
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a series of benzothiazole amide derivatives and their biological evaluation as potent hemostatic agents†
Wenqian Nong a,
Anran Zhaob,
Jinrui Weic,
Hui Chenga,
Xuan Luoa and
Cuiwu Lin*a
a,
Anran Zhaob,
Jinrui Weic,
Hui Chenga,
Xuan Luoa and
Cuiwu Lin*a
aGuangxi Colleges and Universities Key Laboratory of Applied Chemistry Technology and Resource Development, School of Chemistry & Chemical Engineer, Guangxi University, Nanning, 530004, China
bDepartment of Chemistry, Cleveland State University, 2121 Euclid Avenue, Cleveland, OH 44115, USA
cGuangxi Scientific Research Center of Traditional Chinese Medicine, Guangxi University of Chinese Medicine, Nanning, Guangxi 530200, PR China
First published on 7th February 2018
Abstract
A series of benzothiazole amide derivatives were synthesized through a facile and efficient method via a nucleophilic acyl substitution reaction between 2-aminobenzothiazole and various cinnamic acid compounds. The obtained products exhibited good thermal stabilities. All compounds were evaluated for their in vitro hemostatic activities using the commercially available standard drug etamsylate as a positive control. The results showed that compound Q2 had a significant partial coagulation activity, reduced capillary permeability at 5, 10 and 50 μmol L−1, activated thrombin activity, and a more potent platelet aggregation activity than the positive control group (etamsylate, up to 1283.9 times in the nanomole range). A molecular modeling study revealed that compound Q2 was a competitive thrombin activator. Therefore, Q2 may be a potential lead for further biological screening and for the generation of drug molecules. Moreover, the structure–activity relationship of the prepared compounds is also discussed herein.
Introduction
Excessive bleeding during surgery remains a main cause of failure of surgical procedures.1 Homologous blood transfusion and hemostatic drugs are commonly employed during surgery to avoid excessive bleeding.2,3 However, homologous blood transfusions can increase the risk of infection, leading to increased morbidity and mortality.4 Thus, the use of hemostatic agents is preferable. To date, commonly used hemostatic drugs, such as p-aminomethylbenzoic acid, tranexamic acid, aminocaproic acid, aprotinin, and etamsylate, confer adverse side effects and safety risks despite their desirable hemostatic effects, and are therefore being re-evaluated for clinical use.4–6 Thus, the search for safer and more effective hemostatic agents remains crucial.A heterocyclic ring containing nitrogen and sulfur atoms represents a unique and universal scaffold for the design of experimental drugs.7 Benzothiazole is an important derivative unit of the thiazole ring, a rare heterocyclic compound found in alkaloids, and plays an important role in drug chemistry as a key template for the development of various therapeutic agents.8 Due to the presence of sulfur atoms in their structure, benzothiazoles exhibit unique properties not possessed by other heterocyclic compounds.9 Benzothiazole amide systems have interesting structural characteristics and strong bioactivity profiles, as well as anticancer,10 anti-inflammatory,11 antifungal,12 and chronic pain suppression properties.13 Further, given their useful molecular scaffolds for the exploration and exploitation of pharmacophore space, they have emerged as particularly promising synthetic targets.
Chemical compounds with a low biological toxicity stemming from natural products can be obtained from a wide range of sources,14,15 and are being used as lead compounds in the preparation of safe and low toxicity hemostatic drugs. In our previous studies,16,17 we evaluated the activity of cinnamic acid amide derivatives synthesized by sulfa drug 2-aminothiazole with cinnamic acid compounds. The results showed that cinnamic acid amide derivatives may function as hemostatic agents. Therefore, we chose to continue our investigation on the synthesis and properties of cinnamic acid compounds.
According to the drug design principles of combination, the covalent bonding of two different compounds would lead to the synergistic effect of their properties. Thus, we designed nine benzothiazole amide derivatives with cinnamic acid structures and assessed their hemostatic activity. Although the synthetic methods of compounds Q5, Q6, Q7, and Q9 have been previously reported,18–20 the synthesis of the other compounds Q1–Q4, Q8 and the evaluation of their related biological activities remain unreported. Thermogravimetric analysis was used to determine the thermodynamic stability of the compounds as a proxy of drug stability. The hemostatic activity of the prepared compounds was evaluated by in vitro experiments based on the hemostatic mechanism (involving normal operation of blood vessels, platelets, and blood coagulation) through the platelet aggregation assay, activation of partial thromboplastin time (APTT), prothrombin time (PT), thrombin time assay (TT), human C-type natriuretic peptide determination, and thrombin activity assay (including molecular docking of compound Q2 with thrombin). Based on the obtained results, the relationship between the given structure and the corresponding hemostatic activity was discussed, providing a theoretical basis for the screening of hemostatic drugs.
Results and discussion
Chemistry
The amide bond formation reaction is an important reaction in organic synthesis, with the amide bond being widely present in pharmaceutical intermediates and biochemical substances.21,22 Benzothiazole amide derivatives (Q1–Q9) were synthesized by the reaction of cinnamic acid and its derivatives (q1–q9) with 2-aminobenzothiazole through the nucleophilic acyl substitution of acyl chloride (Schemes 1 and 2). NaHCO3 was used as an acid binding agent and the reaction was performed in CH2Cl2 solvent at 0–5 °C. The pH value was kept between 6 and 7 by addition of NaHCO3 to induce precipitation. The reactants were vigorously mixed until solidification of the reaction mixture. Reaction progress was monitored by thin layer chromatography (TLC). Once the reaction was complete, the upper layer solid was collected by filtration of the mixture. The solid product was washed with excess distilled ethanol and water to obtain the pure products. All tested reactions proceeded smoothly under optimal conditions, producing the desired products in good yields (Table 1). However, a trend was observed wherein using cinnamic acid derivatives with substituents of increasing electron-donating strength on the aromatic ring led to products with higher yields, likely due to a decrease in electron density in the aromatic ring causing an acceleration of the reaction. Compounds Q1–Q9 were structurally characterized by various spectroscopic techniques, including 1H and 13C nuclear magnetic resonance (NMR), electron ionization mass spectroscopy, and Fourier-transform infrared spectroscopy.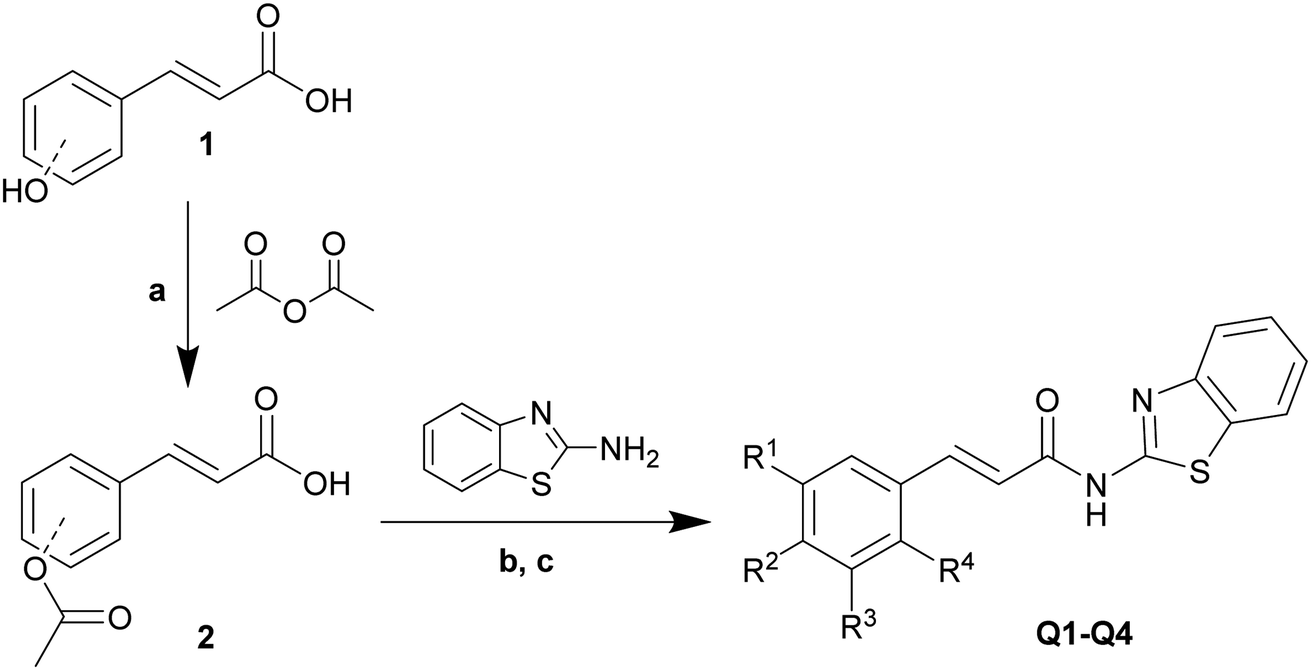 | ||
| Scheme 1 Synthesis of compounds Q1–Q4. Reagents and conditions: (a) acetic anhydride/pyridine, 120 °C, 5 h, 98–100%. (b) SOCl2, DMF, 70 °C, 5 min, 100%. (c) NaHCO3, pH = 6–7, 0–5 °C to room temperature, 3 h, 70–82%. | ||
 | ||
| Scheme 2 Synthesis of compounds Q5–Q9. Reagents and conditions: (b) SOCl2, DMF, 70 °C, 5 min, 100%. (c) NaHCO3, pH = 6–7, 0–5 °C to room temperature, 3 h, 88–93%. | ||
|
||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | Yieldb (%) | EC50c (μmol L−1) |
| a Reaction conditions: the acid chloride of cinnamic acid compounds (1.5 mmol), 2-aminobenzothiazole (1.0 mmol) in 10.0 mL of CH2Cl2, 0–5 °C to room temperature, 3 h.b Isolated yield after flash chromatography.c The EC50 values are expressed as the concentration required to promote ADP-induced human platelet aggregation response by 50%; n = 3.d Reaction conditions: cinnamic acid compounds (ferulic acid, p-hydroxy-cinnamic acid, m-hydroxycinnamic acid, and o-hydroxy-cinnamic acid, 25.0 g), pyridine (5 mL) in 100 mL of acetic anhydride at 120 °C for 5 h.e q5: cinnamic acid; q6: 4-methoxycinnamic acid; q7: 3,4-dimethoxycinnamic acid; q8: 3,4,5-trimethoxycinnamic acid; q9: 3-phenylpropionic acid. | ||||||
| 2-Aminobenzothiazole | 513.7 | |||||
| q1d | H | OCOCH3 | OCH3 | H | 100 | 476.3 |
| q2d | H | OCOCH3 | H | H | 99 | 98.76 |
| q3d | H | H | OCOCH3 | H | 98 | 129.4 |
| q4d | H | H | H | OCOCH3 | 98 | 108.7 |
| q5e | H | H | H | H | 322.3 | |
| q6e | H | OCH3 | H | H | 280.5 | |
| q7e | H | OCH3 | OCH3 | H | 189.4 | |
| q8e | OCH3 | OCH3 | OCH3 | H | 120.9 | |
| q9e | — | — | — | — | 227.8 | |
| Q1 | H | OCOCH3 | OCH3 | H | 82 | 93.87 |
| Q2 | H | OCOCH3 | H | H | 73 | 0.036 |
| Q3 | H | H | OCOCH3 | H | 77 | 1.246 |
| Q4 | H | H | H | OCOCH3 | 70 | 0.119 |
| Q5 | H | H | H | H | 88 | 73.71 |
| Q6 | H | OCH3 | H | H | 90 | 54.58 |
| Q7 | H | OCH3 | OCH3 | H | 92 | 18.09 |
| Q8 | OCH3 | OCH3 | OCH3 | H | 93 | 1.867 |
| Q9 | — | — | — | — | 90 | 35.47 |
| Etamsylate | 46.22 | |||||

Crystal structure description
Single crystals of compounds Q7 (CCDC: 1582748) and Q9 (CCDC: 1582749) were grown from dimethyl sulfoxide and the corresponding structures were confirmed by single crystal X-ray diffraction analysis (Fig. 1). Furthermore, integrals in the NMR spectra confirmed the crystal structures for the two compounds Q7 and Q9. The crystal lattice of compound Q7 belongs to the triclinic system. The crystal lattice of compound Q9 belongs to the monoclinic system, containing two N-(benzo[d]thiazol-2-yl)-3-phenylpropanamide molecules. Selected bond lengths (Å) and angles (°) are provided in the ESI† section.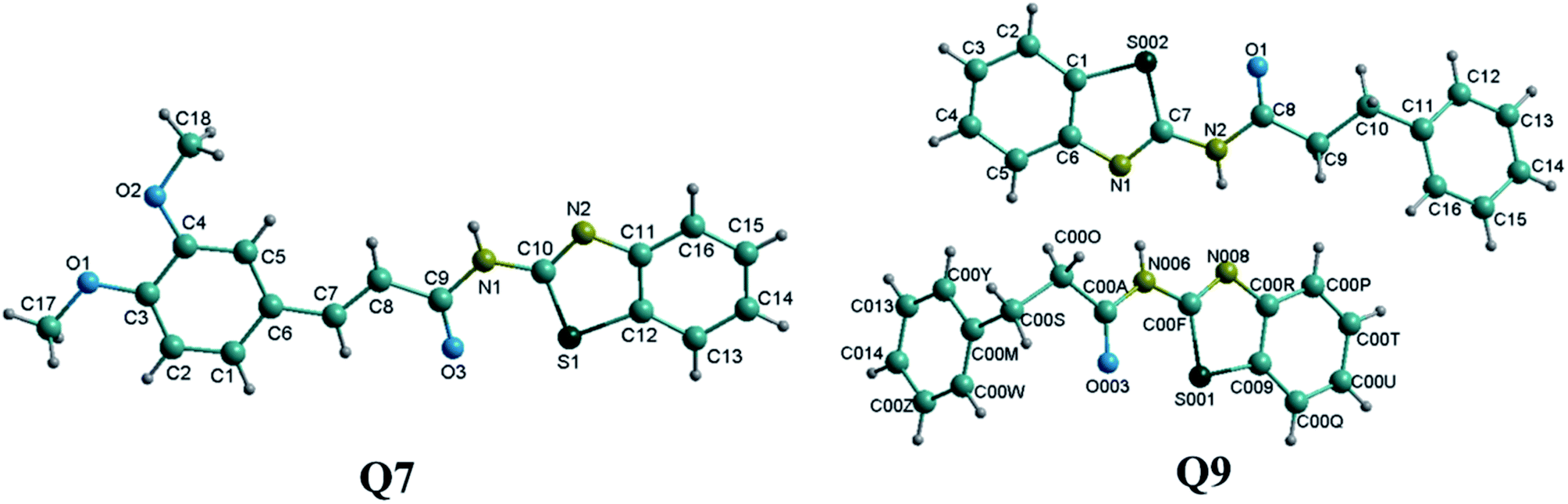 | ||
| Fig. 1 Ball-and-stick structures of compounds Q7 and Q9. | ||
Thermogravimetric analysis
The mass loss and heat flow of the benzothiazole amide derivatives upon increasing temperature are shown in Fig. 2. When the compounds were heated to below 100 °C, no significant changes in mass were observed. With the increase in temperature, the mass loss gradually increased, with the heat flow curve showing obvious fluctuations at approximately 150 °C, indicating the initiation of oxidation in air. The mass of Q4 declined considerably at 180 °C, with the heat flow curve showing a large fluctuation due to sample decomposition. | ||
| Fig. 2 Thermogravimetric and heat flow curves of compounds Q1–Q9 recorded at a constant heating rate. The mass loss is expressed as the fraction M/M0 of the mass at temperature T to the initial mass of the samples equilibrated at laboratory atmosphere. | ||
Thus, the stable nature of the thermogravimetric analysis results indicated that the prepared compounds had a suitable thermal stability at 100 °C. Therefore, these compounds were further screened as hemostatic drugs.
Platelet aggregation activity
When the vascular wall is broken, platelets undergo a series of functional responses, including adhesion, diffusion, release, and accumulation, thus promoting coagulant surface exposure, particle formation, and clot retraction.23 The platelet accumulation response leads to platelet adhesion at the site of injury as well as the action of endogenous agonists such as adenosine diphosphate (ADP), collagen, and thrombin, followed by the release of TXA2, forming a hemostatic plug to obstruct the severed area and prevent bleeding.24,25 Compound Q2 showed the highest efficacy as a platelet aggregation promoter, with the lowest EC50 value (0.036 μmol L−1) in the nanomole range (10 nmol L−1), demonstrating a better platelet aggregation activity than etamsylate (positive control, EC50 = 46.22 μmol L−1; Table 1). Compounds Q4, Q3, Q8, Q7, and Q9 (EC50 = 0.119, 1.246, 1.867, 18.09, and 35.47 μmol L−1) all showed a better platelet aggregation-promoting activity than etamsylate. Thus, adding methoxy groups to N-(benzo[d]thiazol-2-yl)-cinnamamide leads to a decrease in platelet aggregation activity. However, introducing an acetoxyl group to N-(benzo[d]thiazol-2-yl) cinnamamide would lead to an increase in platelet aggregation activity. Thus, the substituent type crucially affects platelet aggregation promotion.Blood coagulation activities
All synthetic benzothiazole amide derivatives Q1–Q9 were screened for in vitro blood coagulant activity, including APTT, PT, and TT assays. APTT tests the intrinsic coagulation activity, providing an approximate indication of coagulation factor II, V, VII, and X activity.26 PT is an ideal and commonly used screening assay for the detection of extrinsic coagulation pathway.27 Finally, TT is a screening test to determine thrombin activity and the ability to convert plasma fibrin into fibrin.28 The coagulation rate (Rxx) was used to evaluate the corresponding coagulation activity of a given compound; negative values may represent partial coagulation activity and smaller values may indicate an improvement in the blood coagulant effects. The corresponding APTT, PT, and TT values of benzothiazole amide derivatives and etamsylate (positive control) are shown in Tables 2–4, while a comparison with p-aminomethylbenzoic acid (another positive control) are provided in the ESI† section.| Compound | Concentration (μmol L−1) | ||||
|---|---|---|---|---|---|
| 1 | 5 | 10 | 50 | 100 | |
| a Data represent mean ± SEM; n = 8. *P < 0.05 vs. etamsylate; **P < 0.01 vs. etamsylate; ***P < 0.001 vs. etamsylate. | |||||
| Etamsylate | −7.12 ± 0.23 | −6.10 ± 0.22 | −4.52 ± 0.46 | −1.01 ± 0.25 | 4.07 ± 0.12 |
| 2-Aminobenzothiazole | −4.87 ± 0.13*** | −5.27 ± 0.35 | −6.19 ± 0.13*** | −4.74 ± 0.23*** | −4.48 ± 0.35*** |
| q1 | −1.72 ± 0.25*** | −0.54 ± 0.16*** | 0.86 ± 0.25*** | 1.58 ± 0.14*** | 1.87 ± 0.38*** |
| q2 | −3.97 ± 0.47*** | −3.56 ± 0.24*** | −1.92 ± 0*** | 0.01 ± 0.27 | 0.82 ± 0.14*** |
| q3 | −2.19 ± 0.36*** | −1.23 ± 0.27*** | 0.55 ± 0.24*** | 0.96 ± 0.24*** | 3.15 ± 0.14 |
| q4 | −3.47 ± 0.12*** | −1.20 ± 0.24*** | −0.96 ± 0.32*** | 0.12 ± 0.24* | 0.24 ± 0.43*** |
| q5 | −2.18 ± 0.39*** | −0.26 ± 0.38*** | 2.05 ± 0.44*** | 2.56 ± 0.26*** | 7.57 ± 0.34*** |
| q6 | −0.96 ± 0.32*** | 0.12 ± 0.11*** | 0.36 ± 0.35*** | 0.60 ± 0.32** | 0.72 ± 0.21*** |
| q7 | −0.73 ± 0.15*** | −0.44 ± 0.44*** | 1.91 ± 0.14*** | 2.20 ± 0.25*** | 2.79 ± 0.15* |
| q8 | −0.48 ± 0.24*** | −0.84 ± 0.60*** | −1.20 ± 0.43*** | −1.67 ± 0.12 | −2.75 ± 0.24*** |
| q9 | 1.03 ± 0.12*** | −0.77 ± 0.26*** | −1.67 ± 0.13*** | −2.31 ± 0.33* | −2.57 ± 0.22*** |
| Q1 | 0.14 ± 0.37*** | −8.58 ± 0.37*** | −10.10 ± 0.14*** | −6.50 ± 0.14*** | −5.26 ± 0.37*** |
| Q2 | −38.84 ± 0.08*** | −38.26 ± 0*** | −36.12 ± 0.22*** | −36.03 ± 0.43*** | −20.25 ± 0.30*** |
| Q3 | 0.42 ± 0.38*** | 0.21 ± 0.21*** | −1.98 ± 0.21*** | −0.73 ± 0.21 | −0.52 ± 0.10*** |
| Q4 | −35.46 ± 0.17*** | −34.96 ± 0.30*** | −34.22 ± 0.22*** | −31.90 ± 0.41*** | −4.80 ± 0.14*** |
| Q5 | −12.06 ± 0.20*** | −10.03 ± 0.34*** | −9.58 ± 0.11*** | −9.47 ± 0.29*** | −7.89 ± 0.11*** |
| Q6 | −9.47 ± 0.11*** | −8.90 ± 0.11*** | −8.46 ± 0.41*** | −7.90 ± 0.11*** | −7.33 ± 0.34*** |
| Q7 | −4.55 ± 0.28*** | −6.21 ± 0.14 | −8.83 ± 0.14*** | −7.31 ± 0.24*** | −4.41 ± 0.24*** |
| Q8 | 2.24 ± 0.11*** | −0.11 ± 0.11*** | −4.69 ± 0.18 | −3.52 ± 0.11*** | 0.85 ± 0.11*** |
| Q9 | −5.75 ± 0.30*** | −4.06 ± 0.46*** | −3.95 ± 0.20 | −1.01 ± 0.30 | 4.51 ± 0.20 |
| Compound | Concentration (μmol L−1) | ||||
|---|---|---|---|---|---|
| 1 | 5 | 10 | 50 | 100 | |
| a Data represent mean ± SEM; n = 8. *P < 0.05 vs. etamsylate; **P < 0.01 vs. etamsylate; ***P < 0.001 vs. etamsylate. | |||||
| Etamsylate | −13.70 ± 0.37 | −10.73 ± 0.11 | −8.03 ± 0.21 | 2.84 ± 0.13 | 4.03 ± 0.46 |
| 2-Aminobenzothiazole | −1.05 ± 0.46*** | −1.22 ± 0.30*** | −1.57 ± 0.17*** | −4.36 ± 0*** | −4.48 ± 0.35*** |
| q1 | 7.60 ± 0.17*** | 4.73 ± 0.45*** | 2.87 ± 0.59** | −1.35 ± 0.45*** | −2.20 ± 0.29*** |
| q2 | −4.78 ± 0.27*** | −3.85 ± 0.27*** | −3.70 ± 0.15*** | −3.39 ± 0.27*** | −2.93 ± 0.27*** |
| q3 | −2.23 ± 0.15*** | −1.92 ± 0.26*** | −0.89 ± 0.39* | −0.30 ± 0.15*** | 0.45 ± 0.14*** |
| q4 | −2.71 ± 0.15*** | −2.56 ± 0.30*** | 0.00 ± 0.15*** | 0.30 ± 0.52*** | 0.90 ± 0.40*** |
| q5 | −5.94 ± 0.45*** | −2.53 ± 0.39*** | −1.78 ± 0.15*** | −0.45 ± 0.39*** | 2.23 ± 0.54** |
| q6 | −4.16 ± 0.15*** | −3.23 ± 0.56*** | −2.31 ± 0.41*** | −1.23 ± 0.15*** | −0.15 ± 0.27*** |
| q7 | −0.77 ± 0.41*** | 0.77 ± 0.27*** | 1.23 ± 0.27*** | 1.69 ± 0.46 | 1.85 ± 0.31** |
| q8 | 1.52 ± 0.45*** | 3.21 ± 0.34**** | 4.39 ± 0.29*** | 5.57 ± 0.45*** | 6.76 ± 0.34*** |
| q9 | −5.42 ± 0.54*** | −1.51 ± 0.26*** | −1.05 ± 0.45*** | −0.45 ± 0.40*** | 1.51 ± 0.15*** |
| Q1 | −2.12 ± 0.28*** | −5.22 ± 0.33*** | −4.73 ± 0.16*** | −3.59 ± 0.57*** | −2.12 ± 0.28*** |
| Q2 | −6.79 ± 0.15*** | −5.91 ± 0.15*** | −4.43 ± 0.15*** | −1.62 ± 0.26*** | −0.59 ± 0.15*** |
| Q3 | −6.57 ± 0.13*** | −5.36 ± 0.13*** | −4.29 ± 0.23*** | −1.21 ± 0.13*** | −0.13 ± 0.13*** |
| Q4 | −9.90 ± 0.15*** | −7.24 ± 0.15*** | −5.47 ± 0.30*** | −4.73 ± 0*** | −2.07 ± 0.44*** |
| Q5 | −11.43 ± 0.13*** | −10.22 ± 0.13 | −9.67 ± 0*** | −4.03 ± 0.23*** | −2.28 ± 0.13*** |
| Q6 | −4.57 ± 0.48*** | −4.70 ± 0.13*** | −6.99 ± 0.13*** | −9.54 ± 0.13*** | −14.79 ± 0.13*** |
| Q7 | −3.57 ± 0.28*** | −4.40 ± 0.16*** | −5.38 ± 0.33*** | −6.85 ± 0.16*** | −7.83 ± 0.33*** |
| Q8 | −1.61 ± 0.13*** | −4.29 ± 0*** | −3.89 ± 0.23*** | −4.83 ± 0.27*** | −1.21 ± 0.13*** |
| Q9 | −9.82 ± 0.27*** | −6.72 ± 0.13*** | −5.19 ± 0.13*** | −4.03 ± 0.23*** | −3.90 ± 0.27*** |
| Compound | Concentration (μmol L−1) | ||||
|---|---|---|---|---|---|
| 1 | 5 | 10 | 50 | 100 | |
| a Data represent mean ± SEM; n = 8. *P < 0.05 vs. etamsylate; **P < 0.01 vs. etamsylate; ***P < 0.001 vs. etamsylate. | |||||
| Etamsylate | −11.75 ± 0.34 | −1.92 ± 0.24 | 5.51 ± 0.44 | 11.51 ± 0.33 | 16.04 ± 0.17 |
| 2-Aminobenzothiazole | 12.57 ± 0.29*** | 11.40 ± 0*** | 9.36 ± 0.29*** | 3.51 ± 0.31*** | 0.58 ± 0.29*** |
| q1 | 0.26 ± 0.26*** | −0.52 ± 0.26* | −1.82 ± 0.26*** | −3.16 ± 0.45*** | −5.47 ± 0.45*** |
| q2 | −2.53 ± 0.51*** | −1.52 ± 0.25 | 1.01 ± 0.44*** | 2.03 ± 0.25*** | 4.30 ± 0.51*** |
| q3 | 0.98 ± 0.42*** | −0.98 ± 0.24 | −4.39 ± 0.24*** | −5.61 ± 0.42*** | −6.10 ± 0.24*** |
| q4 | −4.95 ± 0.26*** | −3.91 ± 0** | −2.87 ± 0.26*** | −1.82 ± 0.52*** | −1.56 ± 0.45*** |
| q5 | −6.58 ± 0.44*** | −5.32 ± 0.25*** | −0.76 ± 0.51*** | 1.77 ± 0.44*** | 3.54 ± 0.25*** |
| q6 | −2.29 ± 0.43*** | −1.02 ± 0.51 | −0.51 ± 0.25*** | 0.51 ± 0.67*** | 2.04 ± 0.25*** |
| q7 | −4.15 ± 0*** | −1.71 ± 0.24 | 2.44 ± 0.42*** | 5.12 ± 0.49*** | 8.29 ± 0*** |
| q8 | −4.30 ± 0.44** | −2.28 ± 0.25 | −1.27 ± 0.44*** | 2.03 ± 0.51*** | 3.04 ± 0.25*** |
| q9 | −4.12 ± 0.45*** | −2.06 ± 0.26 | −1.55 ± 0.26*** | 0.01 ± 0.52*** | 1.03 ± 0.25*** |
| Q1 | −7.38 ± 0.22*** | −11.50 ± 0.38*** | −12.15 ± 0.38*** | −12.80 ± 0.01*** | −14.53 ± 0.22*** |
| Q2 | −6.67 ± 0.24*** | −11.43 ± 0.41*** | −17.14 ± 0.01*** | −24.52 ± 0.24*** | −16.43 ± 0.41*** |
| Q3 | −3.11 ± 0.19*** | −4.47 ± 0*** | −16.70 ± 0.34*** | −14.18 ± 0.19*** | −9.13 ± 0.34*** |
| Q4 | −21.91 ± 0.28*** | −20.24 ± 0.24*** | −18.10 ± 0.24*** | −15.00 ± 0.41*** | −13.33 ± 0.24*** |
| Q5 | −10.56 ± 0.10* | −11.97 ± 0.41*** | −13.62 ± 0.23*** | −20.19 ± 0.23*** | −20.66 ± 0.47*** |
| Q6 | −14.46 ± 0.24*** | −10.9 ± 0.24*** | −5.21 ± 0.47*** | −4.98 ± 0.47*** | −0.95 ± 0.24*** |
| Q7 | −19.31 ± 0.38*** | −17.57 ± 0.22*** | −17.14 ± 0.22*** | −16.05 ± 0.38*** | −13.23 ± 0.22*** |
| Q8 | −29.13 ± 0.39*** | −28.35 ± 0.01*** | −27.96 ± 0.19*** | −16.31 ± 0.39*** | −13.20 ± 0.34*** |
| Q9 | −2.61 ± 0.41*** | −3.79 ± 0.24*** | −11.61 ± 0.24*** | −17.54 ± 0.41*** | −18.48 ± 0.24*** |
Compound Q2 exhibited the lowest APTT value (RAPTT = −38.84 ± 0.08% at 1 μmol L−1; Table 2), a significantly shortened TT (P < 0.001, except 1 μmol L−1; Table 4), and a somewhat shortened PT compared to the positive control etamsylate. The blood coagulant mechanism is most likely due to compound Q2 primarily coagulating blood through the activation of intrinsic pathways and partly through thrombin-mediated promotion of fibrinogen conversion to fibrin in plasma.
Compound Q6 exhibited the lowest PT value (RPT = −14.79 ± 0.13% at 100 μmol L−1; Table 3), with its activity decreasing with decreasing concentration. Nevertheless, Q6 showed a lower activity than etamsylate at 1, 5 and 10 μmol L−1, and a higher activity at 50 and 100 μmol L−1. Further, it showed the lowest RPT value at 1 μmol L−1 (−13.70 ± 0.37%). The PT values of other compounds were higher than −10%, indicating that the extrinsic pathways were less affected.
Compound Q8 exhibited the lowest TT value (RTT = −29.13 ± 0.39%; Table 4). However, a rare effect on APTT and PT was observed, wherein the RAPTT and RPT values of Q8 were all lower than −5% (which can be regarded as no activity), indicating that compound Q8 mainly achieved a blood coagulation effect through stimulation of fibrinogen or an increase of fibrin levels. However, coagulation was not observed to be due to intrinsic or extrinsic pathways.
Combined with blood coagulant activity, N-(benzo[d]thiazol-2-yl)-cinnamamide with a methoxy group substitution lead to a decrease in APTT and PT. Conversely, introducing an acetoxyl group to N-(benzo[d]thiazol-2-yl) cinnamamide lead to an increase in APTT, but a decrease in PT. Moreover, the position of the acetoxyl group was also observed to influence the relevant hemostatic activity, where APTT and PT were increased when the acetoxyl group was located in the ortho or para positions in the benzene ring compared to the meta position.
Thrombin activity assay
Thrombin is a key component in blood coagulation, positioned in the coagulation cascade to cleave fibrinogen into fibrin, resulting in blood clot formation.29 Q2 and Q8, which had a good coagulation activity, were selected for thrombin activity assays (Fig. 3). The EC50 of thrombin activity of Q2 (135.9 μmol L−1) and Q8 (232.1 μmol L−1) were both lower than for etamsylate (126.5 μmol L−1). In combination with blood coagulant activity results and the fact that Q2 significantly shortened APTT and TT compared to etamsylate, we can infer that the coagulation activity mechanism of Q2 functioned through its interaction with thrombin as well as with other proteins or factors in the blood coagulation cascade (Fig. 4). | ||
| Fig. 3 The EC50 values for the activation of thrombin activity by compounds Q2 and Q8. aThe EC50 values are expressed as the concentration required for a thrombin activity response of 50%; n = 3. | ||
 | ||
| Fig. 4 The classical systems for blood coagulation including extrinsic, intrinsic, and/or the common coagulation pathways.30 | ||
Docking of compound Q2 with thrombin
To further assess the interaction mode of compound Q2 with thrombin, a molecular docking study was performed using Autodock vina 1.1.2 software.31 The theoretical binding mode of Q2 in the thrombin binding site is illustrated in Fig. 5. Q2 adopted a compact conformation to bind inside the thrombin pocket. The benzothiazolyl group of Q2 was located within the hydrophobic pocket, surrounded by Tyr-60A, Leu-99, Ile-174, and Trp-215 residues, forming stable hydrophobic bonding. Detailed analysis showed that the phenyl group of Q2 formed anion–π interactions with Asp-189 and Glu-192 residues. Importantly, three key polar interactions were observed between the carbonyl “O” of Q2 and Asp-189 (bond distance: 3.1 Å), Ala-190 (bond distance: 2.5 Å), and Gly-219 (bond distance: 3.0 Å), representing the main interactions between thrombin and Q2 and facilitating Q2 anchoring within the thrombin binding site. The above molecular simulations elucidated the underlying interactions between Q2 and thrombin, providing valuable information for the further development of thrombin activators. | ||
| Fig. 5 Compound Q2 docking into the thrombin binding pocket. | ||
In vitro determination of human C-type natriuretic peptide (CNP)
CNP, a local vascular regulator, is a member of the natriuretic family,32 with potent vasodilator properties.33 The level of CNP in plasma and tissue has been shown to increase upon vascular injury, with a decrease in CNP content suggesting that vascular injury has improved.34 At low concentrations (5, 10, and 50 μmol L−1), the plasma CNP level of Q2 was significantly lower than that of etamsylate (P < 0.05 or P < 0.01; Fig. 6). Yet, at high concentrations (100 and 200 μmol L−1), the reverse was observed. Etamsylate is able to reduce capillary permeability.35–38 Thus, we could infer that Q2 may lead to a reduction in capillary permeability at low concentrations (at 5, 10, and 50 μmol L−1). | ||
| Fig. 6 Effects of compounds Q2 and etamsylate on capillary permeability. The results were expressed as the concentration of CNP (pg mL−1). Values are reported as the mean ± SEM of three independent experiments. | ||
Conclusions
We have developed a facile and efficient method for the synthesis of benzothiazole amide derivatives via a nucleophilic acyl substitution reaction of 2-aminobenzothiazole with cinnamic acid compounds. Products bearing methoxy groups were easily obtained in high yields (up to 93%) and had suitable thermal stabilities as demonstrated by thermogravimetric analysis. Compound Q2 showed good hemostatic activity, with a significantly shortened APTT and TT, reduced CNP level, activated thrombin activity, and more potent platelet aggregation activity compared to etamsylate (up to 1283.9 times), indicating that these benzothiazole amide derivatives, and particularly Q2, should be further screened for the possible generation of potential hemostatic drug molecules.Experimental
Materials and methods
A series of cinnamic acid derivatives of analytical grade were purchased from Shengshun Trading Co., Ltd. (Zhuhai, China). 2-Aminobenzothiazole was purchased from Aladdin reagents (Shanghai, China). Etamsylate was purchased from Hubei Tianyao Pharmaceutical Co., Ltd. APTT, PT, and TT test kits were purchased from Heall Bio-Science Technology, Qingdao, China. ADP was purchased from Sigma-Aldrich (Shanghai, China). Human CNP ELISA Kit were purchased from Shanghai mlbio Biotechnology Co. (Shanghai, China). Thrombin (≥1000 NIH U mg−1 protein) prepared from human plasma was purchased from Sigma-Aldrich (USA). Chromozym-TH was purchased from Roche (Swiss). All other reagents were commercial materials of analytical purity and were used without further purification.1H and 13C NMR spectra were performed in dimethylsulfoxide-d6 on a Bruker Ascend 600 at 600 MHz for 1H and 150 MHz for 13C, respectively. X-ray crystallographic studies were performed using a Nonius Kappa CCD diffractometer equipped with a graphite monochromator and an Oxford cryostream, using Mo-Kα radiation (λ = 0.7107 Å). Infrared spectra were performed as KBr sheets on a Tensor 27 FTIR spectrometer (Bruker, Germany). ESI-MS were collected using a LC-MS 2010A instrument (Shimadzu, Japan).
Plasma coagulation assays and platelet aggregation determination were performed using a blood coagulation analyzer (LG-PABER-I, GTM-STEELLEX Instrument Co., Ltd., Beijing, China). The enzyme–drug reaction was monitored using a BIO-RAD iMark microplate reader.
Synthesis of compound Q1–Q4
As shown in Scheme 1, cinnamic acid compounds (25.0 g; compound 1) and pyridine (5 mL) were added to a solution of acetic anhydride (100 mL). After the addition, the solution was stirred at 120 °C for 5 h using an oil bath. Deionized water (800 mL) was added and the crude acetoxy intermediate (compound 2) was precipitated and separated.39 Compound 2 (1.5 mmol) was dissolved in N,N-dimethylformamide (1 mL) and thionyl chloride (10 mL), and stirred at 70 °C for 5 min. Through distillation under reduced pressure, the liquid residue was collected and dispersed in 20 mL of CH2Cl2, to which 2-aminobenzothiazole (1.0 mmol) was added, and stirred at 0–5 °C. The pH value was kept between 6 and 7 by addition of NaHCO3 to induce precipitation. The upper layer solid was then collected by filtration of the mixture, and washed with excess distilled ethanol and water to obtain the pure products. Data for compounds Q1–Q4 are given below.Synthesis of compound Q5–Q9
Cinnamic acid compounds (1.5 mmol) and N,N-dimethylformamide (1 mL) were added to 10 mL of thionyl chloride, and allowed to react at 70 °C for 5 min. The liquid residue obtained following distillation under reduced pressure was retained. A solution of the liquid residue in CH2Cl2 was added dropwise to a CH2Cl2 solution of 2-aminobenzothiazole (1.0 mmol) under ice cooling. The mixture was stirred at 0–5 °C for 3 hours. TLC was used to monitor whether the reaction had completed. The pH value was kept between 6 and 7 by addition of NaHCO3 to induce precipitation. After the reaction, the amides were isolated using filtration of the mixture and then washed with excess distilled ethanol and water to obtain the pure products. The synthesis route is shown in Scheme 2 and the data for compounds Q5–Q9 are indicated below.Crystallographic data collection and structure determination
Single-crystal X-ray diffraction data of compounds Q7 and Q9 was collected on a CCD area detector diffractometer with graphite monochromated Mo/Kα radiation (λ = 0.071073 nm). The structures were solved by direct methods using Olex2 software40 and refined with full-matrix least squares on F2 using the ShelXL41 program package. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms attached to all carbon and nitrogen atoms were geometrically fixed and the positional and temperature factors were refined isotropically. Structural illustrations were generated using Diamond 3.1 for Windows.Thermogravimetric analysis
The thermogravimetric experiments were performed using a NETZSCH STA 409 PC/PG thermogravimetric analyzer under air atmosphere at a flow rate of 20 mL min−1 for the sample. The explored temperature interval ranged between 30 °C and 250 °C, at a heating rate of 0.5 °C min−1. The decomposition temperature (Td) was taken at the maximum of the heat flow peaks42 and the maximum heat flux changes occurred at the same point.Human plasma samples
Healthy volunteers (aged 20–25) were recruited for this study. Whole blood was drawn from the volunteer's hand vein. The fresh vein blood samples were collected in the morning with overnight fasting using routine procedures. Blood was collected in plastic tubes with 3.8% sodium citrate (citrate/blood: 1/9, v/v) to ensure plasma anticoagulation.43 Samples were centrifuged for 15 minutes at 1000 × g at 2–8 °C within 30 minutes of collection.Platelet aggregation determination in vitro
Functional promotion activity of the nine synthesized compounds was evaluated by measuring the promotion of ADP-induced platelet aggregation in human platelet-rich plasma (PRP) by Born's method.44 PRP was prepared from collected blood by low speed centrifugation at 800 rpm for 10 min at 24 °C, whereas platelet-poor plasma (PPP) was prepared by centrifuging PRP at 3000 rpm for a further 10 min. ADP was selected as an agonist for platelet aggregation test.Light transmission of 100% was calibrated by 280 μL PPP and 20 μL sample solution (samples were dissolved and diluted with 80% macrogol 400), and then platelet aggregation was induced by the addition of ADP (final concentration: 5 μmol L−1) to 280 μL PRP and 20 μL sample solution. Platelet aggregation kinetics were measured and platelet aggregation was expressed as a percentage of the PPP transmission value. The changes in light transmission were recorded. Experiments were completed within 3 h after blood collection.
Blood coagulation assay in vitro
The activation of APTT, PT, and TT assays were examined by commercial kits following the manufacturer's instructions using a hematology analyzer. Samples were dissolved in and diluted with 80% macrogol 400. A 100 μL aliquot of the sample solution was then mixed with 900 μL of plasma and incubated at 37 °C for 5 min. APTT was determined by incubating 50 μL of the plasma solution with 50 μL of APTT activating agent for 3 min at 37 °C, followed by addition of 50 μL of CaCl2 (25 mM). PT was determined by incubating 50 μL of the plasma solution for 3 min at 37 °C, followed by addition of 100 μL of thromboplastic agent. TT was determined by incubating 50 μL of the plasma solution for 3 min at 37 °C, followed by addition of 100 μL of APTT activating agent.The coagulation activity was assessed by assaying the plasma clotting time of TT and APTT, and the decrease in International Normalized Ratio (INR) of PT.
The RAPTT, RPT, or RTT were calculated as follows:
 | (1) |
 | (2) |
 | (3) |
In this set of equations, APTTC, PTC, and TTC correspond to the APTT, PT, and TT at determined sample concentrations. The factors APTT0, PT0, and TT0 correspond to the control assays in which the concentration of the sample was 0 μM. All the data were obtained as mean (n = 8) values.
Determination of thrombin activity
The thrombin activation assay was conducted using the chromogenic substrate method. The substrate Tos–Gly–Pro–Arg–pNA (chromozym-TH) can be hydrolyzed by thrombin and cleaved into two parts, a residual peptide and a yellow substance, p-nitroaniline, which can be measured with a microplate reader. A 20 μL aliquot of the thrombin solution (1 U mL−1, in 0.1% bovine serum albumin, pH 6.5) was mixed with 20 μL of the test compound solution (dissolved in 50 mM Tris buffer, pH 8.3). The mixture was incubated at 37 °C for 10 min before the addition of 20 μL of chromozym-TH solution (2 mM). After a further 10 min of incubation at 37 °C, 50 μL of acetic acid (50% w/v) and 40 μL of water were successively added to terminate the reaction. For each concentration, three replicate wells were used. The absorbance was monitored at 405 nm. The Tris-treated control was considered the blank control. The EC50 values represent the concentration that caused a 50% activation relative to the Tris-treated control.Molecular docking
A molecular docking study was performed to investigate the binding mode between Q2 and human thrombin using Autodock vina 1.1.2.31 The three-dimensional (3D) structure of human thrombin (PDB ID: 3RLW) was downloaded from Protein Data Bank (http://www.rcsb.org/pdb/home/home.do). The 3D structure of Q2 was drawn by ChemBioDraw Ultra 14.0 and ChemBio3D Ultra 14.0 software. The AutoDockTools 1.5.6 package45,46 was employed to generate the docking input files. The search grid of the thrombin was identified as center_x: 16.154, center_y: −14.375, and center_z: 23.051 with dimensions size_x: 15, size_y: 15, and size_z: 15. The value of exhaustiveness was set to 20. For Vina docking, the default parameters were used unless specifically mentioned. The best-scoring pose as judged by the Vina docking score was chosen and visually analyzed using PyMoL 1.7.6 software (http://www.pymol.org/).Human CNP determination
Samples were dissolved in and diluted with 80% polyethylene glycol 400. A 20 μL aliquot of the sample solution was then mixed with 80 μL of plasma and incubated at 37 °C for 15 min. The plasma mixed solution was assayed with a Human CNP ELISA kit according to the manufacturer's instructions. The enzymatic activity was examined at 450 nm using a microplate reader. All samples were assayed in triplicate. Finally, the contents were calculated according to the standard curves.Statistical analysis
Significant differences between the groups were determined using a one-way analysis of variance (ANOVA) in SPSS 22.0 (SPSS Inc., Chicago, Illinois, USA), followed by Dunnett's test with P < 0.05. The EC50 values were determined using linear regression in SPSS 22.0.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21362001), and the Guangxi Key Laboratory of Traditional Chinese Medicine Quality Standards (guizhongzhongkai201104). We acknowledge MogoEdit (a professional English editing company) for English revision of the manuscript.References
- W. C. Wu, A. Trivedi, P. D. Friedmann, W. G. Henderson, T. S. Smith, R. M. Poses, G. Uttley, M. Vezeridis, C. B. Eaton and V. Mor, Ann. Surg., 2012, 255, 708 CrossRef PubMed.
- E. Schindler, J. Photiadis, N. Sinzobahamvya, A. Döres, B. Asfour and V. Hraska, Eur. J. Cardiothorac. Surg., 2011, 39, 495–499 CrossRef PubMed.
- Can Drugs Reduce Surgical Blood Loss?, The Lancet, 331(8578), 155–156 Search PubMed.
- B. L. Erstad, Ann. Pharmacother., 2001, 35, 925 CrossRef CAS PubMed.
- M. Verstraete, Drugs, 1985, 29, 236–261 CrossRef CAS PubMed.
- D. T. Mangano, I. C. Tudor and C. Dietzel, N. Engl. J. Med., 2006, 354, 353 CrossRef CAS PubMed.
- S. Durgamma, P. R. Reddy, V. Padmavathi and A. Padmaja, J. Heterocycl. Chem., 2015, 53, 738–747 CrossRef.
- N. B. Patel and F. M. Shaikh, Sci. Pharm., 2010, 78, 753–765 CrossRef PubMed.
- D. L. N. Surleraux, P. T. B. P. Wigerinck and D. P. Getman, WO 2004/014371 Al, 2010.
- C. R. Gardner, B. B. Cheung, J. Koach, D. S. Black, G. M. Marshall and N. Kumar, Bioorg. Med. Chem., 2012, 20, 6877–6884 CrossRef CAS PubMed.
- P. K. Deb, R. Kaur, B. Chandrasekaran, M. Bala, D. Gill, V. R. Kaki, R. R. Akkinepalli and R. Mailavaram, Med. Chem. Res., 2014, 23, 2780–2792 CrossRef CAS.
- K. Yamazaki, Y. Kaneko, K. Suwa, S. Ebara, K. Nakazawa and K. Yasuno, Bioorg. Med. Chem., 2005, 13, 2509–2522 CrossRef CAS PubMed.
- O. R. Thiel, C. Bernard, T. King, M. Dilmeghaniseran, T. Bostick, R. D. Larsen and M. M. Faul, J. Org. Chem., 2008, 73, 3508–3515 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed.
- B. Aneja, B. Kumar, M. A. Jairajpuri and M. Abid, ChemInform, 2016, 47, 18364–18406 CrossRef.
- W. Nong, A. Zhao, J. Wei, X. Lin, L. Wang and C. Lin, Bioorg. Med. Chem. Lett., 2017, 4506–4511 CrossRef CAS PubMed.
- C. W. Lin, F. Y. Meng, F. Q. Lin, J. R. Wei and C. R. Du, China Pat., CN103288752A, 2013.
- S. Durgamma, A. Muralikrishna, V. Padmavathi and A. Padmaja, Med. Chem. Res., 2014, 23, 2916–2929 CrossRef CAS.
- R. F. Pellon and M. L. Docampo, Synth. Commun., 2013, 43, 537–552 CrossRef CAS.
- H. Youn, L. Hyun-Joo, K. Eun-Kyung, P. Jin-Hwi and J. Jae-Eun, WO2013043002A1, 2013.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2006, 37, 10827–10852 Search PubMed.
- R. J. Watson, D. Batty, A. D. Baxter, D. R. Hannah, D. A. Owen and J. G. Montana, Tetrahedron Lett., 2002, 43, 683–685 CrossRef CAS.
- A. D. Michelson, The Platelets, Academic Press, 2002 Search PubMed.
- P. Harrison, Blood Rev., 2005, 19, 111–123 CrossRef PubMed.
- M. L. Rand, R. Leung and M. A. Packham, Transfus. Apher. Sci., 2003, 28, 307 CrossRef PubMed.
- L. Lv, F. Tang and G. Lan, RSC Adv., 2016, 6, 95358–95368 RSC.
- L. Liu, J. A. Duan, Y. Tang, J. Guo, N. Yang, H. Ma and X. Shi, J. Ethnopharmacol., 2012, 139, 381 CrossRef CAS PubMed.
- W. E. Dager, R. C. Gosselin, S. Kitchen and D. Dwyre, Ann. Pharmacother., 2012, 46, 1627–1636 CrossRef PubMed.
- N. Ayres, D. J. Holt, C. F. Jones, L. E. Corum and D. W. Grainger, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7713–7724 CrossRef CAS PubMed.
- C. Pallister and M. Watson, Haematology, Scion Publishing Ltd.: Banbury UK, 2nd edn, 2010 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- N. Hama, H. Itoh, G. Shirakami, S. Suga, Y. Komatsu, T. Yoshimasa, I. Tanaka, K. Mori and K. Nakao, Biochem. Bioph. Res. Co., 1994, 198, 1177–1182 CrossRef CAS PubMed.
- I. Pham, S. Sediame, G. Maistre, F. Roudotthoraval, P. E. Chabrier, A. Carayon and S. Adnot, Am. J. Physiol., 1997, 273, 1457–1464 Search PubMed.
- M. Furuya, K. Aisaka, T. Miyazaki, N. Honbou, K. Kawashima, T. Ohno, S. Tanaka, N. Minamino, K. Kangawa and H. Matsuo, Biochem. Bioph. Res. Co., 1995, 748, 517 CAS.
- A. Borchers, Hemostatic Drugs, Small Animal Critical Care Medicine, 2nd edn, 2015, ch. 170, pp. 893–898 Search PubMed.
- Q. Sun, Y. U. Hong-Qi and X. Z. Zhao, Chinese & Foreign Medical Research, 2012, 32, 1–2 Search PubMed.
- A. Z. Liu and H. E. Zhe-Sheng, J. Fourth Mil. Med. Univ., 1993, 14, 221–222 Search PubMed.
- A. Z. Liu and Z. L. Zhang, Chin. J. Phys. Med. Rehabil., 2002, 23, 32–34 CAS.
- J. Petersen, K. Hedberg and B. Christensen, Ind. Eng. Chem., Anal. Ed., 1943, 15, 225–226 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2015, 71, 3 CrossRef PubMed.
- D. I. Donato, G. Lazzara and S. Milioto, J. Therm. Anal. Calorim., 2010, 101, 1085–1091 CrossRef CAS.
- M. Hussain, RSC Adv., 2015, 5, 54963–54970 RSC.
- G. V. Born, Nature, 1962, 194, 927 CrossRef CAS PubMed.
- M. F. Sanner, J. Mol. Graphics Modell., 1999, 17, 57–61 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1582748 and 1582749. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra13397a |
| This journal is © The Royal Society of Chemistry 2018 |