | DBU | DMSO | 45 | 13 | 90 |
| 28 | CuBr2 | DBU | DMSO | 65 | 9 | 84 |
| 29 | CuBr2 | DBU | DMSO | 85 | 6 | 76 |
Subsequently, we attempted the CuBr2-catalyzed oxidative conversion of variously substituted THβCs 1 to β-carboline 2 in DMSO with DBU as the base, and the results are summarized in Table 2. The scope of the reaction is very wide; almost all of the tested THβCs could be smoothly converted to β-carbolines in good to excellent yields. All the conversions were performed under the standard reaction conditions (see footnote of the Table 2), and a total of twenty-four variously substituted β-carbolines 2a–2x were obtained in 78–96% yields.


A possible mechanism for the CuBr2-catalyzed oxidative conversion of THβCs 1 to β-carbolines 2 is proposed in Scheme 1. As can be seen from Scheme 1, THβCs 1 would first undergo CuBr2-catalyzed aerobic oxidation of C–N single bond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond to produce 3,4-dihydro-β-carbolines I-A according to Adimurthy's reports.19 3,4-Dihydro-β-carbolines I-A would then undergo DBU-promoted reversible tautomerization20 to form unstable enamine intermediates I-B, which would also undergo the CuBr2-catalyzed aerobic oxidation of C–N single bond to afford β-carbolines 2. Actually, dihydro-β-carbolines I-A could be isolated if the reaction was stopped in the midway. For example, when the CuBr2-catalyzed oxidative conversion of 1-phenyl-THβC 1a to 1-phenyl-β-carboline 2a was stopped at 8 h, 1-phenyl-3,4-dihydro-β-carboline was obtained in 36% yield.
N double bond to produce 3,4-dihydro-β-carbolines I-A according to Adimurthy's reports.19 3,4-Dihydro-β-carbolines I-A would then undergo DBU-promoted reversible tautomerization20 to form unstable enamine intermediates I-B, which would also undergo the CuBr2-catalyzed aerobic oxidation of C–N single bond to afford β-carbolines 2. Actually, dihydro-β-carbolines I-A could be isolated if the reaction was stopped in the midway. For example, when the CuBr2-catalyzed oxidative conversion of 1-phenyl-THβC 1a to 1-phenyl-β-carboline 2a was stopped at 8 h, 1-phenyl-3,4-dihydro-β-carboline was obtained in 36% yield.
 | ||
| Scheme 1 Possible mechanism for the CuBr2-catalyzed oxidative conversion of THβCs 1 to β-carbolines 2. | ||
To showcase the synthetic utility of the above-described method for the CuBr2-catalyzed oxidative conversion of THβCs 1 to β-carbolines 2, we have successively applied the methodology to the efficient and practical total syntheses of two β-carboline alkaloids perlolyrine 3 and flazin 4.
Perlolyrine 3 and flazin 4 are both strongly fluorescent 1-furanyl-β-carboline alkaloids. These two particular alkaloids are widespread in nature, and have been isolated from plants, bacteria, Japanese sake and soy sauce.1h,3b,21 Several total syntheses of perlolyrine 3 and flazin 4 have been reported hitherto,22 we herein report novel practical total syntheses of them, which are depicted in Scheme 2. Pictet–Spengler reaction of tryptamine and tryptophan methyl ester with 2-furaldehyde first gave THβCs 5 and 6 in 82% and 83% yields, respectively. CuBr2-catalyzed oxidation of compounds 5 and 6 with air then furnished β-carbolines 7 and 8 in 89% and 90% yields, respectively. Next, hydroxymethylation of compounds 7 and 8 with HCHO in AcOH produced perlolyrine 3 and compound 9 in 86% and 85% yields, respectively. Hydrolysis of the ester 9 afforded flazin 4 in 92% yield. Thus, perlolyrine 3 was synthesized from tryptamine in 3 steps in 63% overall yield; and flazin 4 was synthesized from tryptophan methyl ester in 4 steps in 58% overall yield.
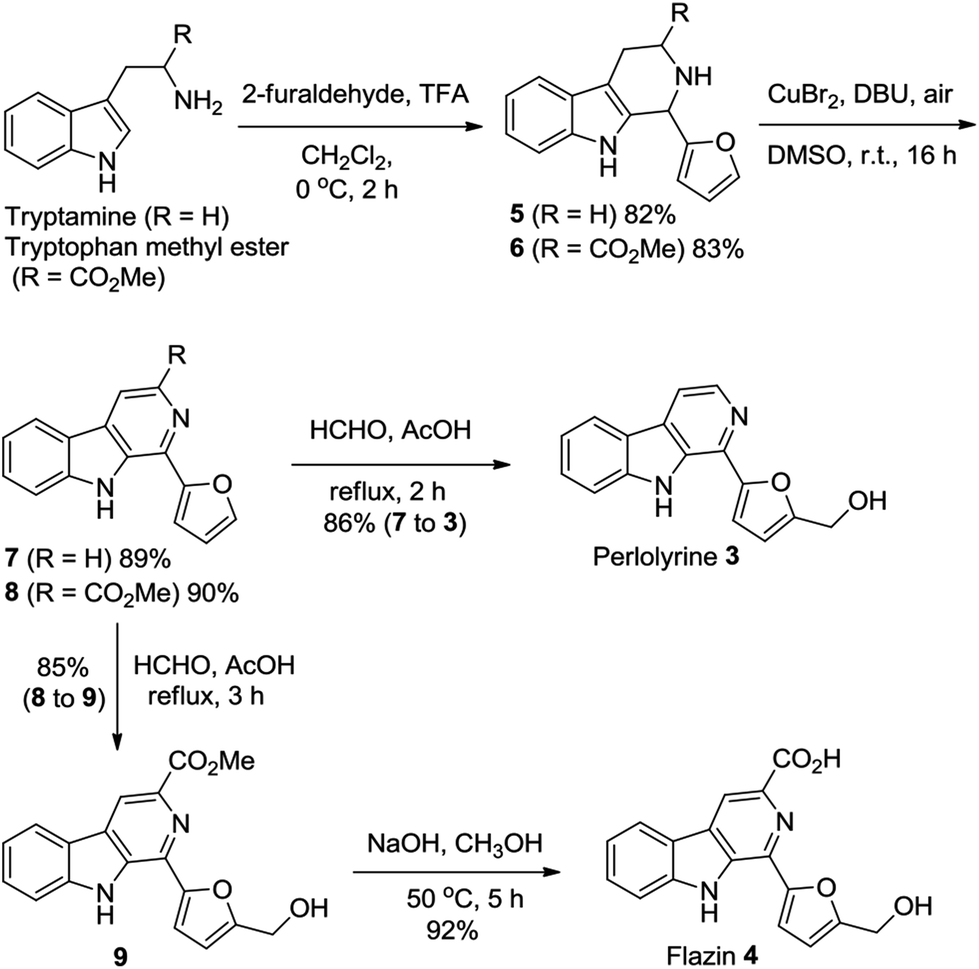 | ||
| Scheme 2 Novel concise total syntheses of β-carboline alkaloids perlolyrine 3 and flazin 4. | ||
Conclusions
In conclusion, we have developed a novel method for the CuBr2-catalyzed oxidative conversion of THβCs to β-carbolines. This method has some advantages as follows: (a) the process was carried out at room temperature with air as the clean oxidant, so it is very mild and environmentally benign; (b) CuBr2 as the catalyst is inexpensive and low-toxic; (c) all products were obtained in good to excellent yields; (d) the reaction has very good tolerance of functional groups, it seemed to be applicable to all the tested substrates; (e) experiment operation is quite easy. In addition, novel practical total syntheses of β-carboline alkaloids perlolyrine and flazin were performed with the above-described CuBr2-catalyzed mild oxidation of THβCs as the key step.Experimental
General
1H NMR and 13C NMR spectra were acquired on a Bruker AM 400 instrument, chemical shifts are given on the δ scale as parts per million (ppm) with tetramethylsilane (TMS) as the internal standard. IR spectra were recorded on a Nicolet Magna IR-550 instrument. Mass spectra were performed with a HP1100 LC-MS spectrometer. Melting points were measured on a Mei-TEMP II melting point apparatus. Column chromatography was performed on silica gel (Qingdao Chemical Factory). All reagents and solvents were analytically pure, and were used as such as received from the chemical suppliers.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4), solvent was removed under vacuum, the residue was then partitioned between EtOAc (30 mL) and an aqueous solution of K2CO3 (10% w/w, 20 mL). Two phases were separated, and the aqueous phase was extracted again with EtOAc (20 mL). Organic extracts were combined, and dried over anhydrous MgSO4. Evaporation of solvent under vacuum gave crude product, which was purified by flash chromatography (eluent: MeOH/CH2Cl2 = 1:20–1:10) to afford THβCs 1a–1x in 70–95% yields.:1 to 1:4), the reaction solution was poured into the mixed solution of EtOAc (30 mL) and aqueous ammonia (5% w/w, 25 mL). After the mixture was stirred for 5 minutes, two phases were separated. The aqueous layer was extracted again with EtOAc (20 mL). The organic extracts were combined and washed with brine (10 mL). The organic solution was dried over anhydrous MgSO4, and then was concentrated under vacuum to give the crude product, which was purified by flash chromatography (eluent: EtOAc/CH2Cl2 = 1:2 to 1:15) to afford pure β-carbolines 2 in 78–96% yield as indicated in the Table 2. Characterization data of the β-carbolines 2a–2x are given in the ESI† of this paper.
:4), solvent was removed under vacuum, the residue was then partitioned between EtOAc (30 mL) and an aqueous solution of K2CO3 (10% w/w, 20 mL). Two phases were separated, and the aqueous phase was extracted again with EtOAc (20 mL). Organic extracts were combined, and dried over anhydrous MgSO4. Evaporation of solvent under vacuum gave crude product, which was purified by flash chromatography (eluent: MeOH/CH2Cl2 = 1:20–1:10) to afford THβCs 1a–1x in 70–95% yields.:1 to 1:4), the reaction solution was poured into the mixed solution of EtOAc (30 mL) and aqueous ammonia (5% w/w, 25 mL). After the mixture was stirred for 5 minutes, two phases were separated. The aqueous layer was extracted again with EtOAc (20 mL). The organic extracts were combined and washed with brine (10 mL). The organic solution was dried over anhydrous MgSO4, and then was concentrated under vacuum to give the crude product, which was purified by flash chromatography (eluent: EtOAc/CH2Cl2 = 1:2 to 1:15) to afford pure β-carbolines 2 in 78–96% yield as indicated in the Table 2. Characterization data of the β-carbolines 2a–2x are given in the ESI† of this paper.Total syntheses of perlolyrine 3 and flazin 4
:3), ethyl acetate (60 mL) and an aqueous solution of K2CO3 (10% w/w, 30 mL) were added. The mixture was vigorously stirred for 5 min, and then two phases were separated. Organic phase was extracted again with ethyl acetate (20 mL). Organic extracts were combined and dried over anhydrous MgSO4, and then was concentrated under vacuum to give the crude product, which was purified by flash chromatography (eluent: MeOH/CH2Cl2 = 1:10) to afford pure compound 5 (1.957 g, 8.213 mmol) in 82% yield as white solid, mp 138–139 °C. 1H NMR (CDCl3, 400 MHz) δ 8.17 (s, 1H, NH in indole), 7.94 (s, 1H), 7.64 (d, J = 7.8 Hz, 1H), 7.48 (d, J = 3.2 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.18 (dd, J1 = 8.0 Hz, J2 = 7.9 Hz, 1H), 7.11 (dd, J1 = 7.8 Hz, J2 = 7.9 Hz, 1H), 6.96 (brs, 1H, NH), 6.66 (d, J = 3.7 Hz, 1H), 6.44 (dd, J1 = 3.7 Hz, J2 = 3.2 Hz, 1H), 3.90 (t, J = 7.3 Hz, 2H), 3.17 (t, J = 7.3 Hz, 2H). 13C NMR (CDCl3, 100 MHz) δ 154.51, 142.52, 135.87, 131.84, 127.23, 121.91, 119.37, 118.34, 111.04, 110.35, 109.94, 107.76, 50.73, 41.46, 22.31. HRMS (ESI) m/z calcd for C15H14N2ONa [M + Na]+: 261.1004, found: 261.1008. IR (KBr, film) 3393, 3145, 2922, 2846, 1450, 1298, 1141, 1010, 739 cm−1.:5) in a combined yield of 83% as offwhite solid. Two epimers can be separated by chromatography, but herein the epimeric mixture was used as such for the next step. Data for cis epimer: white solid, mp 141–142 °C. 1H NMR (CDCl3, 400 MHz) δ 7.98 (s, 1H, NH in indole), 7.50 (d, J = 7.6 Hz, 1H), 7.41 (d, J = 3.5 Hz, 1H), 7.25 (d, J = 7.8 Hz, 1H), 7.15 (dd, J1 = 7.6 Hz, J2 = 7.7 Hz, 1H), 7.11 (dd, J1 = 7.7 Hz, J2 = 7.8 Hz, 1H), 6.32–6.36 (m, 2H), 5.38 (s, 1H), 3.91 (dd, J1 = 9.8 Hz, J2 = 4.5 Hz, 1H), 3.79 (s, 3H), 3.17 (dd, J1 = 15.0 Hz, J2 = 4.5 Hz, 1H), 2.95 (dd, J1 = 15.0 Hz, J2 = 9.8 Hz, 1H), 2.44 (s, 1H, NH). 13C NMR (CDCl3, 100 MHz) δ 173.06, 153.02, 142.85, 136.03, 131.79, 127.01, 122.18, 119.68, 118.28, 111.07, 110.48, 108.61, 107.78, 56.51, 52.33, 51.64, 25.46. HRMS (ESI) m/z calcd for C17H16N2O3Na [M + Na]+: 319.1059, found: 319.1057. IR (KBr, film) 3393, 2950, 1735, 1436, 1351, 1271, 1218, 1174, 1141, 1009, 740 cm−1. Data for trans epimer: white solid, mp 145–146 °C. 1H NMR (CDCl3, 400 MHz) δ 8.30 (s, 1H, NH in indole), 7.49 (d, J = 7.5 Hz, 1H), 7.34 (d, J = 3.4 Hz, 1H), 7.16 (d, J = 7.8 Hz, 1H), 7.11 (dd, J1 = 7.5 Hz, J2 = 7.6 Hz, 1H), 7.06 (dd, J1 = 7.6 Hz, J2 = 7.8 Hz, 1H), 6.23 (dd, J1 = 4.2 Hz, J2 = 3.4 Hz, 1H), 5.96 (d, J = 4.2 Hz, 1H), 5.25 (s, 1H), 3.88 (dd, J1 = 9.5 Hz, J2 = 4.7 Hz, 1H), 3.69 (s, 3H), 3.13 (dd, J1 = 15.2 Hz, J2 = 4.7 Hz, 1H), 2.88 (J1 = 15.2 Hz, J2 = 9.5 Hz, 1H), 2.59 (brs, 1H, NH). 13C NMR (CDCl3, 100 MHz) δ 173.82, 154.40, 142.67, 136.26, 130.97, 126.78, 122.14, 119.49, 118.27, 111.20, 110.28, 108.65, 108.14, 52.24, 52.23, 49.10, 25.17. HRMS (ESI) m/z calcd for C17H16N2O3Na [M + Na]+: 319.1059, found: 319.1055. IR (KBr, film) 3395, 2945, 1736, 1435, 1352, 1271, 1218, 1175, 738 cm−1.:1), the reaction solution was poured into the mixed solution of EtOAc (50 mL) and aqueous ammonia (5% w/w, 20 mL). After the mixture was stirred for 5 minutes, two phases were separated. The aqueous layer was extracted again with EtOAc (20 mL). The organic extracts were combined and washed with brine (10 mL). The organic solution was dried over anhydrous MgSO4, and then was concentrated under vacuum to give the crude product, which was purified by flash chromatography (eluent: EtOAc/CH2Cl2 = 1:3) to afford pure compound 7 (0.842 g, 3.594 mmol) in 89% yield as white solid, mp 166–167 °C. 1H NMR (DMSO-d6, 400 MHz) δ 11.50 (s, 1H, NH in indole), 8.38 (d, J = 5.2 Hz, 1H), 8.25 (d, J = 7.8 Hz, 1H), 8.08 (d, J = 5.2 Hz, 1H), 7.99 (d, J = 1.6 Hz, 1H), 7.77 (d, J = 8.2 Hz, 1H), 7.58 (dd, J1 = 7.6 Hz, J2 = 7.8 Hz, 1H), 7.23–7.32 (m, 2H), 6.79 (dd, J1 = 3.5 Hz, J2 = 3.2 Hz, 1H). 13C NMR (CDCl3, 100 MHz) δ 155.09, 143.34, 141.09, 139.47, 134.09, 131.97, 130.83, 129.17, 122.25, 121.87, 120.72, 114.26, 112.92, 112.25, 109.32. HRMS (ESI) m/z calcd for C15H11N2O [M + H]+: 235.0871, found: 235.0872. IR (KBr, film) 3458, 2998, 2920, 1624, 1557, 1493, 1424, 1377, 1235, 997, 754, 459 cm−1.:3), the solvent was removed by vacuum distillation to give a viscous residue, which was partitioned between ethyl acetate (30 mL) and an aqueous solution of potassium carbonate (10% w/v, 20 mL). Two phases were separated, and aqueous phase was extracted again with ethyl acetate (20 mL). The organic extracts were combined and washed twice with brine (2 × 10 mL). After having been dried with anhydrous MgSO4, the organic solution was concentrated under vacuum to give crude solid, which was purified by flash chromatography (eluent: MeOH/CH2Cl2 = 1:10) to give pure perlolyrine 3 (0.457 g, 1.729 mmol) in 86% yield as pale yellow solid, mp 165–166 °C. 1H NMR (DMSO-d6, 400 MHz) δ 11.24 (s, 1H, NH in indole), 8.38 (d, J = 5.1 Hz, 1H), 8.27 (d, J = 7.8 Hz, 1H), 8.08 (d, J = 5.1 Hz, 1H), 7.78 (d, J = 8.2 Hz, 1H), 7.61 (dd, J1 = 7.8 Hz, J2 = 8.0 Hz, 1H), 7.29 (dd, J1 = 8.0 Hz, J2 = 8.2 Hz, 1H), 7.23 (d, J = 3.3 Hz, 1H), 6.60 (d, J = 3.3 Hz, 1H), 5.50 (t, J = 6.0 Hz, 1H, OH), 4.68 (d, J = 6.0 Hz, 2H). 13C NMR (DMSO-d6, 100 MHz) δ 156.73, 152.14, 140.92, 138.16, 133.14, 130.47, 129.42, 128.39, 121.59, 120.61, 119.68, 113.60, 112.40, 109.62, 109.04, 55.95. HRMS (ESI) calcd for C16H13N2O2 [M + H]+: 265.0977, found: 265.0974. IR (KBr, film) 3370, 2920, 1629, 1568, 1426, 1317, 1234, 1014, 799, 743, 634 cm−1.:1), ethyl acetate (25 mL) and water (20 mL) were added. The mixture was vigorously stirred, and pH value of the aqueous solution was adjusted to 4–5 by adding citric acid. Two phases were separated, and the aqueous solution was extracted twice with ethyl acetate (2 × 25 mL). Organic extracts were combined and washed twice with brine (2 × 10 mL). After having been dried over anhydrous MgSO4, the organic solution was concentrated under vacuum to give a crude solid product, which was purified by flash chromatography (eluent: MeOH/CH2Cl2 = 1:5) to afford pure flazin 4 (0.284 g, 0.921 mmol) in 92% yield as pale yellow solid, mp 239–240 °C. 1H NMR (DMSO-d6, 400 MHz) δ 11.69 (s, 1H, NH in indole), 8.84 (s, 1H), 8.42 (d, J = 7.8 Hz, 1H), 7.87 (d, J = 8.2 Hz, 1H), 7.65 (dd, J1 = 7.8 Hz, J2 = 8.0 Hz, 1H), 7.42 (d, J = 3.2 Hz, 1H), 7.34 (dd, J1 = 8.0 Hz, J2 = 8.2 Hz, 1H), 6.62 (d, J = 3.2 Hz, 1H), 4.69 (s, 2H). 13C NMR (DMSO-d6, 100 MHz) δ 166.64, 157.45, 151.40, 141.53, 137.13, 132.63, 132.06, 129.98, 129.05, 122.17, 121.11, 120.69, 115.85, 112.94, 111.22, 109.37, 56.11. HRMS (ESI) m/z calcd for C17H12N2O4Na [M + Na]+: 331.0695, found: 331.0697. IR (KBr, film) 3242, 2922, 1735, 1624, 1612, 1576, 1360, 1321, 1018, 742 cm−1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 20972048) for the financial support of this work.Notes and references
- (a) W.-H. Jiao, G.-D. Chen, H. Gao, J. Li, B.-B. Gu, T.-T. Xu, H.-B. Yu, G.-H. Shi, F. Yang, X.-S. Yao and H.-W. Lin, J. Nat. Prod., 2015, 78, 125 CrossRef PubMed; (b) K. Ohishi, K. Toume, M. A. Arai, T. Koyano, T. Kowithayakorn, T. Mizoguchi, M. Itoh and M. Ishibashi, J. Nat. Prod., 2015, 78, 1139 CrossRef CAS PubMed; (c) A. Furusato, H. Kato, T. Nehira, K. Eguchi, T. Kawabata, Y. Fujiwara, F. Losung, R. E. P. Mangindaan, N. J. d. Voogd, M. Takeya, H. Yokosawa and S. Tsukamoto, Org. Lett., 2014, 16, 3888 CrossRef CAS PubMed; (d) M. Kitajima, S. Ohara, N. Kogure, D. Santiarworn and H. Takayama, Tetrahedron, 2013, 69, 9451 CrossRef CAS; (e) H. Huang, Y. Yao, Z. He, T. Yang, J. Ma, X. Tian, Y. Li, C. Huang, X. Chen, W. Li, S. Zhang, C. Zhang and J. Ju, J. Nat. Prod., 2011, 74, 2122 CrossRef CAS PubMed; (f) W. D. Inman, W. M. Bray, N. C. Gassner, R. S. Lokey, K. Tenny, Y. Y. Shen, K. TenDyke, T. Suh and P. Grews, J. Nat. Prod., 2010, 73, 255 CrossRef CAS PubMed; (g) W.-H. Jiao, H. Gao, C.-Y. Li, F. Zhao, R.-W. Jiang, Y. Wang, G.-X. Zhou and X.-S. Yao, J. Nat. Prod., 2010, 73, 167 CrossRef CAS PubMed; (h) S. Fotso, R. P. Maskey, D. Schroder, A. S. Ferrer, I. Grun-Wollny and H. Laatsch, J. Nat. Prod., 2008, 71, 1630 CrossRef CAS PubMed; (i) R. Cao, W. Peng, Z. Wang and A. Xu, Curr. Med. Chem., 2007, 14, 479 CrossRef CAS PubMed.
- (a) A. Kamal, M. Sathish, V. L. Nayak, V. Srinivasulu, B. Kavitha, Y. Tangella, D. Thummuri, C. Bagul, N. Shankaraiah and N. Nagesh, Bioorg. Med. Chem., 2015, 23, 5511 CrossRef CAS PubMed; (b) F. C. Savariz, M. A. Foglio, A. L. T. G. Ruiz, W. F. d. Costa, M. d. M. Silva, J. C. C. Santos, I. M. Figueiredo, E. Meyer, J. E. d. Carvalho and M. H. Sarragiotto, Bioorg. Med. Chem., 2014, 22, 6867 CrossRef CAS PubMed; (c) B. Shi, R. Cao, W. Fan, L. Guo, Q. Ma, X. Chen, G. Zhang, L. Qiu and H. Song, Eur. J. Med. Chem., 2013, 60, 10 CrossRef CAS PubMed.
- (a) H. Song, Y. Liu, Y. Liu, L. Wang and Q. Wang, J. Agric. Food Chem., 2014, 62, 1010 CrossRef CAS PubMed; (b) Y. Li, M. Zhao and K. L. Parkin, J. Agric. Food Chem., 2011, 59, 2332 CrossRef CAS PubMed; (c) T. Herraiz, J. Agric. Food Chem., 2007, 55, 8534 CrossRef CAS PubMed.
- (a) Y. Im and L. Y. Lee, Chem. Commun., 2013, 49, 5948 RSC; (b) B. K. Paul, N. Ghosh and S. Mukherjee, RSC Adv., 2016, 6, 9984 RSC.
- (a) F. Nissen, V. Richard, C. Alayrac and B. Witulski, Chem. Commun., 2011, 47, 6656 RSC; (b) S. Ding, Z. Shi and N. Jiao, Org. Lett., 2010, 12, 1540 CrossRef CAS PubMed; (c) G. Verniest, D. England, N. D. Kimpe and A. Padawa, Tetrahedron, 2010, 66, 1496 CrossRef CAS; (d) H. Zhang and R. C. Larock, J. Org. Chem., 2002, 67, 7048 CrossRef CAS PubMed; (e) N. Kanekiyo, T. Kuwada, T. Choshi, J. Nobuhiro and S. Hibino, J. Org. Chem., 2001, 66, 8793 CrossRef CAS PubMed.
- (a) E. D. Cox and J. M. Cook, Chem. Rev., 1995, 95, 1797 CrossRef CAS; (b) J. Stockigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538 CrossRef CAS PubMed.
- (a) R. S. Kusurkar, N. A. H. Alkobati, A. S. Gokule and V. G. Puranik, Tetrahedron, 2008, 64, 1654 CrossRef CAS; (b) A. Kulkarni, M. Abid, B. Torok and X. Huang, Tetrahedron Lett., 2009, 50, 1791 CrossRef CAS; (c) S. Eagon and M. O. Anderson, Eur. J. Org. Chem., 2014, 1653 CrossRef CAS.
- D. Ge, L. Hu, J. Wang, X. Li, F. Qi, J. Lu, X. Cao and H. Gu, ChemCatChem, 2013, 5, 2183 CrossRef CAS.
- S. Muthaiah and S. H. Hong, Adv. Synth. Catal., 2012, 354, 3045 CrossRef CAS.
- J. Wu, D. Talwar, S. Johnston, M. Yan and J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 6983 CrossRef CAS PubMed.
- (a) M. Zheng, Y. Yang, M. Zhao, X. Zhang, J. Wu, G. Chen, L. Peng, Y. Wang and S. Peng, Eur. J. Med. Chem., 2011, 46, 1672 CrossRef CAS PubMed; (b) P. Ashok, S. Chander, J. Balzarini, C. Pannecouque and S. Murugesan, Bioorg. Med. Chem. Lett., 2015, 25, 1232 CrossRef CAS PubMed.
- W. Yin, P. V. V. S. Sarma, J. Ma, D. Han, J. L. Chen and J. M. Cook, Tetrahedron Lett., 2005, 46, 6363 CrossRef CAS.
- M. Cain, O. Campos, F. Guzman and J. M. Cook, J. Am. Chem. Soc., 1983, 105, 907 CrossRef CAS.
- K. Kondo, H. Shigemori, Y. Kikuchi, M. Ishibashi, T. Sasaki and J. Kobayashi, J. Org. Chem., 1992, 57, 2480 CrossRef CAS.
- B. Bai, L. Shen, J. Ren and H. J. Zhu, Adv. Synth. Catal., 2012, 354, 354 CrossRef CAS.
- J. D. Panarese and S. P. Waters, Org. Lett., 2010, 12, 4086 CrossRef CAS PubMed.
- (a) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (b) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed; (c) S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756 CrossRef CAS PubMed; (d) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (e) A. Shaabani, S. Shaabani and H. Afaridoun, RSC Adv., 2016, 6, 48396 RSC; (f) C. Li, S. An, Y. Zhu, J. Zhang, Y. Kang, P. Liu, Y. Wang and J. Li, RSC Adv., 2014, 4, 49888 RSC; (g) H. Tian, H. Qiao, C. Zhu and H. Fu, RSC Adv., 2014, 4, 2694 RSC; (h) A. E. Fernandes, O. Riant, A. M. Jonas and K. F. Jensen, RSC Adv., 2016, 6, 36602 RSC; (i) C. Zhang, P. Zhao, Z. Zhang, J. Zhang, P. Yang, P. Gao, J. Gao and D. Liu, RSC Adv., 2017, 7, 47366 RSC.
- (a) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (b) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525 RSC; (c) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed.
- (a) R. D. Patil and S. Adimurthy, Adv. Synth. Catal., 2011, 353, 1695 CrossRef CAS; (b) R. D. Patil and S. Adimurthy, RSC Adv., 2012, 2, 5119 RSC.
- (a) R. A. Clark and D. C. Parker, J. Am. Chem. Soc., 1971, 93, 7257 CrossRef CAS; (b) J. J. Eisch and F. J. Gadek, J. Org. Chem., 1971, 36, 2065 CrossRef CAS.
- (a) T. Liu, W. Liang and G. Tu, Planta Med., 1988, 54, 472 CrossRef CAS PubMed; (b) B.-N. Su, L. C. Chang, E. J. Park, M. Guendet, B. D. Santarsiero, A. D. Mesecar, R. G. Mehta, H. H. S. Fong, J. M. Pezzuto and A. D. Kinghorn, Planta Med., 2002, 68, 730 CrossRef CAS PubMed; (c) J. A. D. Jeffreys, J. Chem. Soc. C, 1970, 1091 RSC; (d) S.-i. Nakatsuka, B.-n. Feng, T. Goto and K. Kihara, Tetrahedron Lett., 1986, 27, 3399 CrossRef CAS; (e) B. H. Han, M. H. Park, Y. N. Han and L. K. Woo, Arch. Pharmacal Res., 1986, 9, 21 CrossRef CAS; (f) B. H. Han, J. H. Park, M. H. Park and Y. N. Park, Arch. Pharmacal Res., 1985, 8, 249 CrossRef.
- (a) N. Wu, F. Song, L. Yan, J. Li and J. You, Chem. – Eur. J., 2014, 20, 3408 CrossRef CAS PubMed; (b) B. Dassonneville, B. Witulski and H. Detert, Eur. J. Org. Chem., 2011, 2836 CrossRef CAS; (c) W. P. Gessner and A. Brossi, Arch. Pharm., 1988, 321, 95 CrossRef CAS; (d) F. Bracher and D. Hildebrand, Liebigs Ann. Chem., 1992, 1315 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13434g |
| This journal is © The Royal Society of Chemistry 2018 |