
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeposition of Ag and Ag–Au nanocrystalline films with tunable conductivity at the water–toluene interface†
Gemma L. Stansfielda,
Helena M. Johnstona,
Sean N. Baxter b and
P. John Thomas*b
b and
P. John Thomas*b
aSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
bSchool of Chemistry, Bangor University, Deniol Road, Bangor, LL57 2UW, UK. E-mail: john.thomas@bangor.ac.uk
First published on 7th February 2018
Abstract
Thin films of Au, Ag and Ag–Au alloy nanocrystals extending to areas of several square centimetres are obtained by deposition at the interface of water and toluene. Toluene containing chlorotris(triphenylphosphine)silver(I) and/or chlorotriphenylphosphine gold(I) is reacted with aqueous tetrakishydroxymethylphosphonium chloride to obtain nanocrystalline films adhered to the interfacial region. Alloying was induced by varying the composition of the toluene layer. The composition change results in regular and reproducible variation in the transport characteristics of the films, with the initially metallic deposits turning non-metallic with increased Au content. The films at the interface were transferred to different substrates and characterised using atomic force microscopy, UV-visible spectroscopy, X-ray diffraction, X-ray photoelectron spectroscopy, scanning and transmission electron microscopy.
1 Introduction
Films of Ag and Au nanocrystals possess unique optical and electronic properties due to the reduced dimensions of the crystallites and the dominance of surface plasmons.1 A number of potential applications have been identified for films of metal nanocrystals including the design of the next generation electrical and electrochemical sensors, solar cell devices,2–4 as well as optoelectronic devices.5,6 Strong local electrical fields present in nanocrystalline Ag films make them ideal substrates for surface enhanced Raman and related spectroscopic techniques.7 Nanocrystalline films of Ag have previously been shown to considerably enhance the performance of Si based solar cells.8,9 In order to be harnessed in different applications uniform nanocrystalline films spread over large areas with strongly interacting nanocrystals acting as robust charge transport network are desirable. The quest for such films dominates current research in this area.10In the past few years, the interface between two immiscible liquids has emerged as a powerful new method to assemble nanocrystals as well as to deposit films of nanostructured material.11,12 The rapid and reversible adsorption of suitable nanocrystals at the water–oil interface has led to more resilient assembly schemes.13,14 Rao and co-workers have been at the vanguard of a revival interest in using the interface of oil–water to deposit inorganic nanostructures.15 A myriad of materials of contemporary interests have been recently obtained using this scheme, including: single crystalline scrolls of CuO,16 nanoscopic pyramids of crystalline PbS with a large proportion of an highly reactive and open {331} surface17 as well as hierarchical anisotropic Cd(OH)2 nanowire structures.18
Interfacial deposition is accomplished by simply layering a dispersion of organometallic precursor in a solvent such as toluene atop an aqueous solution containing the anionic counterpart of the material of interest. When the right precursors are chosen, the reaction proceeds at the region of interface between the two liquids and produces a thin film that adheres to the interface. The film can subsequently be transferred to a wide variety of substrates. Deposition at the water–oil interface usually features simple chemistry, as the interfacial barrier eliminates the need for masking agents. The method provides a remarkably low-cost, green and scalable route to complex solids. The deposition scheme requires little or no energy input. Furthermore, the nature of properties of interfacial deposits can be varied by controlling parameters such as deposition temperature, growth time and precursor concentrations.10,19
We have shown that device quality films of Au nanocrystals can be directly obtained from interfacial deposits using a novel transfer process.20,21 Herein, we have been successful in obtaining interfacial deposits of Ag nanocrystals with highly reproducible transport characteristics, indicative of robust coupling between the constituent nanocrystals. Further, we show that the transport properties can be systematically varied across a large range by co-reducing Ag and Au precursors yielding Au–Ag nanoalloys.
2 Experimental
2.1 Chemicals
All chemicals were purchased from Aldrich, with the exception of chloro(triphenylphosphine)gold(I) (Acros Organics), they were all used as received with no further purification or analysis.2.2 Synthesis of chlorotris(triphenylphosphine)silver(I)
The complex was prepared following a method previously reported.22 Briefly, to a stirred dispersion of AgCl (1.43 g, 10 mmol) in dichloromethane (150 ml), a solution of triphenylphosphine (TPP) (10.97 g, 4 mmol), in dichloromethane (50 mL), was added drop wise and the stirring continued for an hour, during which time the solids almost completely dissolved. The clear liquid obtained was filtered and, to the filtrate, pentane was added to precipitate a white crystalline solid. The solid was then washed with pentane and air dried. Elemental analysis (%) found: C, 69.45; H, 4.84; Cl, 4.23; P, 9.69; Ag, 11.02. Calcd: C, 69.71; H, 4.84; Cl, 3.82; P, 10.00; Ag, 11.62.2.3 Interfacial deposition of nanocrystalline films
A 10 ml 1.5 mM toluene solution of chlorotris(triphenylphosphine)silver(I) (Ag(PPh3)3Cl) was layered atop 16 ml of 6.25 mM aqueous sodium hydroxide in a 100 ml beaker (diameter 4.8 cm). To initiate the reaction, 330 μL of 50 mM aqueous tetrakishydroxymethylphosphonium chloride (THPC) was injected into the lower layer. Almost immediately a yellow colouration was observed in the lower aqueous layer. The beaker was then quickly transferred to an oven set at the desired temperature and at the end of the reaction; a film with a lustrous grey sheen was seen at the interface between toluene and water. The film was then transferred onto glass substrates for further analysis. To obtain alloyed films, the reaction was carried out with equimolar solutions of (Ag(PPh3)3Cl) and Au(PPh3)Cl, mixed in different propositions. For example, to produce a Au0.7Ag0.3 alloy, a toluene layer with 7 ml 1.5 mM Au(PPh3)Cl solution and 3 ml 1.5 mM (Ag(PPh3)3Cl) was employed.2.4 Characterization methods
X-ray diffraction was carried out on films deposited on glass substrates using a Bruker AXS D8 diffractometer with a Cu Kα source. Atomic force microscopy was carried out using a Nanoscope IIIa microscope operating in tapping mode on interfacial deposits transferred to mica substrates whose surface was freshly cleaved. Scanning and transmission electron microscopy was carried out on Delong Instruments LVEM5 microscope with a FEG gun operating at 5 keV. High resolution SEM and energy dispersive X-ray analysis was carried out using a Phillips XL30 FEG SEM. Samples for TEM were prepared by dispersing the films in ethanol by sonication, followed by drop casting on holey carbon coated Cu grids. Charge transport measurements were carried out using a home-built system composed of a closed-cycle He cryostat, Keithley 2400 series source-measure unit and a Lakeshore 325 temperature controller. The samples were then mounted onto the cold finger of the closed-cycle cryostat and electrodes contacted using copper wires and conductive silver paint. Charge transport studies were carried out on samples held in a vacuum of 10−6 mbar from room temperature down to 15 K. FT-IR was carried out using a Bruker Alpha II spectrometer fitted with an ATR attachment. Proton NMR was carried out using Bruker 400 MHz spectrometer using a dispersion of the sample in CDCl3. Film thickness were measured using Dektak stylus profilometer.3 Results and discussion
In aqueous solutions, THPC hydrolyses in the presence of NaOH to yield HCHO and also possibly H2:23–26| PCl(CH2OH)4 + NaOH → P(CH2OH)3 + HCHO + NaCl + H2O |
P(CH2OH)3 + NaOH → O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P(CH2OH)3 + H2(g) P(CH2OH)3 + H2(g)
| (1) |
The nascent formaldehyde and hydrogen as well as tetrakishydroxymethylphosphine (THP) are all capable of reducing the bound Ag and Au ions present in toluene.27 Following reduction, the continual growth of reduced ions into large crystallites is prevented by the phosphine based ligands: THP, triphenylphosphine (TPP) and tetrakishydroxymethylphosphine oxide (THPO) bind to the surfaces formed by Ag(0) and Au(0) species resulting in the formation of nanocrystals.27 Proton NMR and IR spectra reveal bands characteristics of aliphatic and aromatic species indicating that the surface of the nanocrystals consists of species such as TPH, THPO and TPP. X-ray photoelectron spectroscopy on Ag and Au films revealed P(2p) peaks consistent with the presence of phosphinyl based ligands in the films (see ESI†).28 The particulates thus obtained in the vicinity of the interface posses high chemical potential and are adsorbed at the interface. Previous studies suggest that the room temperature adsorption energies of such nanoparticles at the water–oil interface are of the order of a few millielectron volts.29 Following detailed mechanistic studies of growth of Au nanocrystals at the water–toluene interface, Dryfe and co-workers suggest that following initial adsorption of nanoparticulates, further growth into films takes place with formaldehyde as main reducing agent.30
Deposits were grown at the interface by changing the mole ratio of the Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ag precursors at the start of the reaction at intervals of 10% (AgxAu1−x, 0 ≤ x ≤ 1.0). Energy dispersive X-ray analysis confirmed that the film compositions were in line with the feed ratio used. Striking differences were seen in the appearance of the films. As the Ag content decreased, the colour of the film became darker, moving from metallic grey to a dull brown with a low metallic sheen (see Fig. 1). Absorption spectra revealed plasmon bands whose position shifted nearly linearly from the initial ca. 390 nm for pure Ag films to 720 nm for pure Au films (see ESI†). Such robust colour changes and peak shifts are expected on the basis of changes to plasmon resonance accompanying composition changes.31,32
:Ag precursors at the start of the reaction at intervals of 10% (AgxAu1−x, 0 ≤ x ≤ 1.0). Energy dispersive X-ray analysis confirmed that the film compositions were in line with the feed ratio used. Striking differences were seen in the appearance of the films. As the Ag content decreased, the colour of the film became darker, moving from metallic grey to a dull brown with a low metallic sheen (see Fig. 1). Absorption spectra revealed plasmon bands whose position shifted nearly linearly from the initial ca. 390 nm for pure Ag films to 720 nm for pure Au films (see ESI†). Such robust colour changes and peak shifts are expected on the basis of changes to plasmon resonance accompanying composition changes.31,32
 | ||
| Fig. 1 Photograph showing the various films studied. The compositions from the left are Ag, Ag0.90Au0.10, Ag0.80Au0.20, Ag0.70Au0.30, Ag0.60Au0.40, Ag0.50Au0.50, Ag0.40Au0.60, Ag0.30Au0.70, Ag0.20Au0.80, Ag0.10Au0.90 and Au. The colour changes from a metallic grey to dull brown as the Ag content falls. | ||
SEM images (Fig. 2) reveal highly uniform and continuous deposits spanning areas extending over several tens of square microns. Folds and cracks are apparent in the deposits. At high resolution, globular clusters with diameters in the range of tens of nanometers can be seen throughout the deposits. The cluster edges are jagged due to the presence of smaller particulates. High-resolution images reveal irregular clusters with diameters of 70 nm. AFM images (obtained in tapping mode) reveal another layer of hierarchy in the morphology. The clusters can be resolved into spherical particulates with diameters of around 10 nm. Interestingly, change in the composition of the films produces little change in the dimensions of the particulates. Profilometric measurements reveal the thickness of the various films to be in the range of 100–150 nm.
 | ||
| Fig. 2 Scanning microscopic images of films obtained at 50 °C over 180 min. (A) High magnification image of films of Au nanocrystals. The cracks are produced by solvent evaporation post-deposition. (B) Low magnification image of the same film, showing continuous and uniform deposits spanning many square millimetres (C & D) low and high magnification images of Ag nanocrystal films. (E & F) Low and high magnification images of Ag0.5Au0.5 films. | ||
X-ray diffraction patterns of Ag, Au and Ag0.5Au0.5 films yield broad peaks characteristic of nanocrystals (see ESI†). The peak widths appear to mask the expected small change in peak positions accompanying the change in lattice parameters (from 408.53 pm for Ag to 407.82 pm in the case of Au). Toluene dispersions of Ag, Au and alloy films were obtained by sonication and used for TEM studies. In the case of Ag films, TEM images reveal rough aggregates with irregular outlines (Fig. 3a). Closer inspection reveals that the aggregates contain nanocrystals with dimensions of ≈10 nm. In the case of Au nanocrystals, sonicating is much more effective in breaking up the aggregates. TEM images reveal well resolved spherical nanocrystals with diameters of 10 nm (Fig. 3b). Lattice planes corresponding to the separation between 〈111〉 planes of Ag0.5Au0.5 alloys are apparent in Fig. 3c. The coarse aggregates (in the case of Ag) as well as the clearly separated nanocrystals (Au) suggest that organic ligands are present at the surface of the particulates. The films herein consist of nanocrystals coated with organic ligands aggregating to form spherical clusters with diameters of several tens of nanometres. Previous studies, have confirmed that (at room temperature) nanocrystals with diameters of ≈1.5 nm rapidly form an initial adlayer at the interface that anchors further growth.30,33 Subsequent reduction happens at the surface of these adsorbed nanocrystals.30 The significantly larger diameters obtained herein (ca. 10 nm) suggest that the higher temperature employed here has resulted in extensive secondary growth at the interface following initial adsorption.
 | ||
| Fig. 3 (A) Transmission electron microscopic image of drop cast dispersions of Ag nanocrystal films. (B) TEM image of a drop cast dispersion of Au nanocrystal films. (C) TEM image of the Ag0.5Au0.5 alloy film & (D) atomic force microscopic images of a film of Au nanocrystals. The area is 250 nm × 250 nm. | ||
In the bulk, Au and Ag form alloys in the entire range of compositions.34 More recent studies have suggested that alloying at the nanoscale follows distinct pathways.35,36 In the specific case of Au–Ag, it has been indicated that compositional inhomogeneities are expected in particles synthesized at temperatures close to room temperature.31 The evidence from HR-TEM suggests that alloy particles have indeed been obtained. However, we believe that further studies based on techniques such as X-ray absorption spectroscopy and related techniques are required to establish the veracity of alloying in these deposits.37
As alluded to earlier, in all cases, the films consist of hierarchical structure whereby nanocrystals with diameters of ≈10 nm form higher order aggregates. The persistence of this organization across the composition range suggests that structural control lies in factors extrinsic to the reaction. We believe that the structure adopted by the films is shaped by the interfacial medium used to grow the films. This argument is further reinforced by the previous observations of similar hierarchical organizations in films of Bi2S3,17 Cd(OH)2 (ref. 18) and Pd38 obtained at the water–toluene interface. Clearly, such organizations are brought about by the water–oil interface, the only common factor in all the studies. However, the complex nature of the interfacial deposition process means that it is currently impossible to identify specific properties of the medium that underpin such control.
We find that the room temperature resistance of Ag nanocrystal deposits was in the region of about 102126 sq.−1 and is only slightly higher than bulk Ag. The resistance increases nearly exponentially with increasing Au content (see Fig. 4). A systematic change of resistance over eight orders of magnitude is seen. The robust and methodical change herein is particularly impressive in the context of films of nanocrystals where a number of factors (discussed later) conspire to render reliable measurements across different films all but impossible.
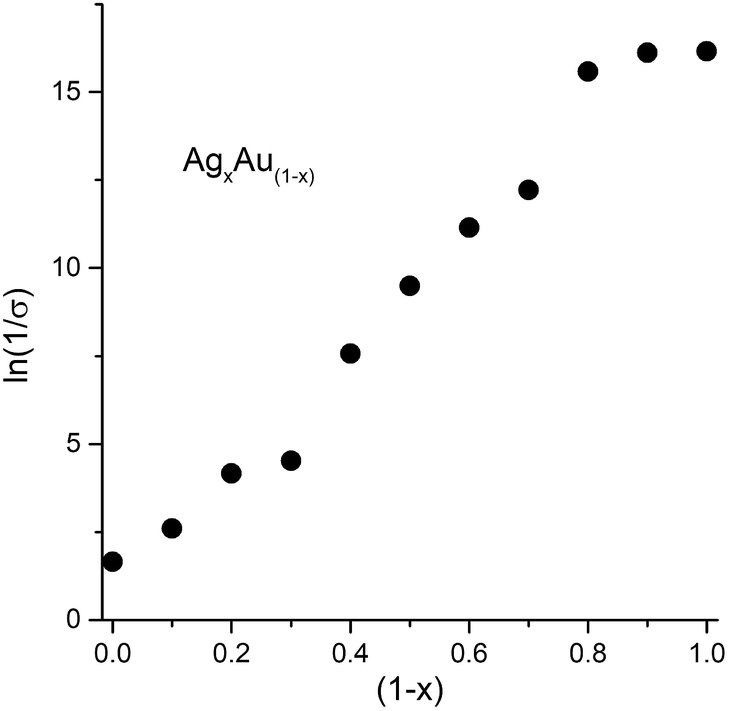 | ||
| Fig. 4 Plot showing the change in the natural log of resistance, ln(1/σ) against the composition of the film of nanocrystals. The resistance is in units of ohms per square. | ||
The electronic properties of films of metal nanocrystals differ from those of the bulk metal.39–41 Charge transport is primarily dependent on the diameter of the crystallites42,43 and separation distance between the grains.44–47 Other pertinent factors include structural disorder in the films48 and monodispersity in the size of grains.47
We studied the dependence of electrical resistance on temperature in detail using different films of Au–Ag nanocrystals. We find that the AgxAu1−x films with x ≤ 0.4 exhibit a negative co-efficient of resistance signalling that deposits are metallic (Fig. 5). Such behaviour is indicative of strong exchange coupling between the particulates. Robust coupling extending over a number of compositions is noteworthy, especially in light of the disorder inherent to these films. When the percentage of Au in the films is increased, the temperature co-efficient becomes positive, signalling that the deposits are non-metallic. The resistance change with fall in temperature increases with Au percentage. The observed behaviour is consistent with the hopping of localized electrons from one nanocrystal to another.21,49–51 Here, the conductivity (σ) is:
| σ ∝ e−2δβe−Ea/kT | (2) |
| σ = Ae−Ea/kT | (3) |
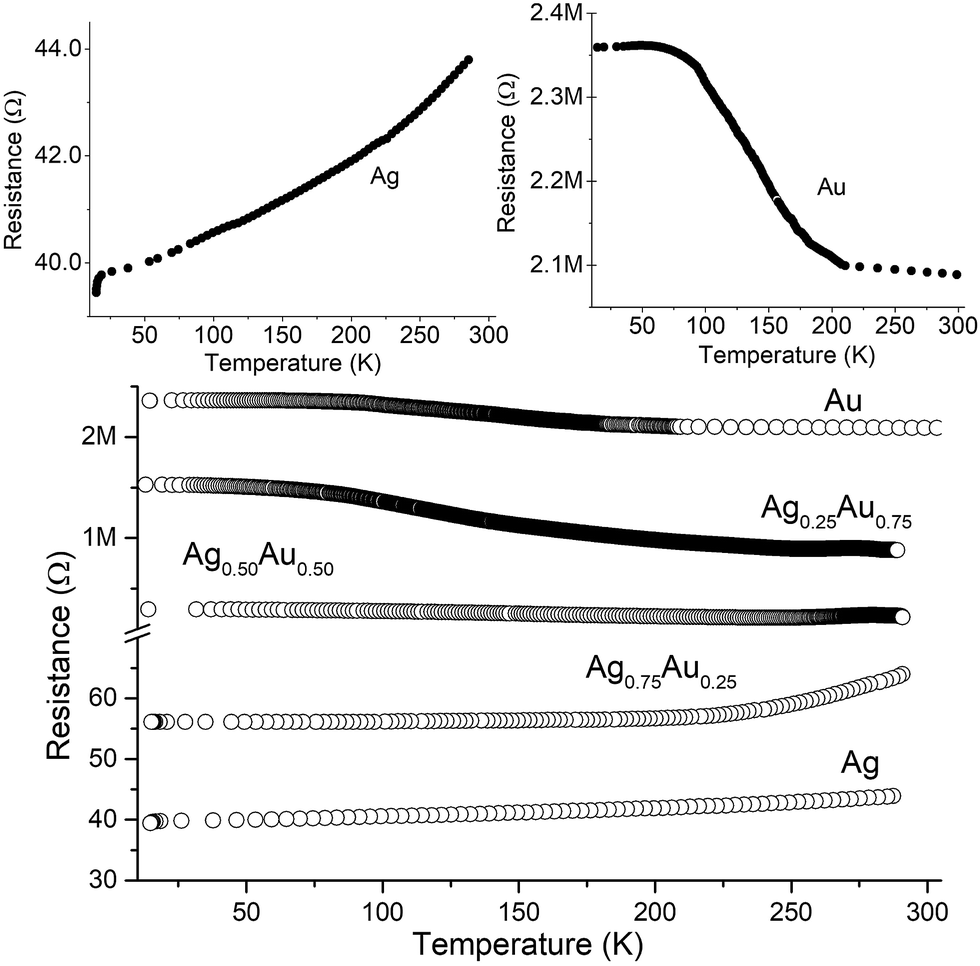 | ||
| Fig. 5 Plot showing the temperature dependence of the film resistance for different compositions (labelled). The plot in top left show resistance vs. temperature data for metallic Ag films, while the top right plot corresponds to Au (non-metallic). Three distinct temperature profiles is apparent in this panel. | ||
The activation energy, Ea corresponds to:
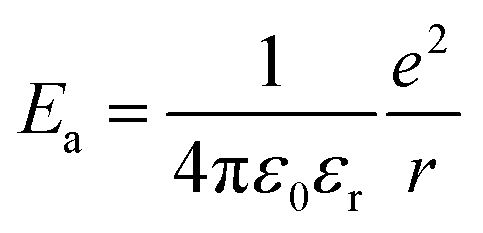 | (4) |
:Ag ratios.
In deposits with non-metallic characteristics, two Arrhenius regimes are apparent, with changeover centred around ≈50 K. The Eas are in the range of a few meV per grain in the high temperature region (100–250 K). At lower temperatures, Eas falls lower (<0.1 meV). Hence, charge transport appears to be mediated by a activated hopping in the high temperature region, while at lower temperatures, tunnelling is dominant. In the case of pure Au, another high temperature regime, above 225 K with very low Ea, mirroring the low temperature region is also seen. Similar multiple domains of activated transport have been previously reported, through the reasons of this behaviour remains elusive.21 Rao and co-workers have studied electronic transport properties of nanocrystalline films obtained at the water–oil interface.53 They found that the charge transport can be metallic when particulate diameters are high (>10 nm) or when particular ligands capable of bridging the gap between particulates are used.
A switch from metallic to non-metallic behaviour can be brought about by factors such as decrease in diameter of the nanocrystals, increasing disorder and separation distance as well as by changes in the nature of interaction with surface ligands. As alluded to earlier, change in composition of the alloy produces little discernible change to the overall dimensions of nanocrystals and therefore unlikely to account for the change to non-metallic behaviour. Given the regular trend with change in composition, we believe that the switch in behaviour can be attributed to the differences in interaction between the particulates and the surface ligands. In the case of Ag rich films, relatively weak binding to surface ligands results in physical contact between neighbouring nanocrystals leading to development of conductive pathways (via partial sintering) which result in overall metallic behaviour. Sintering is expected to be extensive, particularly during the secondary growth phase when the main site of reduction is the surface of interfacially adsorbed particulates. In the case of Au rich films, stronger interactions lead to better defined surface structure and robust interparticle repulsion, resulting in the collapse of conductive pathways, leading to semiconducting behaviour. This argument is supported by TEM images in 3 which show that following sonication, Au films are broken down to well defined spherical nanocrystals while Ag films yield larger aggregates.
Previously, thin Ag films have been obtained at the aqueous–organic interface by reducing an adduct of AgNO3 and p-methoxybenzoic acid in dichloromethane with aqueous hydrazine, in the presence of different surfactants.54 Both Efrima and Rao have reported the synthesis and properties of silver films that have been at the interface between two immiscible liquids using other precursors.55,56 The charge transport characteristics of these films are unknown.
4 Conclusions
Highly uniform films of Ag and Ag–Au nanocrystals with phosphinyl surface ligands have been obtained at the water–toluene interface. The films posses a hierarchical structure consisting of large aggregates of 10 nm sized nanocrystals. The ubiquity of this structure across the range of compositions studied suggests that the medium shapes the films. Remarkable differences were seen in the optical and electronic properties of the films as the composition is varied. Strong electronic coupling between Ag nanocrystals and possibly weak interaction with surface ligands yields metallic films with resistance of a few ohms. On the other hand, films comprising of Au nanocrystals exhibit behaviour akin to semiconductors with resistance values in the range of MΩs. The resistance of the films scales exponentially with composition of the films, changing from metallic to non-metallic behaviour when the percentage of Ag falls below 70%.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Graham Smith and Dr Simon Hodgson of Chester University for help with X-ray photoelectron spectroscopic measurement.References
- G. Xu, M. Tazawa, P. Jin and S. Nakao, Appl. Phys. A: Mater. Sci. Process., 2005, 80, 1535–1540 CrossRef CAS.
- D. B. Mitzi, M. Yuan, W. Liu, A. J. Kellock, S. J. Chey, V. Deline and A. G. Schrott, Adv. Mater., 2008, 20, 3657–3662 CrossRef CAS.
- J. Tang, K. W. Kemp, S. Hoogland, K. S. Jeong, H. Liu, L. Levina, M. Furukawa, X. Wang, R. Debnath, D. Cha, K. W. Chou, A. Fischer, A. Amassian, J. B. Asbury and E. H. Sargent, Nat. Mater., 2011, 10, 765–771 CrossRef CAS PubMed.
- M. Berginc, U. Opara Krašovec and M. Topič, J. Nanomater., 2014, 2014, 1–11 CrossRef.
- J. Wei, N. Schaeffer and M. P. Pileni, J. Phys. Chem. B, 2014, 118, 14070–14075 CrossRef CAS PubMed.
- Z. Chen, B. Nadal, B. Mahler, H. Aubin and B. Dubertret, Adv. Funct. Mater., 2014, 24, 295–302 CrossRef CAS.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- Z. Ouyang, S. Pillai, F. J. Beck, O. Kunz, S. Varlamov, K. R. Catchpole, P. Campbell and M. A. Green, Appl. Phys. Lett., 2010, 96, 261109 CrossRef.
- F. J. Beck, S. Mokkapati and K. R. Catchpole, Opt. Express, 2011, 19, 25230 CrossRef PubMed.
- P. J. Thomas, E. Mbufu and P. O'Brien, Chem. Commun., 2013, 49, 118–127 RSC.
- R. A. W. Dryfe, A. O. Simm and B. Kralj, J. Am. Chem. Soc., 2003, 125, 13014–13015 CrossRef CAS PubMed.
- J. K. Sakata, A. D. Dwoskin, J. L. Vigorita and E. M. Spain, J. Phys. Chem. B, 2005, 109, 138–141 CrossRef CAS PubMed.
- F. Reincke, S. G. Hickey, W. K. Kegel and D. Vanmaekelbergh, Angew. Chem., Int. Ed., 2004, 43, 458–462 CrossRef CAS PubMed.
- Y. Lin, H. Skaff, T. Emrick, A. D. Dinsmore and T. P. Russell, Science, 2003, 299, 226–229 CrossRef CAS PubMed.
- C. N. R. Rao and K. P. Kalyanikutty, Acc. Chem. Res., 2008, 41, 489–499 CrossRef CAS PubMed.
- U. K. Gautam, M. Ghosh and C. N. R. Rao, Langmuir, 2004, 20, 10775–10778 CrossRef CAS PubMed.
- D. Fan, P. J. Thomas and P. O'Brien, Chem. Phys. Lett., 2008, 465, 110–114 CrossRef CAS.
- S. N. Mlondo, E. M. Andrews, P. J. Thomas and P. O'Brien, Chem. Commun., 2008, 2768 RSC.
- D. Fan, P. J. Thomas and P. O'Brien, J. Mater. Chem., 2007, 17, 1381–1386 RSC.
- G. L. Stansfield, P. V. Vanitha, H. M. Johnston, D. Fan, H. AlQahtani, L. Hague, M. Grell and P. J. Thomas, Philos. Trans. R. Soc., A, 2010, 368, 4313–4330 CrossRef CAS PubMed.
- G. L. Stansfield and P. J. Thomas, J. Am. Chem. Soc., 2012, 134, 11888–11891 CrossRef CAS PubMed.
- D. V. Sanghani, P. J. Smith, D. W. Allen and B. F. Taylor, Inorg. Chim. Acta, 1982, 59, 203–206 CrossRef CAS.
- A. Hoffman, J. Am. Chem. Soc., 1921, 43, 1684–1688 CrossRef CAS.
- A. Hoffman, J. Am. Chem. Soc., 1930, 52, 2995–2998 CrossRef CAS.
- M. Grayson, J. Am. Chem. Soc., 1963, 85, 79–83 CrossRef CAS.
- W. J. Vullo, J. Org. Chem., 1968, 5, 9–11 Search PubMed.
- J. L. Hueso, V. Sebastián, Á. Mayoral, L. Usón, M. Arruebo and J. Santamaría, RSC Adv., 2013, 3, 10427 RSC.
- K. Luo, C. Hu, Y. Luo, D. Li, Y. Xiang, Y. Mu, H. Wang and Z. Luo, RSC Adv., 2017, 7, 51605–51611 RSC.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21–41 CrossRef CAS.
- K. Luo, S. L. M. Schroeder and R. A. W. Dryfe, Chem. Mater., 2009, 21, 4172–4183 CrossRef CAS.
- C. Gao, Y. Hu, M. Wang, M. Chi and Y. Yin, J. Am. Chem. Soc., 2014, 136, 7474–7479 CrossRef CAS PubMed.
- S. Pal and G. De, Chem. Mater., 2005, 17, 6161–6166 CrossRef CAS.
- M. K. Sanyal, V. V. Agrawal, M. K. Bera, K. P. Kalyanikutty, J. Daillant, C. Blot, S. Kubowicz, O. Konovalov and C. N. R. Rao, J. Phys. Chem. C, 2008, 112, 1739–1743 CAS.
- M. J. Hostetler, C.-J. Zhong, B. K. H. Yen, J. Anderegg, S. M. Gross, N. D. Evans, M. Porter and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 9396–9397 CrossRef CAS.
- C. Yan and T. Wang, Chem. Soc. Rev., 2017, 46, 1483–1509 RSC.
- M. Cui, H. Lu, H. Jiang, Z. Cao and X. Meng, Sci. Rep., 2017, 7, 41990 CrossRef CAS PubMed.
- I. J. Godfrey, A. J. Dent, I. P. Parkin, S. Maenosono and G. Sankar, J. Phys. Chem. C, 2017, 121, 1957–1963 CAS.
- L. Zheng and J. Li, J. Phys. Chem. B, 2005, 109, 1108–1112 CrossRef CAS PubMed.
- D. V. Talapin, J.-s. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed.
- A. Zabet-Khosousi and A.-A. Dhirani, Chem. Rev., 2008, 108, 4072–4124 CrossRef CAS PubMed.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- M. Brust, D. Bethell, C. J. Kiely and D. J. Schiffrin, Langmuir, 1998, 14, 5425–5429 CrossRef CAS.
- M. Brust, D. J. Schiffrin, D. Bethell and C. J. Kiely, Adv. Mater., 1995, 7, 795–797 CrossRef CAS.
- F. P. Zamborini, L. E. Smart, M. C. Leopold and R. W. Murray, Anal. Chim. Acta, 2003, 496, 3–16 CrossRef CAS.
- J. B. Pełka, M. Brust, P. Gierłowski, W. Paszkowicz and N. Schell, Appl. Phys. Lett., 2006, 89, 063110 CrossRef.
- R. C. Doty, H. Yu, C. K. Shih and B. A. Korgel, J. Phys. Chem. B, 2001, 105, 8291–8296 CrossRef CAS.
- R. Parthasarathy, X.-M. Lin and H. M. Jaeger, Phys. Rev. Lett., 2001, 87, 4 CrossRef.
- A. L. Efros and B. I. Shklovskii, J. Phys. C: Solid State Phys., 1975, 8, L49–L51 CrossRef CAS.
- C. A. Neugebauer and M. B. Webb, J. Appl. Phys., 1962, 33, 74 CrossRef CAS.
- N. J. Hardy, M. D. Hanwell and T. H. Richardson, J. Mater. Sci.: Mater. Electron., 2007, 18, 943–949 CrossRef CAS.
- A. B. Buckman and N. M. Bashara, Phys. Rev., 1968, 174, 719–721 CrossRef CAS.
- V. V. Agrawal, G. U. Kulkarni and C. N. R. Rao, J. Phys. Chem. B, 2005, 109, 7300–7305 CrossRef CAS PubMed.
- I. Farbman and S. Efrima, J. Phys. Chem., 1992, 96, 8469–8473 CrossRef CAS.
- D. Yogev and S. Efrima, J. Phys. Chem., 1988, 92, 5754–5760 CrossRef CAS.
- R. Krishnaswamy, S. Majumdar, R. Ganapathy, V. V. Agrawal, A. K. Sood and C. N. R. Rao, Langmuir, 2007, 23, 3084–3087 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13370g |
| This journal is © The Royal Society of Chemistry 2018 |