
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, and single crystal structure of fully-substituted polynitrobenzene derivatives for high-energy materials†
Wei Yang b,
Huanchang Lua,
Longyu Liaoa,
Guijuan Fan*a,
Qing Ma*a and
Jinglun Huang*a
b,
Huanchang Lua,
Longyu Liaoa,
Guijuan Fan*a,
Qing Ma*a and
Jinglun Huang*a
aInstitute of Chemical Materials, China Academy of Engineering Physics, Mianshan Road 64, Mianyang, China. E-mail: fanguijuan@caep.cn; maq@caep.cn; huangjinglun@caep.cn
bDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, USA
First published on 9th January 2018
Abstract
New energetic fully-substituted polynitrobenzene derivatives were synthesized via the reaction of 1,3,5-trichloro-2,4,6-trinitrobenzene (TCTNB) with 1,2,4-triazole followed by the nucleophilic substitution of the halo groups by aqueous ammonia or sodium azide. These compounds were charactered by 1H NMR, 13C NMR and HRMS. Additionally, the structures of amino derivatives 6, 8 and azido derivative 7 were further confirmed by single crystal X-ray diffraction analysis. Their decomposition temperatures and impact sensitivities were also determined. The derivative 1,2-di-1H-triazol-4,6-diamino-3,5-dinitrobenzene (8) exhibits good thermal stability (Td = 314 °C) and low impact sensitivity (IS = 30 J), which are both superior to that of TNT (Td = 295 °C, IS = 15 J). These synthesized compounds showed high heat of formation ranging from 31.77 to 1014.68 kJ mol−1, and reasonable detonation velocities and pressures.
Introduction
The synthesis and characterization of polynitrobenzene derivatives have attracted continuous attention both theoretically and experimentally as potential intermediates in the development of pharmaceuticals, polymers, organic electronics and energetic materials.1a–e The appropriate introduction of different energetic functional groups to the polynitrobenzene skeleton can afford relatively better energetic performance in different aspects of detonation, thermal stability, density and sensitivity, for example the fully substituted polynitrobenzene energetic molecule 2,4,6-triamino-1,3,5-trinitrobenzene (TATB, vD = 8108 m s−1, d = 1.94 g cm−3).2 Nitro derivatives of benzene and aminobenzene always demonstrate excellent heat-resistance and insensitivity, because of their well known π–π stacking3,4 and strong intramolecular resonance-assisted hydrogen bonds with a binding energy of 7–14 kcal mol−1,5,6 which also increase the crystal density.The main strategies for introducing the energetic groups to polynitrobenzene fall into two categories (Scheme 1). The first and most common method is nitration of benzene skeletons pre-functionalized with energetic groups, providing products such as TNT and 1-(3,4,5-trinitrophenyl)-1H-tetrazole (1).7,8 The other method is exchange of leaving groups on polynitrobenzenes (Picryl Chloride, TCTNB, 2,4,6-trifluoro-1,3,5-trinitroben-zene/TFTNB, etc.) by direct substitution with energetic groups, providing products such as TATB and 2,4,6-tri-1H-triazol-1-yl-1,3,5-trinitrobenzene (2).9,10 Theoretically, fully substitution by energetic groups bearing high heat of formation can increase the density and detonation performance of energetic materials to the utmost extent. However, the nitration of energetic benzenes always leads to non-fully substituted molecules because nitration becomes more difficult after the introduction of nitro groups to the system. Furthermore, direct substitution of halogenated polynitrobenzenes with energetic groups also result in uncontrolled replacement or incomplete substitution of halogens due to different nucleophilicity of azoles and steric-hindrance between substituted groups, which make the reaction unpractical and difficult to purify.11–13 In general, the synthesis and characterization of fully substituted polynitrobenzenes as energetic materials are still challenging.
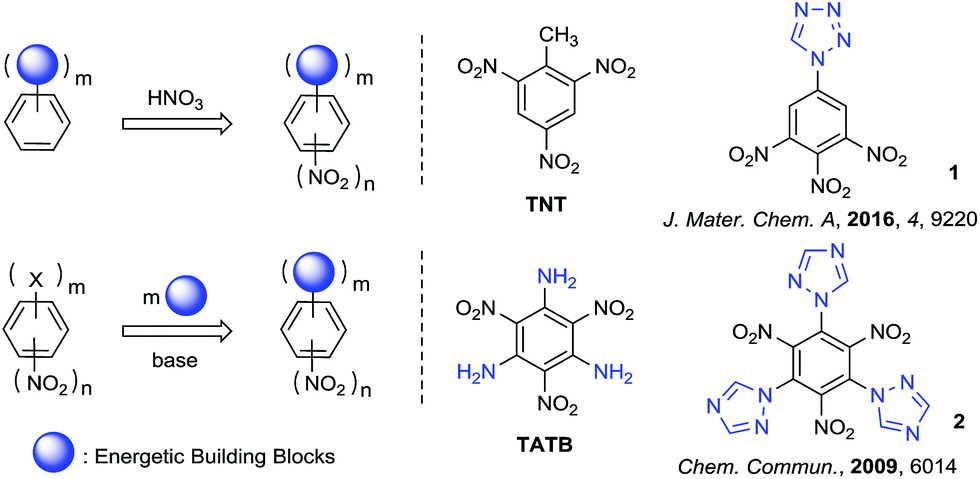 | ||
| Scheme 1 Synthetic strategy of polynitrobenzene derivatives for energetic materials. | ||
The five-membered azoles are invariably popular building blocks in energetic materials due to their high-nitrogen content and multiple C–N/N–N bonds, providing them with high positive heat of formation as well as good detonation performance.14 Although Shreeve and co-workers have reported the synthesis of energetic molecule (2) from TFTNB and 1-trimethylsilyltriazole,10 syntheses of fully substituted polyazole polynitrobenzenes from TCTNB are rarely reported because of its uncontrollable replacement with azoles.
In this work, we discuss a reliable and practical synthetic method for the construction of mono-/di-triazole substituted dinitrobenzene and their fully-substituted derivatives bearing –NH2 or –N3 groups by using the more economical and widely available starting material TCTNB. The compounds were analysed by single crystal X-ray diffraction and the energetic properties were evaluated by density functional theory (DFT) method. Some of the molecules show good thermal stability (up to 314 °C) along with comparable energetic performance, establishing these compounds as useful high-explosive molecules.
Results and discussion
Synthesis
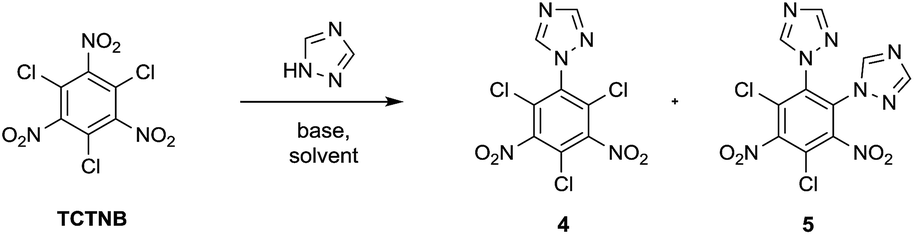
Tetrazole and 1,2,4-triazole were investigated in the nucleophilic substitution of TCTNB owing to their good energetic properties (Scheme 2). When 3 mmol tetrazole was reacted with 1 mmol TCTNB at room temperature to synthesize 2,4,6-tri-1H-triazol-1-yl-1,3,5-trinitrobenzene (3), only starting material was recovered. Reaction temperature, basic additives and solvent were optimized systematically to increase the nucleophilicity of tetrazole, disappointingly leading to same result or a complex mixture due to the decomposition of TCTNB. However, 1,2,4-triazole smoothly underwent substitution at room temperature and afforded the unexpected products 1-1H-triazol-2,4,6-trichloro-3,5-trinitrobenzene (4) and 1,2-di-1H-triazol-4,6-trichloro-3,5-dinitrobenzene (5) with very good reproducibility. Both of the structures were further confirmed by single crystal X-ray diffraction (Fig. 1).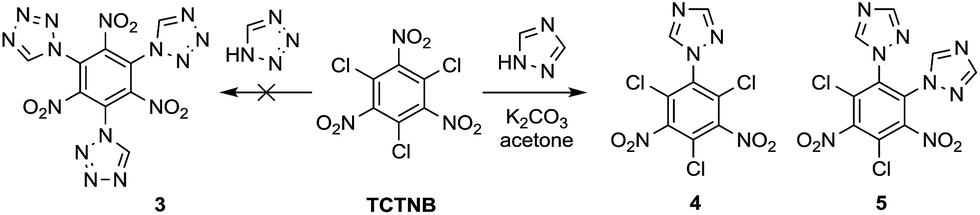 | ||
| Scheme 2 Substitution of TCTNB with azole groups. | ||
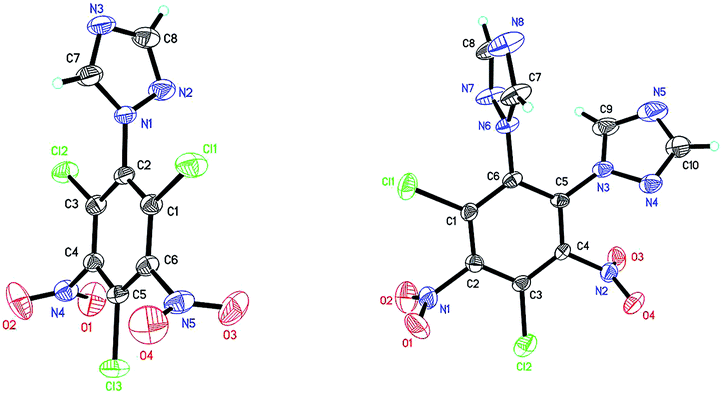 | ||
| Fig. 1 Molecular structure of 4 (left) and 5 (right). | ||
Subsequently, substitutions of TCTNB with 1,2,4-triazole were carried out varying solvents, reactant concentration and bases to optimize the reaction. As shown in Table 1, use of K2CO3 with a concentration of 0.1 M in dioxane afforded trace amount of target molecules (entry 1). Switching to more polar solvents, CH3CN or acetone, resulted in better yields of 28% (4) and 27% (5), respectively (entry 2–3). However, use of weakly basic DMF or DMSO only led to the decomposition of TCTNB (entry 4–5). After a survey of different reactant concentrations (ranging from 0.05 M to 1.0 M), 0.2 M was found to be optimal, which afforded 36% of 4 and 34% of 5 (entry 6–9). Other bases were also investigated in the reaction. Similar yields were achieved using KHCO3, Na2CO3 and NaHCO3 (entry 10–12), while the use of a stronger base CsCO3 or organic bases (TEA and DBU) led to decomposition of TCTNB (entry 13–15). Delightfully, a combined yield of 70% for 4 and 5 was achieved by using K2CO3 as basic additive and a reaction concentration of 0.2 M in acetone.
|
||||
|---|---|---|---|---|
| Entry | Base (3.0 equiv.) | Solvent | Conc. (M) | Yield (4/5%) |
| a Reactions were carried out with 3 equiv. of 1,2,4-triazole at room temperature.b No base was added.c Most of TCTNB decomposed under the corresponding conditions. | ||||
| 1 | K2CO3 | Dioxane | 0.1 | 6/4 |
| 2 | K2CO3 | CH3CN | 0.1 | 20/21 |
| 3 | K2CO3 | Acetone | 0.1 | 28/27 |
| 4 | —b | DMF | 0.1 | Messc |
| 5 | —b | DMSO | 0.1 | Messc |
| 6 | K2CO3 | Acetone | 0.05 | 27/29 |
| 7 | K2CO3 | Acetone | 0.2 | 36/34 |
| 8 | K2CO3 | Acetone | 0.5 | 19/17 |
| 9 | K2CO3 | Acetone | 1.0 | 15/16 |
| 10 | KHCO3 | Acetone | 0.2 | 29/30 |
| 11 | Na2CO3 | Acetone | 0.2 | 34/35 |
| 12 | NaHCO3 | Acetone | 0.2 | 28/29 |
| 13 | CsCO3 | Acetone | 0.2 | Messc |
| 14 | TEA | Acetone | 0.2 | Messc |
| 15 | DBU | Acetone | 0.2 | Messc |
The preferred substitution of the nitro group rather than the better leaving chloro group may be caused by steric hindrance between triazole and adjacent nitro groups upon approach of triazole at the chloro position in the transition state.
Further modification to improve the energetic properties and inter-/intra-molecular hydrogen bonds was realized by substituting the chloro groups with explosophoric groups NH3 and N3 (Scheme 3). Initial attempts to aminate 4 with aqueous ammonia at room temperature only led to partially substituted compounds as a mixture of regioisomers and degrees of substitutions. When refluxed with aqueous ammonia in ethanol, the chloro groups of 4 were completely converted to amino groups to provide 1-1H-triazol-2,4,6-triamino-3,5-trinitrobenzene (6). The same amination condition was applied to 1,2-di-1H-triazol-4,6-trichloro-3,5-dinitrobenzene (5), however, one of the triazoles was replaced by either a ethoxy or an amino group during reflux. 1,2-Di-1H-triazol-4,6-diamino-3,5-dinitrobenzene (8) was successfully synthesized by a mixture of 5 and aqueous ammonia in acetone at ambient temperature. As expected, a significant increase in thermal stability of 6 (Td = 307 °C) and 8 (Td = 314 °C) relative to 4 (Td = 256 °C) and 5 (Td = 254 °C) was observed, which clearly reflects the existence of strong inter-/intra-molecular hydrogen bonds between the amino groups and nitro groups. Next, azidation of 4 and 5 was conducted with NaN3 in the mixed solution of MeOH/H2O or DMF/H2O at room temperature, producing 1-1H-triazol-2,4,6-triazido-3,5-trinitrobenzene (7) and 1,2-di-1H-triazol-4,6-diazido-3,5-dinitrobenzene (9) respectively. X-ray diffraction analysis unambiguously elucidated the structures of compounds 6, 7 and 8. Compound 9 was characterized with 1H, 13C NMR and HRMS.
 | ||
| Scheme 3 Synthesis of fully-substituted polynitrobenzene derivatives 6–9. | ||
Crystal structures
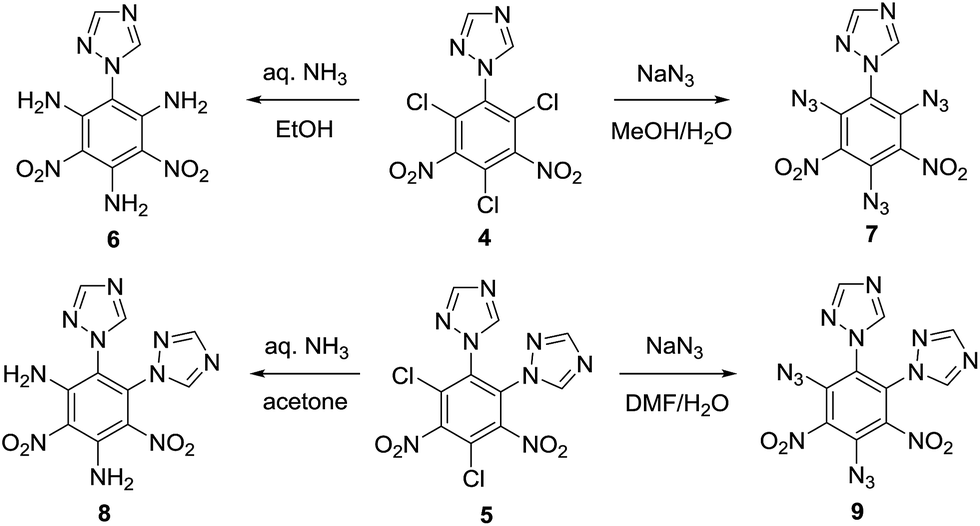
Compound 6 (Fig. 2) crystallizes as a dimer in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with four molecules per unit cell (crystalline parameters of 6 can be found in the ESI†). The triazole ring is twisted out of the benzene plane, which is further revealed by the C(2)–N(3)–C(3)–C(8) torsion angle of 80.7(3)°. Additionally, both of the nitro groups are considered to be approximately in the benzene plane, which can be illustrated by the O(3)–N(7)–C(7)–C(8) torsion angle of 177.6(2)° and the O(2)–N(5)–C(5)–C(4) torsion angle of 176.4(2)°, respectively. Nitro groups (N5, N7) and amino group (N4, N6, N8) are presumably held coplanar with the benzene plane by intramolecular hydrogen bonding (N4–H4B⋯O1 = 1.902 Å, N6–H6A⋯O2 = 1.857 Å, N6–H6B⋯O3 = 1.846 Å, N8–H8B⋯O4 = 1.890 Å). Intermolecular hydrogen bonds can also be seen in Fig. 2 as dashed lines between nitro and amino groups (N8–H8A⋯O5 = 2.731 Å, N4–H4A⋯O8 = 2.775 Å), and between azole and nitro groups (C1–H1A⋯O5 = 2.960 Å, C2–H2A⋯O3 = 2.565 Å). Along with the hydrogen bonds, π–π interaction results in stacking planes with an interplanar distance of 3.876 Å, which contributes to the good thermal stability and sensitivity of compound 6.
with four molecules per unit cell (crystalline parameters of 6 can be found in the ESI†). The triazole ring is twisted out of the benzene plane, which is further revealed by the C(2)–N(3)–C(3)–C(8) torsion angle of 80.7(3)°. Additionally, both of the nitro groups are considered to be approximately in the benzene plane, which can be illustrated by the O(3)–N(7)–C(7)–C(8) torsion angle of 177.6(2)° and the O(2)–N(5)–C(5)–C(4) torsion angle of 176.4(2)°, respectively. Nitro groups (N5, N7) and amino group (N4, N6, N8) are presumably held coplanar with the benzene plane by intramolecular hydrogen bonding (N4–H4B⋯O1 = 1.902 Å, N6–H6A⋯O2 = 1.857 Å, N6–H6B⋯O3 = 1.846 Å, N8–H8B⋯O4 = 1.890 Å). Intermolecular hydrogen bonds can also be seen in Fig. 2 as dashed lines between nitro and amino groups (N8–H8A⋯O5 = 2.731 Å, N4–H4A⋯O8 = 2.775 Å), and between azole and nitro groups (C1–H1A⋯O5 = 2.960 Å, C2–H2A⋯O3 = 2.565 Å). Along with the hydrogen bonds, π–π interaction results in stacking planes with an interplanar distance of 3.876 Å, which contributes to the good thermal stability and sensitivity of compound 6.
 | ||
| Fig. 2 Molecular structure of 6 (left) and packing diagram of 6 (right). Dashed lines indicate strong hydrogen bonding. | ||
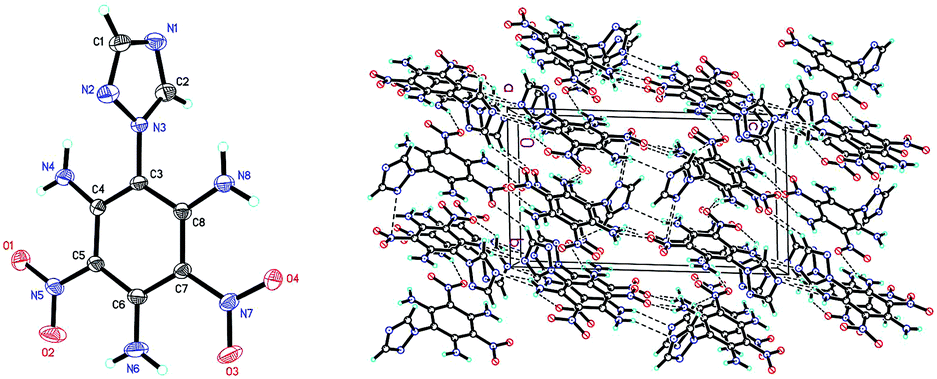
Compound 7 (Fig. 3a) crystallizes in the monoclinic space group P21/c with four molecules per unit cell (crystalline parameters of 7 can be found in the ESI†). The triazole ring is twisted out of the benzene plane, which is further revealed by the C(2)–N(3)–C(3)–C(8) torsion angle of 73.4(3)°. Neither of the nitro groups are coplanar with the benzene ring, which is illustrated by the O(1)–N(4)–C(2)–C(3) torsion angle of 126.8(2)° and the O(3)–N(8)–C(4)–C(5) torsion angle of 101.1(2)°. The three nitrogen atoms of the azido groups show a slightly bent arrangement with the N(3)–N(2)–N(1) angle of 170.4(3)°, the N(7)–N(6)–N(5) angle of 171.2(2)° and the N(11)–N(10)–N(9) angle of 169.8(2)°, respectively. Further investigation on hydrogen bonds in the crystal structure of compound 7 shows one intermolecular hydrogen bond, (C8–H8⋯O2 = 2.830 Å) shown in Fig. 3a. Besides, nitro–π stacking interactions are also found in the crystal (Fig. 3b), where the distances between the oxygen atoms of nitro groups and the centroids of the benzene rings lie in the range of 3.130–3.544 Å. Each molecule accepts and donates two nitro–π interactions. Similar nitro–π stacking has been reported before and may be due to the interaction between the electron-deficient nitro benzene ring and the electron-rich nitro group.15,16
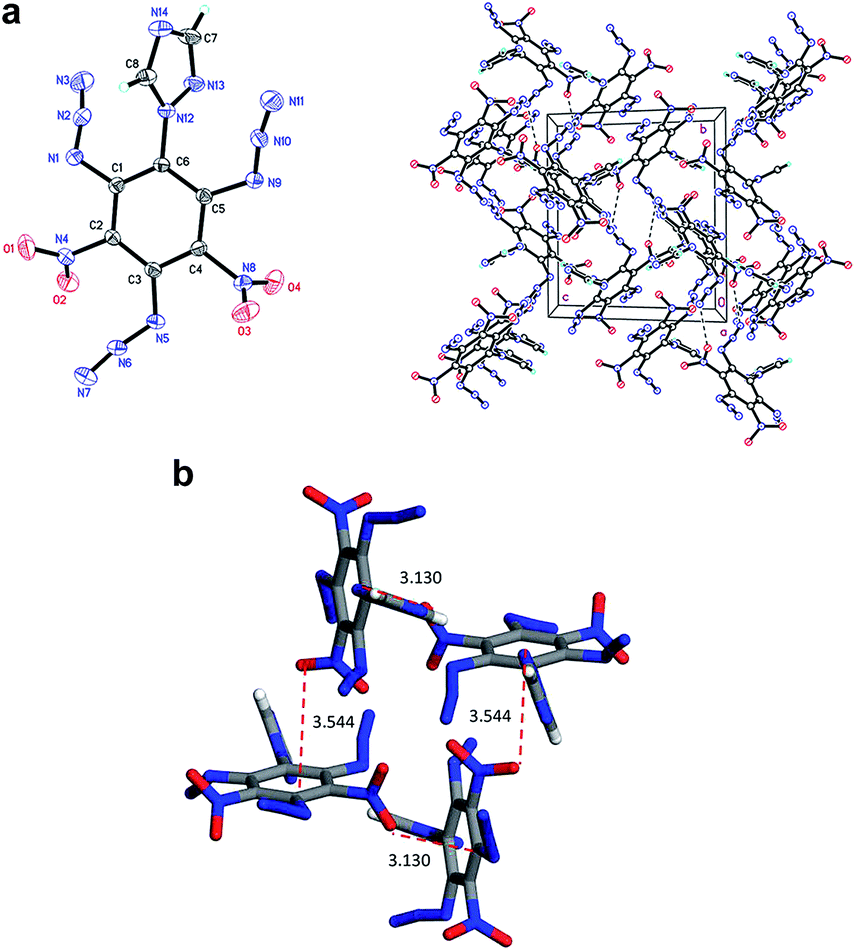 | ||
| Fig. 3 (a) Molecular structure of 7 (left) and packing diagram of 7 (right). Dashed lines indicate strong hydrogen bonding. (b) The nitro–π interactions (dashed red lines between nitro oxygen atoms and ring centroids) for each molecule of 7. | ||
Compound 8 (Fig. 4) crystallizes in the monoclinic space group P21/c with four molecules per unit cell (crystalline parameters of 8 can be found in the ESI†). Both of the triazole rings are twisted out of the benzene plane, which is further revealed by the C(7)–N(1)–C(1)–C(6) torsion angle of 53.7(4)° and the C(9)–N(8)–C(6)–C(1) torsion angle of 66.7(4)°, respectively. The nitro group adjacent to the triazole group is not coplanar with the benzene plane (∠O(3)–N(7)–C(5)–C(6): −159.7(3)°), while the other nitro group is approximately in the benzene plane (∠O(2)–N(5)–C(3)–C(2): −175.0(3)°). Nitro groups (N5, N7) and amino group (N4, N6) are presumably held coplanar with the benzene plane by intramolecular hydrogen bonding (N4–H4B⋯O1 = 1.872 Å, N6–H6B⋯O2 = 1.884 Å, N6–H6A⋯O3 = 1.936 Å). Intermolecular hydrogen bonds can also be seen in Fig. 4 as dashed lines between nitro and amino groups (N4–H4A⋯O4 = 2.583 Å), and between azole and nitro groups (C8–H8⋯O2 = 2.859 Å). Fig. 4 shows the π–π interactions that result in stacking planes with an interplanar distance of 3.870 Å. The “V-shaped” packing of compound 8 can prevent interlayer sliding within the crystal lattice, which improves the insensitivity.17
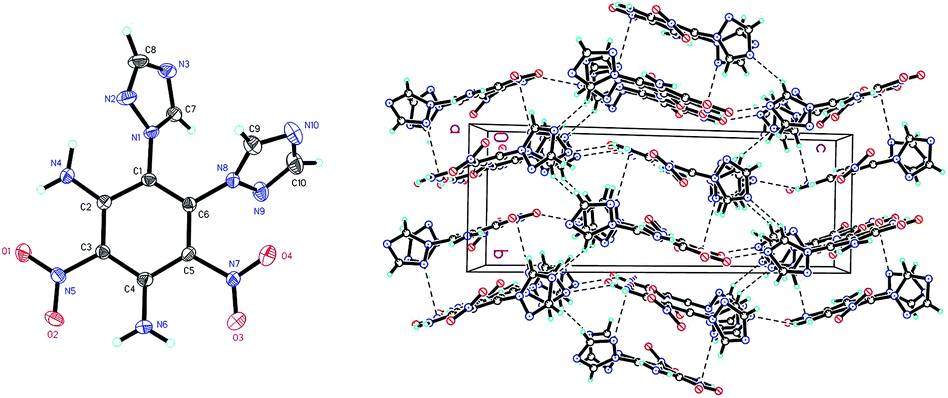 | ||
| Fig. 4 Molecular structure of 8 (left) and packing diagram of 8 (right). Dashed lines indicate strong hydrogen bonding. | ||
Physical and detonation properties
The energetic properties and some key characteristics of 6–9 are shown in Table 2. To evaluate the potential of these novel polynitrobenzene derivatives, their energetic properties were compared to TNT and TATB. Although the oxygen balance values of 6, 8 and 9 are more negative than TATB and TNT, the introduction of azido groups to energetic molecules certainly improves the negative oxygen balance better than that of amino groups.| Comp. | OBa | ΔfHb | dc | vDd | Pe | Tdf | ISg |
|---|---|---|---|---|---|---|---|
| a Oxygen balance (%).b Calculated solid-phase HOF (kJ mol−1).c Calculated density (at 298 K), the crystal density obtained by X-ray crystallographic analysis is shown in parenthesis (g cm−3).d Detonation velocity (km s−1).e Detonation pressure (GPa).f Thermal decomposition temperature (°C).g Impact sensitivity (BAM fall hammer) (J). | |||||||
| 6 | −91.36 | 31.77 | 1.73(1.67) | 6.55 | 18.53 | 307 | 7 |
| 7 | −58.07 | 1014.68 | 1.77(1.77) | 7.50 | 24.67 | 132 | 1 |
| 8 | −96.32 | 316.24 | 1.73(1.66) | 6.56 | 18.67 | 314 | 30 |
| 9 | −74.95 | 974.62 | 1.75 | 7.13 | 22.19 | 134 | 2 |
| TNT | −73.97 | −67.36 | 1.65 | 6.88 | 19.5 | 295 | 15 |
| TATB | −55.80 | −139.8 | 1.93 | 8.11 | 31.15 | 330 | 50 |
The heat of formation plays a very important role in designing energetic materials. Compound 6–9 have positive heats of formation which are superior to those of TNT and TATB. Compound 7 and 9 exhibited relatively high positive heat of formation at 1014.68 kJ mol−1 and 974.62 kJ mol−1 among the title compounds, respectively. The calculated densities of compound 6–9 lie in the range from 1.73 to 1.77 g cm−3 and the experimental densities range between 1.66 and 1.77 g cm−3, as obtained by X-ray crystallographic analysis. The calculated detonation pressures (P) and detonation velocities (vD) were estimated based on traditional Chapman–Jouget thermodynamic detonation theory using EXPLO5 V6.02.18 The calculated detonation velocities for the polynitrobenzene derivatives fall in the range between 6.55 and 7.50 km s−1 (compared with 6.88 km s−1 for TNT and 8.11 km s−1 for TATB), and the detonation pressures lie in the range of 18.53 to 24.67 GPa (compared with 19.5 GPa for TNT and 31.15 GPa for TATB).
These polynitrobenzene derivatives show excellent thermal stabilities, ranging from 132 to 314 °C. Obviously, amino derivatives exhibit much better stabilities than azido derivatives. Compound 8 has the highest thermal stability (314 °C) due to strong intramolecular hydrogen bonding. Impact sensitivities were tested using the standard BAM fall hammer method. Azido derivatives 7 (1 J) and 9 (2 J) are very sensitive compared with TNT (15 J) and TATB (50 J), while the amino derivatives 6 (7 J) and 8 (30 J) have improved sensitivities to different extents. Overall, the amino polynitrobenzene derivatives exhibit excellent thermal stabilities, low impact sensitivity and high heat of formation that make them potential energetic materials.
Conclusions
In summary, a serial of novel energetic fully-substituted polynitrobenzene derivatives (6–9) were obtained by the reliable substitution of TCTNB with 1,2,4-triazole, followed by the displacement of the halogen groups with aqueous ammonia or sodium azide. Their structures were characterized by 1H, 13C NMR spectroscopy and HRMS. Single crystal X-ray diffraction analysis was accomplished for compounds 4–8 and delivers insights into their structural characteristics. The energetic derivatives 6 and 8 exhibit good thermal stabilities ranging from 307 to 314 °C due to the introduction of amino groups and enhancement of intra-/inter-molecular hydrogen bonds. Theoretical detonation values for compound 6–9 are comparable to those of explosives such as TNT, which indicate 6 and 8 may be candidates for future applications as thermally stable energetic materials.Experimental section
Safety cautions
Although none of the above energetic polynitrobenzene derivatives described herein have exploded or detonated in the course of this research, small scale and safety training are strongly encouraged. Mechanical actions such as scratching and scraping must be avoided. Manipulations should be performed behind a safety shield with standard thickness of 7 mm or 12 mm. Face shield, eye protection and leather gloves must be strictly worn.General method
All chemicals or reagents used in this research were analytical grade materials purchased from Alfa Aesar or J&K, if not stated otherwise. 1H and 13C spectra were recorded using a 600 MHz (Bruker AVANCE III 600) nuclear magnetic resonance spectrometer operating at 600 and 150.85 MHz respectively. Chemical shifts in the 1H and 13C spectra are reported relative to Me4Si as external standards. The decomposition (onset) points were obtained on a differential scanning calorimeter-thermal gravity (TGA/DSC1, METTLER TOLEDO LF/1100) at a heating rate of 5 °C min−1.X-ray crystallography
A light yellow plate crystal (4) of dimensions 0.21 × 0.16 × 0.13 mm3, a light yellow plate crystal (5) of dimensions 0.22 × 0.17 × 0.13 mm3, a light yellow plate crystal (6) of dimensions 0.20 × 0.17 × 0.11 mm3, a light yellow plate crystal (7) of dimensions 0.18 × 0.16 × 0.13 mm3 and a light yellow plate crystal (8) of dimensions 0.17 × 0.13 × 0.07 mm3 were mounted on a MiteGen MicroMesh using a small amount of Cargille Immersion Oil. Data were collected on a Bruker three-circle platform diffractometer equipped with a SMART APEX II CCD detector. A Kryo-Flex low-temperature device was used to keep the crystals at a constant 293 K and 273 K during data collection. Data collection was performed and the unit cell was initially refined using APEX2. Data reduction was carried out using SAINT and XPREP. Corrections were applied for Lorentz, polarization, and absorption effects using SADABS. The structures were further solved and refined with the aid of the programs using direct methods and least-squares minimization by SHELXS-97 and SHELXL-97 code.1 The full-matrix least-squares refinement on F2 involved atomic coordinates and anisotropic thermal parameters for all non-H atoms. The H atoms were included using a riding model. The non-H atoms were refined anisotropically. The finalized CIF files were checked with checkCIF, and deposited at the Cambridge Crystallographic Data Centre as supplementary publications (4), (5), (6), (7) and (8). Intra- or intermolecular hydrogen-bonding interactions were analyzed with Diamond software (version 3.2K) as well as the illustrations of molecular structures.Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15–1:1) to give the product 4 (Rf = 0.7, EtOAc/hexanes 1:1) and product 5 (Rf = 0.4, EtOAc/hexanes 1:1). Product 4 (385 mg, 1.14 mmol, 36%, mp. = 184 °C) as a pale yellow solid. 1H (DMSO-d6): δ = 8.97 (s, 1H), 8.51 (s, 1H) ppm; 13C (DMSO-d6): δ = 154.18, 147.21, 147.15, 135.62, 129.46, 121.63 ppm; HRMS (ESI/[M − H]−) calcd for C8HCl3N5O4: 335.9100, found 334.9232. Product 5 (397 mg, 1.07 mmol, 34%, mp. = 200 °C) as a pale yellow solid. 1H (DMSO-d6): δ = 8.86 (s, 1H), 8.51 (s, 1H), 8.35 (s, 1H), 8.33 (s, 1H) ppm; 13C (DMSO-d6): δ = 154.38, 154.13, 148.53, 147.85, 146.86, 144.56, 133.62, 130.95, 129.44, 122.06 ppm; HRMS (ESI/[M − H]−) calcd for C10H3Cl2N8O4: 368.9660, found 368.9853.:5) to give the product 7 (0.18 g, 0.50 mmol, 71%) as a pale yellow solid. 1H (DMSO-d6): δ = 9.11 (s, 1H), 8.57 (s, 1H) ppm; 13C (DMSO-d6): δ = 154.34, 151.17, 135.75, 132.35, 129.23, 120.82 ppm; HRMS (ESI/[M + Na]+) calcd for C8H2N14NaO4: 381.0276, found 381.0501.:1) to give the product 8 (0.30 g, 0.92 mmol, 68%, mp. = 306 °C) as a pale yellow solid. 1H (DMSO-d6): δ = 8.63 (s, 1H), 8.44 (s, 1H), 8.05 (s, 1H), 8.04 (s, 1H), 7.60 (br, 2H), 6.90 (br, 2H) ppm; 13C (DMSO-d6): δ = 153.23, 152.61, 148.02, 146.42, 144.71, 143.44, 138.14, 121.98, 108.86, 102.87 ppm; HRMS (ESI/[M − H]−) calcd for C10H7N10O4: 331.0657, found 331.0651.
:15–1:1) to give the product 4 (Rf = 0.7, EtOAc/hexanes 1:1) and product 5 (Rf = 0.4, EtOAc/hexanes 1:1). Product 4 (385 mg, 1.14 mmol, 36%, mp. = 184 °C) as a pale yellow solid. 1H (DMSO-d6): δ = 8.97 (s, 1H), 8.51 (s, 1H) ppm; 13C (DMSO-d6): δ = 154.18, 147.21, 147.15, 135.62, 129.46, 121.63 ppm; HRMS (ESI/[M − H]−) calcd for C8HCl3N5O4: 335.9100, found 334.9232. Product 5 (397 mg, 1.07 mmol, 34%, mp. = 200 °C) as a pale yellow solid. 1H (DMSO-d6): δ = 8.86 (s, 1H), 8.51 (s, 1H), 8.35 (s, 1H), 8.33 (s, 1H) ppm; 13C (DMSO-d6): δ = 154.38, 154.13, 148.53, 147.85, 146.86, 144.56, 133.62, 130.95, 129.44, 122.06 ppm; HRMS (ESI/[M − H]−) calcd for C10H3Cl2N8O4: 368.9660, found 368.9853.:5) to give the product 7 (0.18 g, 0.50 mmol, 71%) as a pale yellow solid. 1H (DMSO-d6): δ = 9.11 (s, 1H), 8.57 (s, 1H) ppm; 13C (DMSO-d6): δ = 154.34, 151.17, 135.75, 132.35, 129.23, 120.82 ppm; HRMS (ESI/[M + Na]+) calcd for C8H2N14NaO4: 381.0276, found 381.0501.:1) to give the product 8 (0.30 g, 0.92 mmol, 68%, mp. = 306 °C) as a pale yellow solid. 1H (DMSO-d6): δ = 8.63 (s, 1H), 8.44 (s, 1H), 8.05 (s, 1H), 8.04 (s, 1H), 7.60 (br, 2H), 6.90 (br, 2H) ppm; 13C (DMSO-d6): δ = 153.23, 152.61, 148.02, 146.42, 144.71, 143.44, 138.14, 121.98, 108.86, 102.87 ppm; HRMS (ESI/[M − H]−) calcd for C10H7N10O4: 331.0657, found 331.0651.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work by the National Natural Science Foundation of China (Grant No. 11402237 and 11302200) is gratefully acknowledged. We acknowledge the suggestions of Ethan Magno in writing this paper.Notes and references
- (a) P. Wardman, Environ. Health Perspect., 1985, 64, 309 CrossRef CAS PubMed; (b) S. Shrivastava and H. Matsuoka, Colloid Polym. Sci., 2016, 294, 879 CrossRef CAS; (c) F. Diness and M. Begtrup, Org. Lett., 2014, 16, 3130 CrossRef CAS PubMed; (d) R. P. Singh, R. D. Verma, D. T. Meshri and J. M. Shreeve, Angew. Chem., Int. Ed., 2006, 45, 3584 CrossRef CAS PubMed; (e) T. B. Brill and K. J. James, Chem. Rev., 1993, 93, 2667 CrossRef CAS.
- H. H. Krause, New Energetic Materials, in Energetic Materials, ed. U. Teipel, Wiley-VCH, Germany, 2005, pp. 1–24 Search PubMed.
- J. Zhang, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 10532 CrossRef CAS PubMed.
- J. Zhang, Q. Zhang, T. T. Vo, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 1697 CrossRef CAS PubMed.
- P. Yin, D. A. Parrish and J. M. Shreeve, J. Am. Chem. Soc., 2015, 137, 4778 CrossRef CAS PubMed.
- I. V. Omelchenko, O. V. Shishkin, L. Gorb, F. C. Hill and J. Leszczynski, Struct. Chem., 2012, 23, 1585 CrossRef CAS.
- W. S. Chen, C. N. Juan and K. M. Wei, J. Hazard. Mater., 2007, 147, 97 CrossRef CAS PubMed.
- N. Kommu, M. Balaraju, V. D. Ghule and A. K. Sahoo, J. Mater. Chem. A, 2017, 5, 7366 CAS.
- (a) A. O. Yigiter, M. K. Atakol, M. L. Aksu and O. Atakol, J. Therm. Anal. Calorim., 2007, 127, 2199 CrossRef; (b) Q. Ma, L. Y. Liao, B. B. Cheng, G. J. Fan, J. L. Huang and J. Wang, Polycyclic Aromat. Compd., 2016, 36, 639 CrossRef CAS; (c) Q. Ma, G. Fan, L. Liao and J.-L. Huang, Polycyclic Aromat. Compd., 2017, 37, 327 CrossRef CAS.
- Z. Zeng, Y. Guo, B. Twamley and J. M. Shreeve, Chem. Commun., 2009, 40, 6014 RSC.
- M. K. William, G. W. Lawrence, E. M. Sitzmann and H. G. Adolph, J. Chem. Soc., Perkin Trans. 1, 1981, 1, 1815 Search PubMed.
- P. H. Gore, S. D. Hammond and D. F. C. Morris, Tetrahedron Lett., 1970, 32, 2747 CrossRef.
- L. A. Kaplan, J. Am. Chem. Soc., 1964, 86, 740 CrossRef CAS.
- H. Gao and J. M. Shreeve, Chem. Rev., 2011, 111, 7377 CrossRef CAS PubMed.
- B. R. Kaafarani, B. Wex, A. G. Oliver, J. A. Krause Bauer and D. C. Neckers, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, o227 CAS.
- J. Y. Shin, B. O. Patrick and D. Dolphin, CrystEngComm, 2008, 10, 960 RSC.
- M. C. Schulze, B. L. Scott and D. E. Chavez, J. Mater. Chem. A, 2015, 3, 17963 CAS.
- M. Sućeska, EXPLO5, version 6.02, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, CCDC 1552452–1552456, Differential Scanning Calorimeter (DSC) figures for 4–9, Computational details. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra13346d |
| This journal is © The Royal Society of Chemistry 2018 |