
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and insecticidal activities of 5-deoxyavermectin B2a oxime ester derivatives†
Guoshao Sun‡
 ,
Jingjing Zhang‡,
Shuhui Jin and
Jianjun Zhang*
,
Jingjing Zhang‡,
Shuhui Jin and
Jianjun Zhang*
Department of Applied Chemistry, College of Science, China Agricultural University, Beijing, 100193, P. R. China. E-mail: zhangjianjun@cau.edu.cn
First published on 22nd January 2018
Abstract
Three series of avermectin B2a oxime ester derivatives were synthesized using avermectin B2a as starting material. All of the compounds were characterized by 1H NMR, 13C NMR, and HRMS. Bioassay results indicated that some of the derivatives (8b, 8c, 8d, 8f, 11k, 11l, 14c, 14j) showed potent insecticidal activities against Myzus persicae, Caenorhabditis elegans, or Tetranychus cinnabarinus. As shown by initial insecticidal activity data, compound 8d showed excellent activities (>90%) against M. persicae and C. elegans, which were more potent than that of avermectin B2a. Compound 8d might be a lead compound for designing new avermectin B2a derivatives.
Introduction
Although chemical pesticides remain the most effective method to control pests in agriculture, the increasing and long-term application of chemical pesticides has resulted in pest resistance and negative impacts on human health and environmental safety.1–3 Considering that the use of natural pesticides may decelerate resistance development and reduce environmental pollution, greater attention has been placed on the development of nature-product-based agrochemicals.4–8 Avermectins, one of the most important classes of natural insecticides, anthelmintics, and acaricides, are a family of 16-membered ring macrocyclic lactones isolated from the culture broth of Streptomyces avermitilis.9–11 Compared with traditional pesticides, avermectins can interact stereoselectively and with a high affinity with the glutamate-gated chloride channel, leading to a chloride ion flux into neurons, causing paralysis and death.12–14 Due to their remarkable biological activity and low mammalian toxicity, avermectins have long been widely commercialized for agricultural use in China.During the industrial production of avermectin, two end-products are obtained – avermectin B1a (Fig. 1) and avermectin ointment.15 Avermectin ointment, composed of approximately 30% avermectin B2a (Fig. 1), is the mother liquor retrieved during the purification of avermectin B1a.16 In 2012, China banned the use of avermectin ointment in the production of avermectin emulsifiable concentrate, which was the primary use of avermectin ointment.16 However, given the high toxicity of avermectin ointment, its disposal requires special treatment and high costs, resulting in a waste of resources. Thus, we developed a purification method to retrieve avermectin B2a from avermectin ointment, which allowed for further molecule structure optimization.
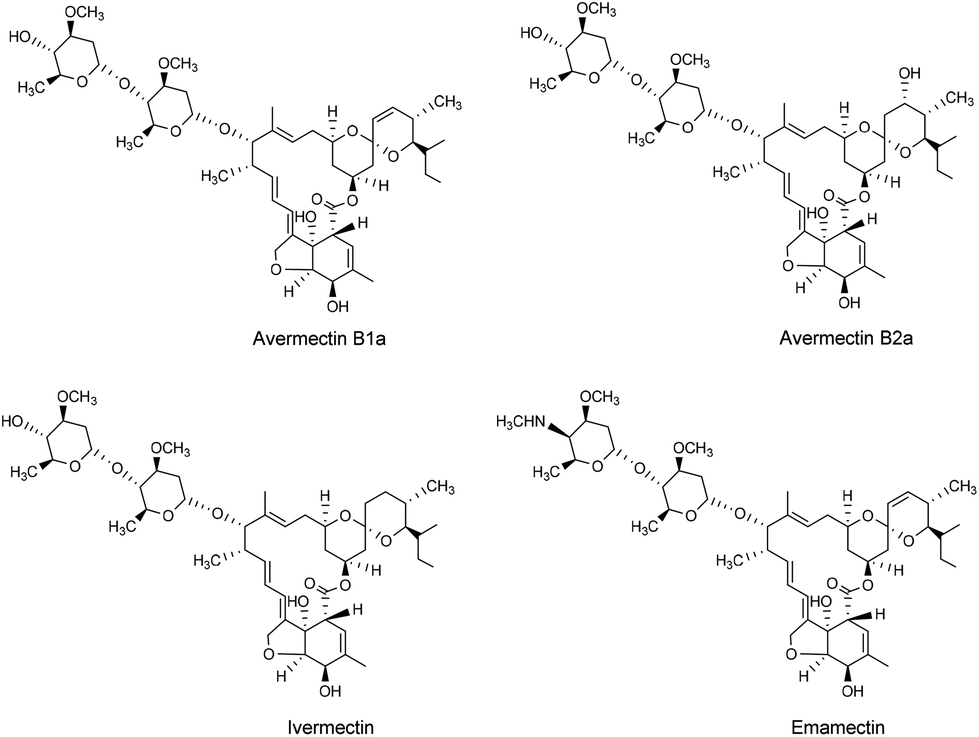 | ||
| Fig. 1 The chemical structures of avermectin B1a, B2a, ivermectin, and emamectin. | ||
Various efforts have been made to obtain compounds with higher potency and broader activity spectra by modifying the avermectin B1a molecule.13,17–19 For example, ivermectin (22,23-dihydro-avermectin B1a, Fig. 1), has the same excellent potency and spectrum against nematode parasites as avermectin B1a, albeit with a greater safety profile.20 Indeed, ivermectin is widely used as an anthelmintic agent for human and animal health. Furthermore, emamectin (Fig. 1), which has a 1500-fold increased insecticidal activity compared to avermectin B1a, has achieved commercial success.13 However, few studies and patents have been concerned with the structural modification of avermectin B2a (Fig. 1).21,22 Avermectin B2a has a slightly lower biological activity than avermectin B1a, but with greater safety,23 making it a favorable candidate for further optimization and development.
Various avermectin and milbemycin (Fig. 2) oximes have been reported to have excellent biological activity.18,24,25 For instance, selamectin (avermectin monosaccharide analogue, Fig. 2), and milbemycin oxime (Fig. 2) are well-known broad-spectrum anthelmintics with outstanding efficacy and safety.26,27 Further, 5-keto-5-oxime ester derivatives of milbemycin have shown a higher efficacy in microfilariae control than milbemycin itself.25
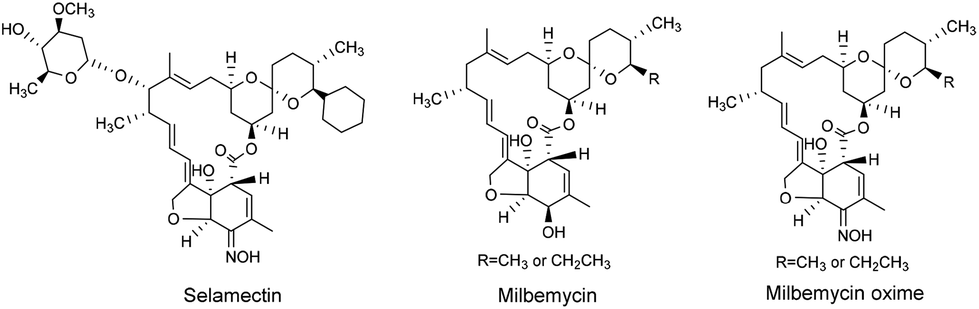 | ||
| Fig. 2 The chemical structures of selamectin, milbemycin, and milbemycin oxime. | ||
Based on the previous considerations, we reported herein on the design and synthesis of three series of avermectin B2a oxime ester derivatives. The insecticidal activities of the target compounds against M. persicae, C. elegans, and T. cinnabarinus were evaluated. Additionally, the impact of changes involving carboxylic group substitutions and saccharide functionality were discussed, potentially providing some useful structural insights for further refinement.
Experimental
Chemicals and instruments
All reagents and solvents were of analytical grade and used without further purification. The melting points were measured on a YRT-3 apparatus and were uncorrected. 1H-NMR and 13C-NMR spectra were recorded on a Bruker Avance 300 spectrometer (300 MHz, 1H; 75 MHz, 13C) in CDCl3 or DMSO-d6 solution with tetramethylsilane as the internal standard. Chemical shifts were reported in ppm (δ). High-resolution mass spectra (ESI-HRMS) were performed on a Bruker Daltonics Bio-TOF-Q III mass spectrometer.Synthesis of 5-o-alloc avermectin B2a (compound 2; Scheme 1)
A solution of allyl chloroformate (AllocCl, 5.3 g, 44.0 mmol) in dried dichloromethane (CH2Cl2, 50 mL) was added dropwise over 30 minutes to a solution of avermectin B2a (30.0 g, 33.7 mmol) and N,N,N′,N′-tetramethylethylene diamine (TEMED, 7.8 g, 67.1 mmol) in CH2Cl2 (100 mL) at −25 °C. The mixture was stirred for 1 h at −25 °C. The reaction mixture was poured into water and extracted with CH2Cl2 (3 × 100 mL). The organic layer was washed with 5% dilute hydrochloric acid (100 mL), 5% aqueous sodium bicarbonate (100 mL), and saturated sodium chloride solution (100 mL), dried over anhydrous sodium sulfate, filtered, and evaporated. The resulting residue was purified by column chromatography on silica gel using a mixture of petroleum ether and ethyl acetate (v/v = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent to afford compound 2 as a pale yellow foamy solid (28.2 g, 86%). Mp 122–125 °C. 1H NMR (300 MHz, CDCl3) δ 5.68–6.06 (m, 4H, H9, H10, H11,
:1) as the eluent to afford compound 2 as a pale yellow foamy solid (28.2 g, 86%). Mp 122–125 °C. 1H NMR (300 MHz, CDCl3) δ 5.68–6.06 (m, 4H, H9, H10, H11, 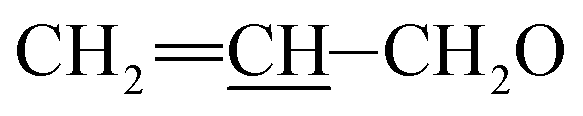 ), 5.53–5.65 (m, 1H, H3), 5.24–5.46 (m, 5H, H5, H1′′, H19,
), 5.53–5.65 (m, 1H, H3), 5.24–5.46 (m, 5H, H5, H1′′, H19,  ), 4.91–5.06 (m, 1H, H15), 4.57–4.86 (m, 5H, H1′, H8a × 2,
), 4.91–5.06 (m, 1H, H15), 4.57–4.86 (m, 5H, H1′, H8a × 2,  ), 4.14 (d, J = 6.1 Hz, 1H, H13), 3.71–4.03 (m, 6H, 7-OH, H23, 23-OH, H5′′, H5′, H6), 3.41–3.71 (m, 10H, H25, H17, H3′′, H3′, 3′′-OCH3, 3′-OCH3), 3.36–3.42 (m, 1H, H2), 3.26 (t, J = 9.0 Hz, 1H, H4′), 3.18 (t, 1H, J = 9.2 Hz, H4′′), 2.48–2.79 (m, 2H, H12, H24), 2.18–2.43 (m, 4H, H16 × 2, H2′, H2′′),1.76–2.10 (m, 5H, H18, Me4a, H20), 1.45–1.69 (m, 11H, Me14a, H20, H26, H27 × 2, H22 × 2, H2′, H2′′), 1.24–1.35 (m, 6H, Me6′, Me6′′), 1.19 (d, J = 6.9 Hz, 3H, Me12a), 0.86–1.03 (m, 10H, H28, Me24a, Me26a, H18); 13C NMR (75 MHz, CDCl3) δ 173.19, 154.79, 139.31, 137.98, 135.66, 133.09, 131.46, 124.72, 121.47, 120.40, 118.66, 117.54, 99.61, 98.47, 94.81, 81.64, 80.88, 80.40, 79.30, 78.18, 77.51, 76.06, 73.51, 70.81, 69.83, 68.69, 68.38, 68.29, 68.12, 67.74, 67.24, 56.42, 56.36, 45.71, 41.16, 40.74, 39.71, 36.37, 35.69, 35.11, 34.46, 34.21, 34.09, 27.24, 20.15, 19.58, 18.35, 17.63, 15.09, 13.72, 12.37, 11.76. HRMS (ESI) calcd for C52H79O17 (M + H)+ 975.5317, found 975.5328.
), 4.14 (d, J = 6.1 Hz, 1H, H13), 3.71–4.03 (m, 6H, 7-OH, H23, 23-OH, H5′′, H5′, H6), 3.41–3.71 (m, 10H, H25, H17, H3′′, H3′, 3′′-OCH3, 3′-OCH3), 3.36–3.42 (m, 1H, H2), 3.26 (t, J = 9.0 Hz, 1H, H4′), 3.18 (t, 1H, J = 9.2 Hz, H4′′), 2.48–2.79 (m, 2H, H12, H24), 2.18–2.43 (m, 4H, H16 × 2, H2′, H2′′),1.76–2.10 (m, 5H, H18, Me4a, H20), 1.45–1.69 (m, 11H, Me14a, H20, H26, H27 × 2, H22 × 2, H2′, H2′′), 1.24–1.35 (m, 6H, Me6′, Me6′′), 1.19 (d, J = 6.9 Hz, 3H, Me12a), 0.86–1.03 (m, 10H, H28, Me24a, Me26a, H18); 13C NMR (75 MHz, CDCl3) δ 173.19, 154.79, 139.31, 137.98, 135.66, 133.09, 131.46, 124.72, 121.47, 120.40, 118.66, 117.54, 99.61, 98.47, 94.81, 81.64, 80.88, 80.40, 79.30, 78.18, 77.51, 76.06, 73.51, 70.81, 69.83, 68.69, 68.38, 68.29, 68.12, 67.74, 67.24, 56.42, 56.36, 45.71, 41.16, 40.74, 39.71, 36.37, 35.69, 35.11, 34.46, 34.21, 34.09, 27.24, 20.15, 19.58, 18.35, 17.63, 15.09, 13.72, 12.37, 11.76. HRMS (ESI) calcd for C52H79O17 (M + H)+ 975.5317, found 975.5328.
Synthesis of 5-o-alloc-4′′,23-bis-o-(tert-butyldimethysilyl) avermectin B2a (compound 3; Scheme 1)
Triethylamine (TEA, 19.6 g, 193.7 mmol) and tert-butyldimethylsilyl chloride (TBDMSCl, 25.0 g, 165.9 mmol) were added to a solution of 5-o-alloc avermectin B2a (27.0 g, 27.7 mmol) in 150 mL of dry N,N-dimethylformamide (DMF). The reaction mixture stirred at −5 °C for 48 h. The reaction mixture was diluted with 300 mL of ether, water (150 mL) was added and the layers were separated. The aqueous layers was extracted twice with ether (2 × 200 mL) and the combined organic layers was dried over magnesium sulfate, filtered, and evaporated to a yellow foam. The foam was purified by column chromatography on silica gel using a mixture of petroleum ether and ethyl acetate (v/v = 10:1) as the eluent to afford compound 3 as a yellow foamy solid (26.9 g, 81%). Mp 128–131 °C. 1H NMR (300 MHz, CDCl3) δ 5.68–6.05 (m, 4H, H9, H10, H11,  ), 5.52–5.63 (m, 1H, H3), 5.22–5.43 (m, 5H, H5, H1′′, H19,
), 5.52–5.63 (m, 1H, H3), 5.22–5.43 (m, 5H, H5, H1′′, H19, 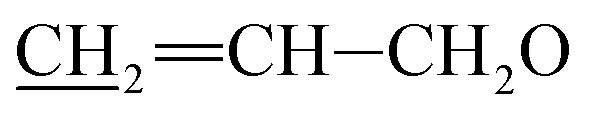 ), 4.92–5.06 (m, 1H, H15), 4.59–4.86 (m, 5H, H1′, H8a × 2,
), 4.92–5.06 (m, 1H, H15), 4.59–4.86 (m, 5H, H1′, H8a × 2,  ), 4.09–4.16 (m, 2H, H13, 7-OH), 3.94 (s, 1H, 23-OH), 3.71–4.03 (m, 7H, H23, H5′′, H5′, H6, H17, H3′′, H3′), 3.30–3.48 (m, 8H, H25, 3′′-OCH3, 3′-OCH3, H2), 3.22 (t, J = 8.9 Hz, 1H, H4′), 3.14 (t, 1H, J = 8.8 Hz, H4′′), 2.45–2.60 (m, 1H, H12), 1.77–2.39 (m, 10H, H16 × 2, H2′, H2′′, H24, H18, Me4a, H20), 1.45–1.69 (m, 11H, Me14a, H20, H26, H27 × 2, H22 × 2, H2′, H2′′), 1.13–1.32 (m, 9H, Me6′, Me6′′, Me12a), 0.84–1.01 (m, 28H, H28, Me24a, Me26a, H18,
), 4.09–4.16 (m, 2H, H13, 7-OH), 3.94 (s, 1H, 23-OH), 3.71–4.03 (m, 7H, H23, H5′′, H5′, H6, H17, H3′′, H3′), 3.30–3.48 (m, 8H, H25, 3′′-OCH3, 3′-OCH3, H2), 3.22 (t, J = 8.9 Hz, 1H, H4′), 3.14 (t, 1H, J = 8.8 Hz, H4′′), 2.45–2.60 (m, 1H, H12), 1.77–2.39 (m, 10H, H16 × 2, H2′, H2′′, H24, H18, Me4a, H20), 1.45–1.69 (m, 11H, Me14a, H20, H26, H27 × 2, H22 × 2, H2′, H2′′), 1.13–1.32 (m, 9H, Me6′, Me6′′, Me12a), 0.84–1.01 (m, 28H, H28, Me24a, Me26a, H18, 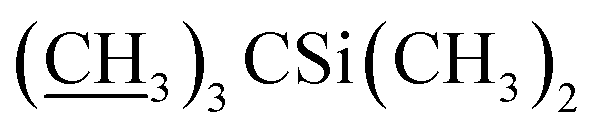 ), 0.04–0.12 (m, 12H,
), 0.04–0.12 (m, 12H, 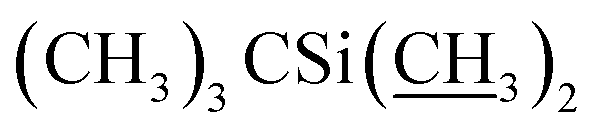 ); 13C NMR (75 MHz, CDCl3) δ 173.47, 154.81, 139.12, 138.17, 134.55, 133.00, 131.50, 124.62, 121.63, 120.47, 118.85, 118.65, 98.57, 97.62, 94.88, 81.87, 79.29, 78.55, 77.20, 77.14, 73.58, 69.90, 69.40, 69.01, 68.88, 68.70, 68.45, 67.34, 67.27, 56.57, 56.26, 45.73, 42.48, 41.69, 39.77, 36.55, 36.36, 35.33, 34.64, 34.15, 29.65, 27.34, 26.01, 25.86, 20.18, 19.61, 18.36, 18.30, 18.27, 18.13, 15.06, 13.96, 12.25, 11.95, −3.99, −4.40, −4.77, −4.99. HRMS (ESI) calcd for C64H107O17Si2 (M + H)+ 1203.7047, found 1203.7083.
); 13C NMR (75 MHz, CDCl3) δ 173.47, 154.81, 139.12, 138.17, 134.55, 133.00, 131.50, 124.62, 121.63, 120.47, 118.85, 118.65, 98.57, 97.62, 94.88, 81.87, 79.29, 78.55, 77.20, 77.14, 73.58, 69.90, 69.40, 69.01, 68.88, 68.70, 68.45, 67.34, 67.27, 56.57, 56.26, 45.73, 42.48, 41.69, 39.77, 36.55, 36.36, 35.33, 34.64, 34.15, 29.65, 27.34, 26.01, 25.86, 20.18, 19.61, 18.36, 18.30, 18.27, 18.13, 15.06, 13.96, 12.25, 11.95, −3.99, −4.40, −4.77, −4.99. HRMS (ESI) calcd for C64H107O17Si2 (M + H)+ 1203.7047, found 1203.7083.
Synthesis of 4′′,23-bis-o-(tert-butyldimethysilyl) avermectin B2a (compound 4; Scheme 1)
Ammonium acetate (33.3 g, 432.0 mmol) was added to a solution of 5-o-alloc-4′′,23-bis-o-(tert-butyldimethysilyl) avermectin B2a (26.0 g, 21.6 mmol) in methanol (MeOH, 40 mL) and tetrahydrofuran (THF, 40 mL) at −10 °C. With vigorous stirring, sodium borohydride (NaBH4, 0.41 g, 10.8 mmol), tetrakis(triphenylphosphine) palladium (Pd[PPh3]4, 1.0 g, 0.9 mmol), and another portion of NaBH4 (0.41 g, 10.8 mmol) were added sequentially and quickly. The reaction was monitored with TLC until completion. The reaction mixture was concentrated under vacuum. The residue was diluted with CH2Cl2 (100 mL) and washed with brine (100 mL), then the organic phase was dried over anhydrous sodium sulfate, filtered, and evaporated to a yellow foam. The foam was purified by column chromatography on silica gel using a mixture of petroleum ether and ethyl acetate (v/v = 6:1) as the eluent to afford compound 4 as a yellow foamy solid (21.3 g, 88%). Mp 145–148 °C. 1H NMR (300 MHz, CDCl3) δ 5.82–5.93 (m, 1H, H9), 5.68–5.80 (m, 2H, H10, H11), 5.26–5.45 (m, 3H, H3, H1′′, H19), 4.93–5.09 (m, 1H, H15), 4.79 (d, J = 2.8 Hz, 1H, H1′), 4.67 (s, 2H, H8a × 2), 4.29 (t, J = 7.1 Hz, 1H, H5), 4.14 (s, 1H, 7-OH), 3.54–4.01 (m, 9H, H13, 23-OH, H23, H5′′, H5′, H6, H17, H3′′, H3′), 3.27–3.48 (m, 8H, H25, 3′′-OCH3, 3′-OCH3, H2), 3.23 (t, J = 9.0 Hz, 1H, H4′), 3.15 (t, 1H, J = 8.8 Hz, H4′′), 2.46–2.68 (m, 2H, H12, H24), 2.14–2.39 (m, 4H, H16 × 2, H2′, H2′′), 1.77–1.97 (m, 5H, H18, Me4a, H20), 1.44–1.67 (m, 11H, Me14a, H20, H26, H27 × 2, H22 × 2, H2′, H2′′), 1.13–1.35 (m, 9H, Me6′, Me6′′, Me12a), 0.84–1.03 (m, 28H, H28, Me24a, Me26a, H18, 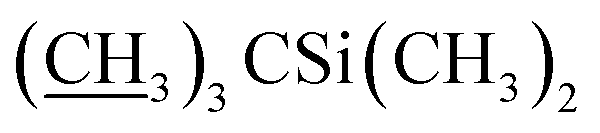 ), 0.02–0.17 (m, 12H,
), 0.02–0.17 (m, 12H, 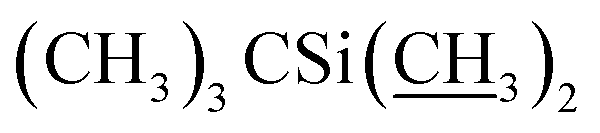 ); 13C NMR (75 MHz, CDCl3) δ 173.60, 139.47, 138.06, 137.78, 134.47, 124.58, 120.38, 118.88, 118.04, 98.52, 97.57, 94.87, 81.88, 80.83, 80.32, 79.24, 79.16, 78.50, 77.20, 77.11, 69.85, 69.38, 68.96, 68.64, 68.30, 67.65, 67.31, 67.23, 56.48, 56.14, 45.66, 42.45, 41.69, 39.77, 36.55, 36.33, 35.29, 34.57, 34.11, 29.58, 27.28, 25.97, 25.82, 25.63, 20.11, 19.83, 18.29, 18.23, 18.07, 14.98, 13.90, 12.19, 11.91, −4.04, −4.45, −4.81, −5.03. HRMS (ESI) calcd for C60H103O15Si2 (M + H)+ 1119.6836, found 1119.6873.
); 13C NMR (75 MHz, CDCl3) δ 173.60, 139.47, 138.06, 137.78, 134.47, 124.58, 120.38, 118.88, 118.04, 98.52, 97.57, 94.87, 81.88, 80.83, 80.32, 79.24, 79.16, 78.50, 77.20, 77.11, 69.85, 69.38, 68.96, 68.64, 68.30, 67.65, 67.31, 67.23, 56.48, 56.14, 45.66, 42.45, 41.69, 39.77, 36.55, 36.33, 35.29, 34.57, 34.11, 29.58, 27.28, 25.97, 25.82, 25.63, 20.11, 19.83, 18.29, 18.23, 18.07, 14.98, 13.90, 12.19, 11.91, −4.04, −4.45, −4.81, −5.03. HRMS (ESI) calcd for C60H103O15Si2 (M + H)+ 1119.6836, found 1119.6873.
Synthesis of 5-oxo-4′′,23-bis-o-(tert-butyldimethysilyl) avermectin B2a (compound 5; Scheme 1)
A solution of phenyl dichlorophosphate (4.9 g, 23.2 mmol) in CH2Cl2 (30 mL) was added dropwise over 20 minutes to a solution of 4′′,23-bis-o-(tert-butyldimethysilyl) avermectin B2a (20.0 g, 17.9 mmol), dimethyl sulfoxide (DMSO, 5.6 g, 71.7 mmol) and TEA (9.06 g, 89.5 mmol) in CH2Cl2 (100 mL) at −15 °C. The mixture was stirred for 1 h at −15 °C and then poured into 1% H3PO4. The aqueous phase was extracted with CH2Cl2 (2 × 100 mL), and the combined organic phase was washed with saturated aqueous solution of sodium bicarbonate (100 mL) and saturated sodium chloride solution (100 mL). Then the organic phase was dried over anhydrous sodium sulfate, filtered, and evaporated to a yellow foam. The foam was purified by column chromatography on silica gel using a mixture of petroleum ether and ethyl acetate (v/v = 10:1) as the eluent to afford compound 5 as a yellow foamy solid (16.2 g, 81%). Mp 134–137 °C. 1H NMR (300 MHz, CDCl3) δ 6.56–6.64 (m, 1H, H3), 5.89–6.00 (m, 1H, H9), 5.66–5.87 (m, 2H, H10, H11), 5.30–5.48 (m, 2H, H1′′, H19), 4.92–5.08 (m, 1H, H15), 4.81 (d, J = 3.0 Hz, 1H, H1′), 4.75 (s, 2H, H8a × 2), 4.29 (t, J = 7.1 Hz, 1H, H5), 4.06 (s, 1H, 7-OH), 3.97 (s, 1H, 23-OH), 3.54–3.91 (m, 9H, H13, H25, H23, H5′′, H5′, H6, H17, H3′′, H3′), 3.29–3.50 (m, 7H, 3′′-OCH3, 3′-OCH3, H2), 3.25 (t, J = 8.9 Hz, 1H, H4′), 3.16 (t, 1H, J = 8.8 Hz, H4′′), 2.49–2.66 (m, 1H, H12), 2.19–2.42 (m, 5H, H24, H16 × 2, H2′, H2′′), 1.81–2.02 (m, 5H, H18, Me4a, H20), 1.42–1.65 (m, 11H, Me14a, H20, H26, H27 × 2, H22 × 2, H2′, H2′′), 1.19–1.33 (m, 9H, Me6′, Me6′′, Me12a), 0.87–1.03 (m, 28H, H28, Me24a, Me26a, H18, 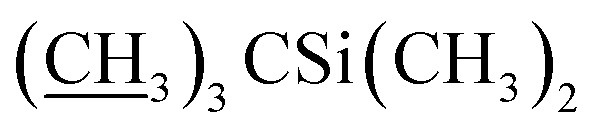 ), 0.02–0.14 (m, 12H,
), 0.02–0.14 (m, 12H, 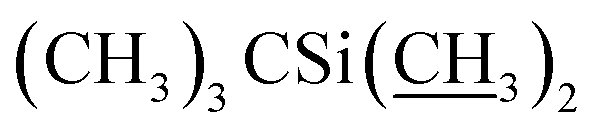 ); 13C NMR (75 MHz, CDCl3) δ 192.03, 172.18, 139.17, 138.01, 137.69, 136.69, 134.60, 124.53, 121.86, 118.83, 98.53, 97.60, 94.87, 81.91, 81.75, 80.79, 79.26, 78.53, 77.13, 69.91, 69.77, 69.39, 69.00, 67.35, 67.28, 56.51, 56.19, 46.60, 42.46, 41.71, 39.90, 36.53, 36.36, 35.30, 34.60, 34.53, 34.10, 29.62, 27.34, 25.99, 25.85, 20.02, 18.33, 18.27, 18.10, 15.38, 15.03, 13.94, 12.22, 11.94, 0.94, −4.01, −4.42, −4.78, −5.01. HRMS (ESI) calcd for C60H101O15Si2 (M + H)+ 1117.6679, found 1117.6682.
); 13C NMR (75 MHz, CDCl3) δ 192.03, 172.18, 139.17, 138.01, 137.69, 136.69, 134.60, 124.53, 121.86, 118.83, 98.53, 97.60, 94.87, 81.91, 81.75, 80.79, 79.26, 78.53, 77.13, 69.91, 69.77, 69.39, 69.00, 67.35, 67.28, 56.51, 56.19, 46.60, 42.46, 41.71, 39.90, 36.53, 36.36, 35.30, 34.60, 34.53, 34.10, 29.62, 27.34, 25.99, 25.85, 20.02, 18.33, 18.27, 18.10, 15.38, 15.03, 13.94, 12.22, 11.94, 0.94, −4.01, −4.42, −4.78, −5.01. HRMS (ESI) calcd for C60H101O15Si2 (M + H)+ 1117.6679, found 1117.6682.
Synthesis of 5-oxo avermectin B2a (compound 6; Scheme 1)
70% hydrogen fluoride–pyridine complex (50 g, 353.1 mmol) was added to a cold mixture of pyridine (25 mL) and THF (55 mL) at 0 °C. A portion (84 mL) of the resulting reagent solution was added to a cold (0 °C) solution of 5-oxo-4′′,23-bis-o-(tert-butyldimethysilyl) avermectin B2a (15.0 g, 13.4 mol) in THF (100 mL). The reaction mixture stirred at room temperature for 4 days. The reaction mixture was concentrated under vacuum. The residue was diluted with CH2Cl2 and washed with 5% dilute hydrochloric acid (100 mL), 5% aqueous sodium bicarbonate (100 mL), and saturated sodium chloride solution (100 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and evaporated. The resulting residue was purified by column chromatography on silica gel using a mixture of petroleum ether and ethyl acetate (v/v = 4:1) as the eluent to afford compound 6 as a pale yellow foamy solid (7.8 g, 66%). Mp 143–145 °C. 1H NMR (300 MHz, CDCl3) δ 6.59–6.64 (m, 1H, H3), 5.92–6.01 (m, 1H, H9), 5.69–5.87 (m, 2H, H10, H11), 5.35–5.48 (m, 2H, H1′′, H19), 4.91–5.08 (m, 1H, H15), 4.67–4.84 (m, 3H, H8a × 2, H1′), 3.95–4.06 (s, 2H, 7-OH, 23-OH), 3.74–3.89 (m, 4H, H23, H13, H5′′, H5′), 3.41–3.68 (m, 11H, H17, H25, H2, H3′′, H3′, 3′′-OCH3, 3′-OCH3), 3.27 (t, J = 9.0 Hz, 1H, H4′), 3.18 (t, 1H, J = 9.2 Hz, H4′′), 2.51–2.62 (m, 2H, H12, 4′′-OH), 2.21–2.41 (m, 5H, H24, H16 × 2, H2′, H2′′), 1.84–2.05 (m, 5H, H18, Me4a, H20), 1.40–1.65 (m, 11H, Me14a, H20, H26, H27 × 2, H22 × 2, H2′, H2′′), 1.15–1.36 (m, 9H, Me6′, Me6′′, Me12a), 0.84–1.03 (m, 10H, H28, Me24a, Me26a, H18); 13C NMR (75 MHz, CDCl3) δ 191.94, 171.71, 138.88, 138.10, 137.81, 136.55, 135.65, 124.58, 121.78, 117.45, 99.54, 98.37, 94.74, 81.99, 81.47, 80.80, 80.27, 79.21, 78.11, 77.20, 75.93, 70.76, 69.73, 69.63, 68.24, 68.16, 68.07, 67.20, 56.33, 56.28, 46.48, 41.04, 40.69, 39.77, 36.24, 35.60, 35.02, 34.16, 33.98, 27.18, 19.96, 18.28, 17.58, 15.34, 15.01, 13.65, 12.31, 11.70. HRMS (ESI) calcd for C48H73O15 (M + H)+ 889.4949, found 889.4950.
Synthesis of 5-oximino-5-deoxyavermectin B2a (compound 7; Scheme 1)
A solution of NH2OH–HCl (2.8 g, 40.3 mmol) in water (12 mL) was added dropwise to a solution of 5-oxoavermectin B2a (7.1 g, 8.0 mmol) in methanol (25 mL) and 1,4-dioxane (25 mL). The mixture was stirred at room temperature for 8 h and then evaporated under reduced pressure. The residue was dissolved in dichloromethane (100 mL) and washed with water (50 mL). The organic phase was then dried over anhydrous sodium sulfate, filtered, and evaporated to a yellow foam, which was purified by silica gel column chromatography with petroleum ether–ethyl acetate (v/v = 1:1) to afford compound 7 as a white solid (6.3 g, 87%). Mp: 164–166 °C. 1H NMR (300 MHz, CDCl3) δ 5.70–6.03 (m, 4H, H3, H9, H10, H11), 5.32–5.49 (m, 2H, H1′′, H19), 4.90–5.10 (m, 1H, H15), 4.68–4.85 (m, 4H, H1′, H8a × 2, H6), 3.73–4.02 (m, 6H, 7-OH, 23-OH, H23, H13, H5′′, H5′), 3.42–3.71 (m, 11H, H2, H25, H17, H3′′, H3′, 3′′-OCH3, 3′-OCH3), 3.28 (t, J = 8.9 Hz, 1H, H4′), 3.19 (t, 1H, J = 9.1 Hz, H4′′), 2.48–2.63 (m, 1H, H12), 2.23–2.42 (m, 5H, H24, H16 × 2, H2′, H2′′), 1.89–2.08 (m, 5H, H18, Me4a, H20), 1.43–1.75 (m, 11H, Me14a, H20, H26, H27 × 2, H22 × 2, H2′, H2′′), 1.26–1.34 (m, 6H, Me6′, Me6′′), 1.21 (d, J = 6.9 Hz, 3H, Me12a), 0.84–1.04 (m, 10H, H28, Me24a, Me26a, H18); 13C NMR (75 MHz, DMSO) δ 170.20, 150.34, 140.08, 136.64, 135.15, 129.57, 126.29, 125.34, 121.38, 117.92, 99.00, 97.68, 94.63, 81.48, 79.74, 79.08, 78.89, 77.65, 75.59, 72.65, 69.46, 68.52, 68.38, 68.06, 67.43, 66.86, 56.68, 55.90, 45.61, 41.21, 40.95, 35.62, 35.25, 34.77, 34.14, 33.62, 26.72, 19.96, 18.24, 17.80, 17.51, 14.62, 13.65, 12.16, 12.09; HRMS (ESI) calcd for C48H74NO15 (M + H)+ 904.5058, found 904.5063.
Synthesis of 5-oxoavermectin B2a monosaccharide (compound 9; Scheme 2)
5-Oxoavermectin B2a (4.0 g, 4.5 mmol) was added to a solution of concentrated sulfuric acid (1.5 mL) in 2-propanol (23.5 mL) and stirred at −5 °C for 36 h. The reaction mixture was diluted with 100 mL of ethyl ether, and the solution was washed with 5% aqueous sodium bicarbonate and water, dried, and concentrated in vacuo to a yellow foam, which was purified by silica gel column chromatography with petroleum ether–ethyl acetate (v/v = 4:1) to afford compound 9 as a yellow foam solid (2.7 g, 81%). Mp 152–155 °C. 1H NMR (300 MHz, CDCl3) δ 6.34–6.78 (m, 1H, H3), 5.64–5.99 (m, 3H, H9, H10, H11), 5.26–5.49 (m, 1H, H19), 4.96 (t, J = 7.6 Hz, 1H, H15), 4.64–4.85 (m, 4H, H1′, H8a × 2, H6), 3.70–4.03 (m, 5H, 7-OH, H23, H13, H5′, H17), 3.41–3.68 (m, 6H, H25, H3′, H2, 3′-OCH3), 3.19 (t, 1H, J = 9.1 Hz, H4′), 2.46–2.67 (m, 2H, H12, H24), 2.20–2.41 (m, 3H, H16 × 2, H2′), 1.84–2.09 (m, 5H, Me4a, H18, H20), 1.42–1.73 (m, 10H, Me14a, H20, H2′, H26, H27 × 2, H22 × 2), 1.29 (d, J = 6.2 Hz, 3H, Me6′), 1.17 (d, J = 6.9 Hz, 3H, Me12a), 0.83–1.04 (m, 10H, H28, Me24a, Me26a, H18); 13C NMR (75 MHz, CDCl3) δ 192.02, 171.65, 138.90, 138.22, 138.09, 137.78, 131.46, 124.58, 121.85, 118.76, 117.45, 99.57, 94.93, 82.02, 81.41, 80.84, 78.21, 75.93, 70.76, 69.77, 69.63, 68.35, 68.26, 68.10, 56.44, 46.49, 41.03, 40.71, 39.78, 36.25, 35.60, 35.03, 33.98, 27.19, 19.97, 17.61, 15.35, 15.04, 13.67, 12.32, 11.71; HRMS (ESI) calcd for C41H61O12 (M + H)+ 745.4163, found 745.4168.
Synthesis of 5-oximino-5-deoxyavermectin B2a monosaccharide (compound 10; Scheme 2)
A solution of NH2OH–HCl (1.0 g, 14.4 mmol) in water (5 mL) was added dropwise to a solution of 5-oxoavermectin B2a monosaccharide (2.2 g, 3.0 mmol) in methanol (10 mL) and 1,4-dioxane (10 mL). The mixture was stirred at room temperature for 8 h and then evaporated under reduced pressure. The residue was dissolved in CH2Cl2 (50 mL) and washed with water (25 mL). The organic phase was then dried over anhydrous sodium sulfate, filtered, and evaporated to a yellow foam, which was purified by silica gel column chromatography with petroleum ether–ethyl acetate (v/v = 1:1) to afford compound 10 as a white solid (1.9 g, 83%). Mp 214–217 °C. 1H NMR (300 MHz, CDCl3) δ 5.66–6.04 (m, 4H, H3, H9, H10, H11), 5.25–5.45 (m, 1H, H19), 5.00 (t, J = 6.9 Hz, 1H, H15), 4.58–4.86 (m, 4H, H8a × 2, H6, H1′), 3.68–4.06 (m, 5H, 7-OH, 23-OH, H23, H13, H5′), 3.37–3.65 (m, 7H, H17, H25, H3′, H2, 3′-OCH3), 3.17 (t, 1H, J = 9.9 Hz, H4′), 2.15–2.63 (m, 4H, H12, H24, H16 × 2), 1.37–2.10 (m, 16H, H2′, Me4a, H18, H20, Me14a, H20, H2′, H26, H27 × 2, H22 × 2), 1.27 (d, J = 6.2 Hz, 3H, Me6′), 1.15 (d, J = 6.9 Hz, 3H, Me12a), 0.80–1.04 (m, 10H, H28, Me24a, Me26a, H18); 13C NMR (75 MHz, DMSO) δ 170.22, 150.37, 140.03, 136.80, 135.17, 129.61, 126.29, 125.29, 121.45, 117.87, 99.04, 94.66, 81.17, 79.24, 79.10, 77.86, 75.65, 72.70, 69.50, 68.55, 68.44, 68.11, 67.44, 56.64, 45.64, 41.22, 40.98, 35.66, 35.27, 34.80, 34.67, 33.64, 26.75, 20.03, 17.85, 17.53, 14.67, 13.67, 12.17, 12.11. HRMS (ESI) calcd for C41H62NO12 (M + H)+ 760.4272, found 760.4278.
Synthesis of 5-oxoavermectin B2a aglycone (compound 12; Scheme 2)
5-Oxoavermectin B2a (4.0 g, 4.5 mmol) was added to a solution of concentrated sulfuric acid (2.5 mL) in methanol (22.5 mL) and stirred at −5 °C for 24 h. The reaction mixture was diluted with 100 mL of ethyl ether, and the solution was washed with 5% aqueous sodium bicarbonate and water, dried, and concentrated in a vacuum to a yellow foam, which was purified by silica gel column chromatography with petroleum ether–ethyl acetate (v/v = 6:1) to afford compound 12 as a yellow solid foam (2.3 g, 85%). Mp 180–183 °C. 1H NMR (300 MHz, CDCl3) δ 6.54–6.63 (m, 1H, H3), 5.67–5.95 (m, 3H, H9, H10, H11), 5.23–5.38 (m, 2H, H15, H19), 4.65–4.72 (m, 2H, H8a × 2), 4.01 (s, 1H, 7-OH), 3.91 (s, 1H, 23-OH), 3.69–3.86 (m, 3H, H23, H13, H6), 3.44–3.62 (m, 3H, H17, H25, H2), 2.45–2.60 (m, 1H, H12), 2.21–2.40 (m, 2H, H24, H20),1.77–2.01 (m, 6H, H16 × 2, Me4a, H18), 1.38–1.72 (m, 9H, Me14a, H20, H26, H27 × 2, H22 × 2), 1.17 (d, J = 7.0 Hz, 3H, Me12a), 0.80–1.03 (m, 10H, H28, Me24a, Me26a, H18); 13C NMR (75 MHz, CDCl3) δ 192.04, 171.46, 139.15, 138.35, 138.22, 137.77, 136.41, 124.48, 121.90, 116.41, 99.48, 81.91, 80.86, 77.25, 71.30, 69.83, 69.66, 68.31, 68.11, 46.44, 41.04, 40.78, 40.08, 36.06, 35.58, 35.06, 34.09, 27.37, 18.96, 15.34, 14.46, 13.70, 12.41, 11.39; HRMS (ESI) calcd for C34H49O9 (M + H)+ 601.3377, found 601.3381.
Synthesis of 5-oximino-5-deoxyavermectin B2a aglycone (compound 13; Scheme 2)
A solution of NH2OH–HCl (1.1 g, 15.8 mmol) in water (5 mL) was added dropwise to a solution of 5-oxoavermectin B2a aglycone (2.0 g, 3.3 mmol) in methanol (10 mL) and 1,4-dioxane (10 mL). The mixture was stirred at room temperature for 6 h and then evaporated under reduced pressure. The residue was dissolved in CH2Cl2 (50 mL) and washed with water (25 mL). The organic phase was then dried over anhydrous sodium sulfate, filtered, and evaporated to a yellow foam, which was purified by silica gel column chromatography with petroleum ether–ethyl acetate (v/v = 2:1) to afford compound 13 as a white solid (1.7 g, 84%). Mp 210–213 °C. 1H NMR (300 MHz, CDCl3) δ 5.60–5.98 (m, 4H, H3, H9, H10, H11), 5.24–5.43 (m, 2H, H19, H15), 4.63–4.81 (m, 3H, H8a × 2, H6), 4.01 (s, 1H, 7-OH), 3.87 (s, 1H, 23-OH), 3.69–3.83 (m, 2H, H23, H13), 3.50–3.66 (m, 2H, H17, H25), 3.32–3.43 (m, 1H, H2), 2.46–2.61 (m, 1H, H12), 2.23–2.42 (m, 2H, H24, H20),1.87–2.07 (m, 5H, H16 × 2, Me4a), 1.40–1.78 (m, 10H, H18, Me14a, H20, H26, H27 × 2, H22 × 2), 1.18 (d, J = 6.9 Hz, 3H, Me12a), 0.83–1.02 (m, 10H, H28, Me24a, Me26a, H18); 13C NMR (75 MHz, CDCl3) δ 172.70, 151.19, 139.18, 138.21, 137.36, 131.88, 125.30, 124.80, 121.42, 116.47, 99.57, 78.58, 77.49, 72.99, 71.35, 70.00, 68.68, 68.39, 67.65, 46.36, 41.08, 40.18, 36.15, 35.65, 35.12, 34.15, 27.41, 19.04, 17.45, 14.53, 13.74, 12.47, 11.45, 0.94; HRMS (ESI) calcd for C34H50NO9 (M + H)+ 616.3486, found 616.3486.
General synthetic procedure for compounds 8a–n, 11a–n, and 14a–l (Schemes 1 and 2)
Dicyclohexylcarbodiimide (DCC, 0.4 g, 1.9 mmol) and carboxylic acid (R1COOH, R2COOH or R3COOH, 1.9 mmol) were added to a solution of 5-oxime-5-deoxyavermectin B2a in CH2Cl2 (15 mL) and the mixture was stirred at room temperature. After the reaction was completed, as monitored with TLC, the reaction mixture was then filtered and the filtrate was evaporated under reduced pressure to yield a residue. The residue was purified by silica gel column chromatography with petroleum ether–ethyl acetate (v/v = 6:1–1:1) to afford the target compounds. The 1H-NMR, 13C-NMR and HRMS spectral data of the compounds 8a–n, 11a–n, and 14a–l are supplied as ESI.†
Insecticidal assay
All bioassays were performed on representative test organisms reared in the laboratory. Each test sample was prepared in methanol and diluted to the required concentration with distilled water. The insecticidal activities of the target compounds against M. persicae, C. elegans, and T. cinnabarinus were tested according to previously reported procedures.24,28,29 Assessments were made on a dead/live basis, and mortality rates were corrected using Abbott's formula.30Results and discussion
Chemistry
In this research, three series of novel avermectin B2a oxime ester derivatives were synthesized and spectroscopic data was obtained, which agreed well with their expected structures. The synthesis of the target compounds 8a–n was shown in Scheme 1. Initially, the 5-hydroxyl group of avermectin B2a (1) was selectively protected with AllocCl in CH2Cl2 to give compound 2. The subsequent reaction, using TBDMSCl as a protective agent for the hydroxyl groups at the 4- and 23-positions, yielded compound 3. Removal of the AllocCl protecting group of compound 3 with NaBH4 and Pd(PPh3)4 in MeOH–THF (1:1) formed compound 4. Oxidation of compound 4 using a PhOPOCl2, Et3N, and DMSO reagent system in dried CH2Cl2 afforded compound 5. The t-butyldimethylsilyl protecting group was removed using a HF/Py complex to form the important intermediate compound 6. Compound 6 was reacted with NH2OH–HCl to yield intermediate 7, which on reaction with various carboxylic acids, gave the target compounds 8a–n.
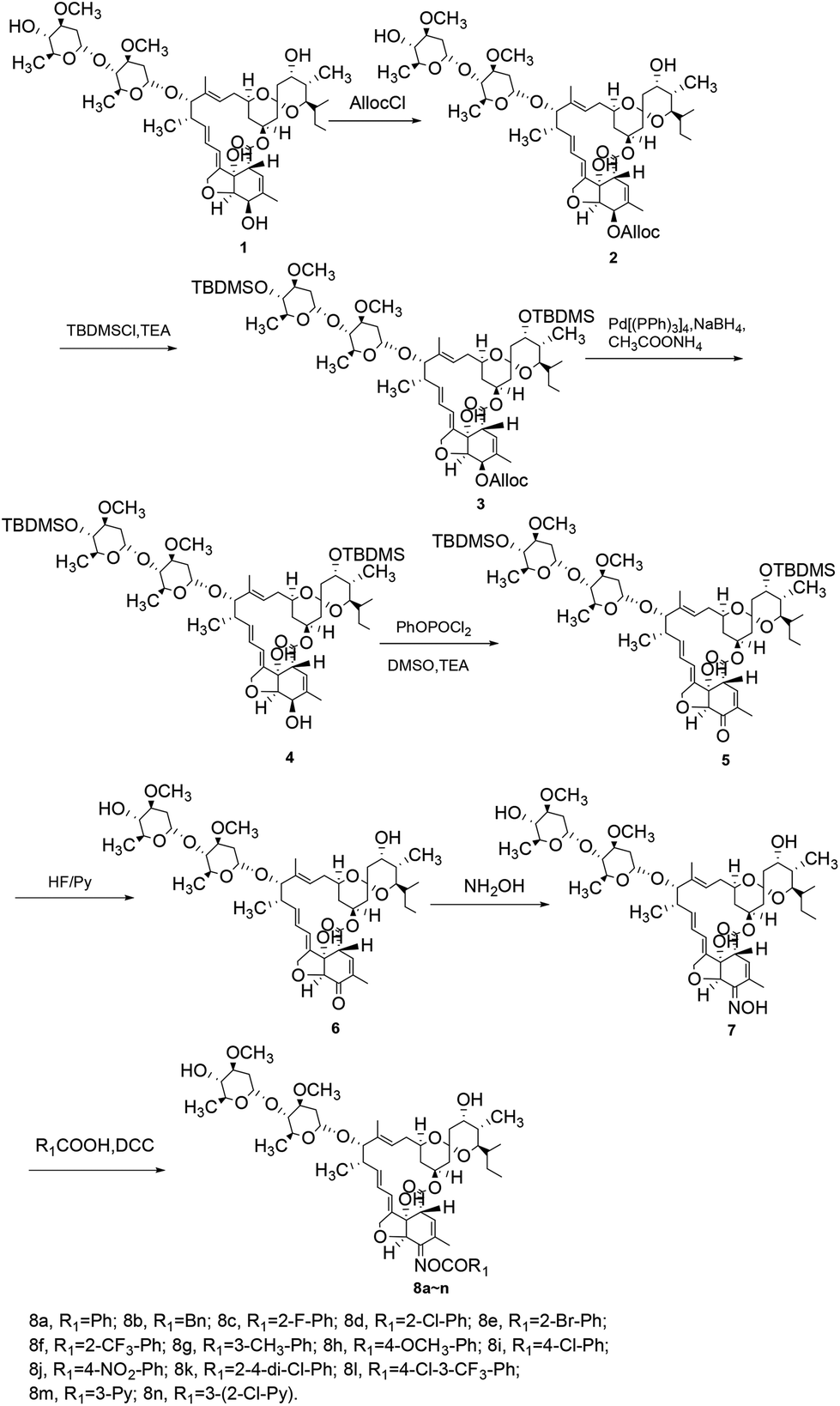 | ||
| Scheme 1 Synthesis of target compounds 8a–n. | ||
The target compounds 11a–n and 14a–l were synthesized from compound 6 as shown in Scheme 2. The intermediate 6 was de-sugared with 3% concentrated H2SO4 in isopropanol as a solvent to afford compound 9, and compound 12 was prepared by a reaction between intermediate 6 and 5% concentrated H2SO4 in methanol. Similarly, the target compounds 11a–n and 14a–l were prepared through oximate and esterification, respectively, via a similar procedure as described above for compounds 8a–n in good yield.
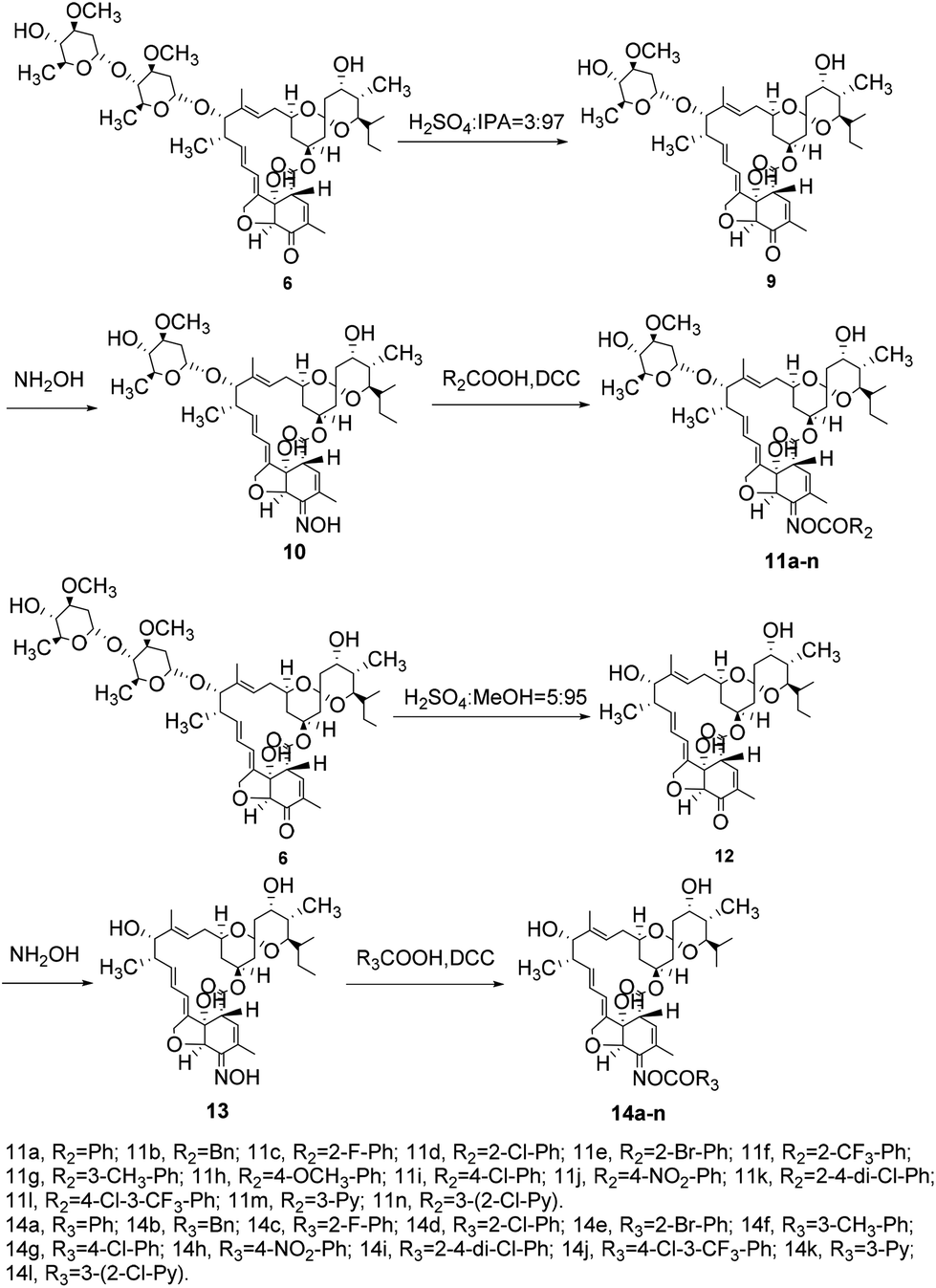 | ||
| Scheme 2 Synthesis of the target compounds 11a–n and 14a–l. | ||
Insecticidal activity
The insecticidal activities against M. persicae, C. elegans, and T. cinnabarinus of the 5-deoxyavermectin B2a oxime ester derivatives were evaluated using avermectin B2a as the positive control. The results were summarized in Table 1.| Compound | M. persicae | C. elegans | T. cinnabarinus | ||
|---|---|---|---|---|---|
| 50 μg mL−1 | 1 μg mL−1 | 10 μg mL−1 | 1 μg mL−1 | 0.1 μg mL−1 | |
| 8a | 85.1 ± 2.4a | 59.1 ± 3.1 | 100 | 100 | 63.7 ± 2.7 |
| 8b | 87.7 ± 3.2 | 82.3 ± 1.8 | 100 | 100 | 79.5 ± 3.2 |
| 8c | 90.3 ± 2.4 | 77.8 ± 3.3 | 100 | 100 | 83.2 ± 3.3 |
| 8d | 94.3 ± 2.3 | 90.4 ± 1.8 | 100 | 93.3 ± 3.1 | 62.2 ± 3.5 |
| 8e | 82.3 ± 2.8 | 73.6 ± 2.8 | 100 | 100 | 73.1 ± 2.3 |
| 8f | 88.6 ± 3.2 | 76.2 ± 2.4 | 100 | 100 | 88.3 ± 3.7 |
| 8g | 63.1 ± 1.2 | 43.2 ± 1.5 | 100 | 91.1 ± 3.0 | |
| 8h | 81.7 ± 4.7 | 61.6 ± 3.1 | 100 | 87.2 ± 2.5 | |
| 8i | 90.6 ± 2.6 | 46.5 ± 2.4 | 100 | 92.6 ± 2.2 | |
| 8j | 71.7 ± 3.4 | 54.8 ± 1.9 | 100 | 70.8 ± 3.1 | |
| 8k | 95.1 ± 2.8 | 61.0 ± 1.3 | 100 | 83.2 ± 1.9 | |
| 8l | 87.0 ± 1.0 | 63.8 ± 2.2 | 100 | 100 | 55.8 ± 2.4 |
| 8m | 51.3 ± 2.7 | 70.9 ± 1.4 | 100 | 83.4 ± 3.1 | |
| 8n | 44.2 ± 4.1 | 70.3 ± 3.2 | 100 | 73.2 ± 2.9 | |
| 11a | 65.0 ± 2.4 | 23.0 ± 2.1 | 93.4 ± 2.1 | ||
| 11b | 70.6 ± 1.7 | 40.9 ± 3.4 | 100 | 72.2 ± 2.1 | |
| 11c | 60.9 ± 2.8 | 75.9 ± 2.7 | 97.1 ± 3.6 | ||
| 11d | 69.4 ± 3.0 | 83.7 ± 3.0 | 90.8 ± 2.3 | ||
| 11e | 50.2 ± 1.8 | 61.6 ± 2.9 | 94.5 ± 3.1 | ||
| 11f | 67.9 ± 0.6 | 63.2 ± 2.5 | 95.4 ± 1.5 | ||
| 11g | 58.8 ± 3.1 | 34.1 ± 2.1 | 90.4 ± 2.0 | ||
| 11h | 72.6 ± 2.4 | 20.9 ± 2.8 | 75.2 ± 1.9 | ||
| 11i | 71.4 ± 5.7 | 20.7 ± 1.5 | 76.7 ± 1.3 | ||
| 11j | 52.1 ± 3.7 | 70.2 ± 2.1 | 71.0 ± 2.1 | ||
| 11k | 67.1 ± 2.1 | 73.7 ± 2.8 | 86.8 ± 2.7 | ||
| 11l | 68.8 ± 2.4 | 74.4 ± 2.1 | 82.2 ± 2.9 | ||
| 11m | 40.8 ± 2.6 | 71.0 ± 1.8 | 95.0 ± 1.3 | ||
| 11n | 38.0 ± 3.7 | 55.7 ± 3.3 | 86.3 ± 1.6 | ||
| 14a | 33.6 ± 3.1 | 7.0 ± 1.0 | 54.1 ± 1.9 | ||
| 14b | 41.6 ± 5.1 | 28.3 ± 3.0 | 84.3 ± 3.1 | ||
| 14c | 23.3 ± 3.1 | 65.9 ± 2.8 | 94.7 ± 2.7 | ||
| 14d | 52.0 ± 1.0 | 62.5 ± 1.5 | 36.3 ± 2.4 | ||
| 14e | 22.6 ± 1.2 | 60.7 ± 1.5 | 53.11 ± 1.6 | ||
| 14f | 27.6 ± 1.8 | 19.6 ± 2.3 | 62.4 ± 2.8 | ||
| 14g | 36.5 ± 2.9 | 15.3 ± 2.5 | 26.1 ± 1.6 | ||
| 14h | 68.7 ± 1.9 | 66.2 ± 2.0 | 21.4 ± 3.1 | ||
| 14i | 44.9 ± 3.8 | 32.7 ± 2.3 | 43.1 ± 2.5 | ||
| 14j | 20.4 ± 2.7 | 69.9 ± 1.5 | 92.9 ± 2.7 | ||
| 14k | 31.8 ± 1.9 | 32.5 ± 2.7 | 55.8 ± 2.6 | ||
| 14l | 24.3 ± 2.9 | 27.4 ± 2.1 | 41.6 ± 1.7 | ||
| 7 | 90.0 ± 3.8 | 91.9 ± 2.4 | 100 | 100 | 74.8 ± 1.6 |
| 10 | 68.4 ± 1.1 | 82.7 ± 1.9 | 94.7 ± 2.6 | ||
| 13 | 45.8 ± 2.1 | 42.0 ± 1.3 | 57.2 ± 2.5 | ||
| B2a | 93.5 ± 2.1 | 87.1 ± 2.7 | 100 | 100 | 78.7 ± 2.9 |
Most of the target compounds 8a–n exhibited moderate to excellent insecticidal activities against the three systems (Table 1). For example, compounds 8b, 8d, and 8k exhibited a 87.7%, 94.3%, and 95.1% insect mortality in M. persicae, similarly to the corresponding parent compound avermectin B2a (93.5%). The insecticidal activities of compounds 8b and 8c against T. cinnabarinus at 0.1 μg mL−1 were 79.5% and 83.2%, respectively, as compared with a 78.7% mortality achieved by avermectin B2a at the same concentration.
In order to investigate whether the disaccharide group can influence insecticidal activity, the corresponding monosaccharide (11a–n) and aglycone (14a–l) oxime ester congeners were also prepared. The insecticidal activities of avermectin disaccharide derivatives (8a–n) were more potent than their corresponding monosaccharide (11a–n) and aglycone (14a–l) analogues (Table 1). For instance, the insecticidal activity of compound 8b against C. elegans at 1 μg mL−1 was 82.3%, and the corresponding compounds 11b and 14b displayed mortalities as high as 40.9% and 28.3%. Thus, the disaccharide moiety played a significant role in the insecticidal activity of these oxime ester analogues.
By changing the position of the phenyl ring substitution and introducing different substituent groups on the phenyl ring in compounds 8a, 11a, and 14a, the monosaccharide (11a–l) and aglycone (14a–l) oxime ester analogues were shown to have a similar structure–activity relationship as the disaccharide derivatives (8a–l). Therefore, compounds 8a–l were used to investigate structure–activity relationship correlations. Compound 8d, with a chlorine atom substitution in the 2-position, showed the best insecticidal activities against M. persicae and C. elegans among compounds 8c–f. With regards to T. cinnabarinus, compounds with trifluoromethyl or fluorine in the 2-position (8c, 8f) showed better insecticidal activity. By introducing a chlorine atom in the 4-position, the activity of compound 8i against M. persicae was slightly increased compared with that of compound 8a, but with a subsequent detrimental impact on activity against C. elegans and T. cinnabarinus. The insecticidal activities of compounds 8k and 8l were affected by the above effects and other reasons (shape etc.) together. Compound 8b, with a methylene group inserted between the phenyl and the formoxyl groups, showed slightly higher insecticidal activities against the three insect systems than compound 8a. In general, introduction of electron donating moieties at the phenyl ring (8g, 8h) resulted in poor control of M. persicae, C. elegans, and T. cinnabarinus. Furthermore, when the substitution group was changed from a phenyl ring to a pyridyl ring, compounds 8m and 8n showed much weaker insecticidal activities against M. persicae and T. cinnabarinus than compound 8a, but possessed superior insecticidal activity against C. elegans. Compounds 11m, 11n, 14k, and 14l showed similar structure–activity relationship correlations as the corresponding compounds 8m and 8n.
Conclusions
In summary, three series of avermectin B2a derivatives bearing oxime ester moieties were synthesized and screened for their insecticidal activity against M. persicae, C. elegans, and T. cinnabarinus. The results showed that lots of the synthesized compounds 8a–n showed excellent insecticidal activity. In particular, compound 8d showed higher insecticidal activities against M. persicae and C. elegans in comparison with avermectin B2a. Significantly, the present study revealed important structure–activity relationship information such as the finding that the disaccharide moiety played a pivotal role in the insecticidal activity of these derivatives or that the position of substitution on the aromatic ring and electronic distribution of substituent groups had substantial effects on insecticidal potency. These results will be of particular value in the further optimization of avermectin B2a.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21772230).Notes and references
- K. Etebari, M. J. Furlong and S. Asgari, Sci. Rep., 2015, 5, 14642 CrossRef CAS PubMed.
- J. K. Kim, S. R. Choi, J. Lee, S. Y. Park, S. Y. Song, J. Na, S. W. Kim, S. J. Kim, I. S. Nou and Y. H. Lee, J. Agric. Food Chem., 2013, 61, 11222–11230 CrossRef CAS PubMed.
- J. L. Huang, M. Lv and H. Xu, RSC Adv., 2017, 7, 15997–16004 RSC.
- X. Yu, Z. P. Che and H. Xu, Chem.–Eur. J., 2017, 23(19), 4467–4526 CrossRef CAS PubMed.
- Y. Guo, R. G. Yang and H. Xu, Sci. Rep., 2016, 6, 35321 CrossRef PubMed.
- Y. Guo, R. G. Yang and H. Xu, RSC Adv., 2017, 7, 28009–28015 RSC.
- Q. Li, X. B. Huang, S. C. Li, J. C. Ma, M. Lv and H. Xu, J. Agric. Food Chem., 2016, 64, 5472–5478 CrossRef CAS PubMed.
- R. Wang, X. Y. Zhi, J. Li and H. Xu, J. Agric. Food Chem., 2015, 63, 6668–6674 CrossRef CAS PubMed.
- T. Yamada, Y. Horimatsu, T. Hirose, A. Sugawara, S. Omura and T. Sunazuka, Tetrahedron Lett., 2017, 58(32), 3119–3124 CrossRef CAS.
- W. X. Guan, W. X. Zhang, L. M. Tang, Y. Wang and H. X. Cui, J. Agric. Food Chem., 2017 DOI:10.1021/acs.jafc.7b01427.
- P. T. Meinke, S. P. Rohrer, E. C. Hayes, J. M. Schaeffer, M. H. Fisher and H. Mrozik, J. Med. Chem., 1992, 35(21), 3879–3884 CrossRef CAS PubMed.
- J. P. Arena, K. K. Liu, P. S. Paress, E. G. Frazier, D. Cully, H. Mrozik and J. M. Schaeffer, J. Parasitol., 1995, 81(2), 286–294 CrossRef CAS PubMed.
- J. Zhang, X. Nan, H. T. Yu and P. L. Cheng, et al., Eur. J. Med. Chem., 2016, 121, 422–432 CrossRef CAS PubMed.
- J. M. Schaeffer and H. W. Haines, Biochem. Pharmacol., 1989, 38(14), 2329–2338 CrossRef CAS PubMed.
- Z. X. Liu, L. H. Wang, H. M. Nie, J. J. Guo, T. Wang, L. B. Song and S. Lu, WO 20170110520 A1, 2017.
- H. Shan, S. K. Liu and Z. M. Wang, CN 1041176991 A, 2015.
- S. Q. Yan, X. M. Liang, J. J. Zhang and D. Q. Wang, Chin. J. Org. Chem., 2009, 29(12), 2005–2016 CAS.
- X. G. Liu, J. P. Wu, X. M. Liang and D. Q. Wang, Pest Manage. Sci., 2004, 60(7), 697–702 CrossRef CAS PubMed.
- E. Callens, O. Hueter, E. Lamy, P. Luethi, T. Winkler and P. M. J. Jung, Tetrahedron Lett., 2007, 48(4), 707–709 CrossRef CAS.
- S. Yamashita, D. Hayashi, A. Nakano, Y. Hayashi and M. Hirama, J. Antibiot., 2016, 69(1), 31–50 CrossRef CAS PubMed.
- H. Mrozik, P. Eskola, M. H. Fisher, J. R. Egerton, S. Cifelli and D. A. Ostlind, J. Med. Chem., 1982, 25(6), 658–663 CrossRef CAS PubMed.
- S. Terashima, T. Katoh and E. Saitoh, WO 9945012 A1, 1999.
- W. L. Shoop, H. Mrozik and M. H. Fisher, Vet. Parasitol., 1995, 59(2), 139–156 CrossRef CAS PubMed.
- J. H. Zhao, X. J. Xu, M. H. Ji, J. L. Cheng and G. N. Zhu, J. Agric. Food Chem., 2011, 59(9), 4836–4850 CrossRef CAS PubMed.
- Y. Tsukamoto, K. Sato, S. Mio, S. Sugai, T. Yanai, N. Kitano, S. Muramatsu, Y. Nakada and J. Ide, Agric. Biol. Chem., 1991, 55(10), 2615–2621 CAS.
- S. Moraesda, F. C. V. Mariade and A. Mendesde, et al., Parasites Vectors, 2016, 9, 407 CrossRef PubMed.
- C. R. Heredia, N. C. Romero and C. L. Miranda, Vet. Parasitol., 2017, 247, 77–79 CrossRef PubMed.
- L. M. Katiki, J. F. S. Ferreira and A. M. Zajac, et al., Vet. Parasitol., 2011, 182(2–4), 264–268 CrossRef CAS PubMed.
- G. S. Sun, X. Xu, S. H. Jin, L. Lin and J. J. Zhang, Molecules, 2017, 22(6), 958 CrossRef PubMed.
- W. S. Abbott, J. Econ. Entomol., 1925, 18, 265–267 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13258a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |