
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methylene violet 3RAX-conjugated porphyrin for photodynamic therapy: synthesis, DNA photocleavage, and cell study†
Si Yao‡
ac,
Yunman Zheng‡b,
Lijun Jiangd,
Chen Xied,
Fengshou Wu *c,
Chi Huang*b,
Xiong Zhangc,
Ka-Leung Wong*de,
Zaoying Lib and
Kai Wang*a
*c,
Chi Huang*b,
Xiong Zhangc,
Ka-Leung Wong*de,
Zaoying Lib and
Kai Wang*a
aHubei Collaborative Innovation Center for Advanced Organic Chemical Materials, College of Chemistry and Chemical Engineering, Hubei University, Wuhan, P. R. China. E-mail: kaiwang@hubu.edu.cn
bCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan, P. R. China. E-mail: chihuang@whu.edu.cn
cKey Laboratory for Green Chemical Process of Ministry of Education, Wuhan Institute of Technology, Wuhan, P. R. China. E-mail: wfs42@126.com
dDepartment of Chemistry, Hong Kong Baptist University, Hong Kong, P. R. China. E-mail: klwong@hkbu.edu.hk
eChangshu HKBU Technology Company Limited, Jiangsu Province, China
First published on 24th January 2018
Abstract
A methylene violet 3RAX-conjugated porphyrin has been designed and synthesized as a potential photosensitizer in photodynamic therapy because of its high DNA binding constant and the efficacy in inhibiting cancer cells with subcellular localization.
Introduction
Photodynamic therapy (PDT) in vivo or in vitro involves light and chemicals as photosensitizers and normally depends on the generation of singlet oxygen.1,2 Singlet oxygen, as a primary damaging agent, induces the apoptosis of cancer cells in photodynamic therapy. Specifically, it could effectively damage the tiny blood vessels in the tumour tissue and then cause cell death and local bleeding. As a result, the tumour tissue will be completely destroyed after a period of time.3 Besides, the reactive singlet oxygen is able to manufacture injury through interacting with biological macromolecules,4 including DNA and proteins.Phenazine compounds with heterocyclic structure have been widely applied in dyes, biomedicine,5–8 pesticide, photoelectric material,9 and chemical analysis.10 Methylene violet 3RAX, as one of the typical phenazine derivatives, has the rigid planar phenazine structure. Methylene violet 3RAX shows high biological activity in the anti-tumour therapy because it is able to change the molecular structure of DNA, undermine the module of DNA, and induce the generation of the reactive singlet oxygen which can cause cells death by cutting off DNA strands in tumour cell.11,12
An extensive attention has been paid on porphyrin-based compounds as photodynamic therapy photosensitizers for anticancer in recent years. Porphyrins have long been of interest because of their high binding affinity to DNA strands.13 There has been reported that porphyrins linked with some anticancer drugs can effectively localize in tumour cells and improve the therapy efficiency through the synergistic effect of porphyrins and anticancer drugs.14 However, to the best of our knowledge, few reported the porphyrin–methylene violet 3RAX conjugate and investigated its application in anti-cancer. To this end, we first time designed and synthesized compound 2 by conjugating porphyrin with synthesized methylene violet 3RAX (1) (Scheme 1). The binding properties of compounds 1 and 2 with DNA were characterized through photophysical studies including absorption and emission spectra. The DNA photocleavage activities of two compounds were investigated by agarose gel electrophoresis. The generation of singlet oxygen upon irradiation was determined by using 1,3-diphenylisobenzofuran (DPBF) as an indicator. Besides, the subcellular localization and the photocytotoxicity of compound 2 against HeLa cells were also evaluated.
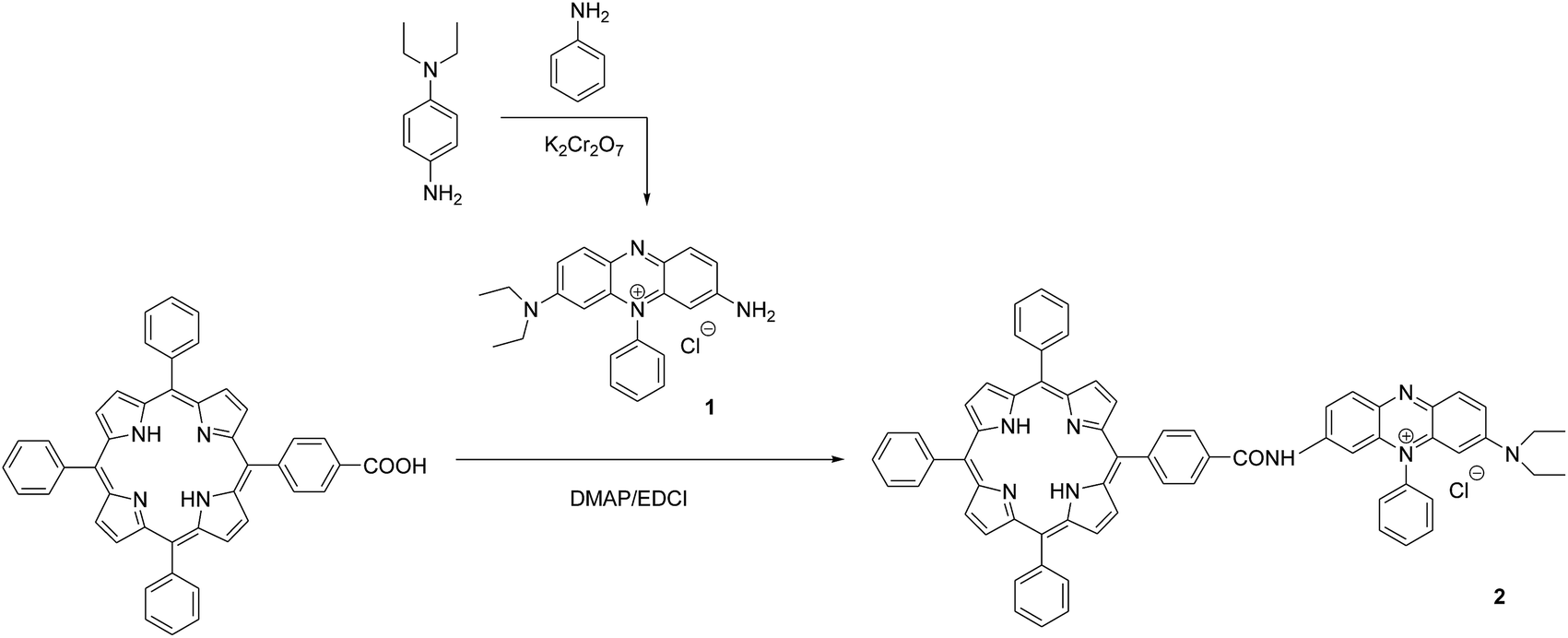 | ||
| Scheme 1 Synthetic route of compound 1 and 2. | ||
Experimental
NMR spectra were recorded on Varian Mercury-VX400 (400 MHz). The IR spectra (KBr pellets) were recorded on a Shimadzu FT-IR 3000 (KBr, V cm−1). Mass spectra were obtained from a Waters ZQ4000 (ESI). Elemental analyses were performed using an Elementar Vario EL. UV-vis spectra were measured on a Shimadzu 1901 spectrometer. Fluorescence spectra were measured on a Perkin Elmer LS-55 spectrometer. Calf-thymus DNA (ct-DNA) was obtained from Sigma company. An extinction coefficient of 6600 M−1 cm−1 at 260 nm was used to determine the concentration of ct-DNA.15 Silica gel 60 (0.04–0.063 mm) for column chromatography was obtained from Chinese medicine Reagent Co., Ltd. Other reagents were not further purified.General procedure for the synthesis of methylene violet 3RAX (1): to a solution of 4-amino-N,N-diethylaniline (5 g, 30.5 mmol), aniline (5.8 g, 61.0 mmol) and concentrated hydrochloric acid (8 mL) in acetic acid/sodium acetate buffer solution (200 mL) was added a large excess of potassium dichromate (19.7 g, 67.1 mmol). The reaction mixture was stirred for 3 h at 105 °C through protecting from light. After cooling to room temperature, the mixture was filtered and sodium chloride (120 g) was added to the filtrate, yielding the precipitate (1). Yield 2.3 g (35%). 1H NMR (400 MHz, CDCl3) δ: 1.09 (t, J = 7.1 Hz, 6H), 3.33 (q, J = 7.1 Hz, 4H), 5.63 (s, 1H), 5.99 (s, 1H), 7.13 (d, J = 7.8 Hz, 1H), 7.32 (d, J = 7.8 Hz, 2H), 7.69–7.80 (m, 4H), 7.85 (d, J = 8.9 Hz, 1H); IR (KBr), ν/cm−1: 3058, 1649 (NH2), 1339, 1198 (C–NH2, N–H), 1601, 1524, 1484 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N), 1069, 1006, 991, 874, 849, 812, 776, 704 (C–H), 1392 (–CH3) cm−1; UV-vis (DMSO), λmax/nm: 560; ESI-MS, m/z: 343.58 [M]+. Elemental analysis: C22H23N4+ calcd: C 69.74, H 6.12, N 14.79. Found: C 69.69; H 6.23; N 14.70.
C, C–N), 1069, 1006, 991, 874, 849, 812, 776, 704 (C–H), 1392 (–CH3) cm−1; UV-vis (DMSO), λmax/nm: 560; ESI-MS, m/z: 343.58 [M]+. Elemental analysis: C22H23N4+ calcd: C 69.74, H 6.12, N 14.79. Found: C 69.69; H 6.23; N 14.70.
General procedure for the synthesis of methylene violet 3RAX-conjugated porphyrin (2): to a solution of methylene violet 3RAX (0.1 g, 0.26 mmol) in dry chloroform (20 mL) was added 5-(4-carboxyphenyl)-10,15,20-triphenylporphyrin (0.2 g, 0.32 mmol),16 DMAP (0.03 g, 0.26 mmol) and EDCI (0.1 g, 0.53 mmol). The solution was heated to 60 °C for 10 h with stirring. After cooling to room temperature, the reaction mixture was poured into saturated brine (15 mL) and then extracted with chloroform (3 × 10 mL). The combined organic layer was dried using anhydrous sodium sulfate and concentrated in vacuum. The residue was purified by silica gel column with chloroform/methanol (v/v 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as an eluent. The second fraction was collected. Yield 0.2 g (85%). 1H NMR (400 MHz, CDCl3) δ: −2.91 (s, 2H), 0.90 (t, J = 7.1 Hz, 6H), 3.44 (q, J = 7.1 Hz, 4H), 5.44 (s, 1H), 7.49 (d, J = 8.1 Hz, 1H), 7.75–7.84 (m, 16H), 8.21 (m, 9H), 8.64–8.85 (m, 11H); IR (KBr), ν/cm−1: 2922 (C–H), 1675 (CO), 1598, 1508 (Ph-H), 1072, 997, 868, 798, 696 (C–H); UV-vis (DMSO), λmax/nm: 419, 516, 551, 588, 647; ESI-MS, m/z: 984.95 [M + H]+. Elemental analysis: C67H51N8O+ calc. C 81.77, H 5.22, N 11.39. Found: C 81.84; H 5.32; N 11.45.
:1) as an eluent. The second fraction was collected. Yield 0.2 g (85%). 1H NMR (400 MHz, CDCl3) δ: −2.91 (s, 2H), 0.90 (t, J = 7.1 Hz, 6H), 3.44 (q, J = 7.1 Hz, 4H), 5.44 (s, 1H), 7.49 (d, J = 8.1 Hz, 1H), 7.75–7.84 (m, 16H), 8.21 (m, 9H), 8.64–8.85 (m, 11H); IR (KBr), ν/cm−1: 2922 (C–H), 1675 (CO), 1598, 1508 (Ph-H), 1072, 997, 868, 798, 696 (C–H); UV-vis (DMSO), λmax/nm: 419, 516, 551, 588, 647; ESI-MS, m/z: 984.95 [M + H]+. Elemental analysis: C67H51N8O+ calc. C 81.77, H 5.22, N 11.39. Found: C 81.84; H 5.32; N 11.45.
The spectral measurements were performed at room temperature. The compounds were first dissolved in a small amount of DMSO (<5% total volume) and then dispersed in buffer media (pH = 7.4, 0.05 M Tris–HCl, 0.1 M NaCl). UV-vis absorption spectra of compound 1 and 2 were recorded within a range of 450–740 nm. Fluorescence spectra of compound 1 and 2 were recorded between 450 nm and 700 nm. Induced CD spectra were recorded within a range of 400–700 nm.
1,3-Diphenylisobenzofuran (DPBF) was used as a selective singlet oxygen (1O2) acceptor,17 which was bleached upon reaction with 1O2.18 The compounds with DPBF in DMSO (50 μM) were prepared in the dark. Each sample container was covered with aluminium foil with a yellow filter (with cut-off wavelength <500 nm) on one side. The samples were then exposed to light (50 watts) through the filter. The normalized absorbance of DPBF at 418 nm in these samples was reported as a function of the photo-irradiation time. By plotting the relationship between absorbance intensity and irradiation time, the rates of 1O2 production of compound 1 and 2 were determined.
The DNA photocleavage activities of compound 1 and 2 were measured using the plasmid DNA relaxation assay. Briefly, the plasmid DNA (pBluescript, 0.5 μg), enriched with the covalently-closed circular or supercoiled conformer (Form I), and the one-phor-all plus buffer (10 mM Tris–acetate, 10 mM magnesium acetate, 50 mM potassium acetate, pH 7.5) was vortexed. Aliquots of the DNA were pipetted into different Eppendorf tubes. Various amounts of autoclaved water (control sample) or compounds 1, 2 were added into the Eppendorf tubes to give a final volume of 20 μL in each sample tube. The sample mixtures were then photo-irradiated at 400–500 nm for 60 min using a transilluminator (Vilber Lourmat) containing 4 × 15 W light tubes (Aqua Lux) with maximum emission at 435 nm. During the irradiation procedure, the compounds did not undergo any photodegradation because of the low power density of light. After photo-irradiation, 2 μL of the 6× sample dye solution (which contained 20% glycerol, 0.25% bromophenol blue and 0.25% xylene cyanol FF) was added to each Eppendorf tube and mixed well by centrifugation. The sample mixtures were loaded onto a 0.8% (v/v) agarose gel (gel dimension: 13 cm × 10 cm), with 1 × TBE buffer (89 mM Tris–borate, 1 mM EDTA, pH 8) used as supporting electrolyte, and electrophoresed at 1.3 V cm−1 for 3 h using a mini gel set (CBS Scientific Co., Model No. MGU-502T). After electrophoresis, the gel was stained with 0.5 μg mL−1 ethidium bromide solution for 30 min and then destained using deionized water for 10 min. The resulting gel image was viewed under 365 nm and captured digitally using a gel documentation system (BioRad).19
The MTT viability assay was performed according to the standard methods.20 In brief, HeLa cells (3 × 103 per well) were seeded in 96-well plates 24 hours prior to exposure to drugs. The cells were treated with samples overnight in the dark. The cytotoxicity was determined by the MTT reduction assay. The cell monolayers were rinsed twice with phosphate-buffered saline (PBS) and then incubated with 50 μL MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] solution (0.5 mg mL−1) at 37 °C for 3 hours. Then the media were removed, and 100 μL of DMSO solubilizing reagent was added and shook for 30 minutes to dissolve the formed formazan crystals in living cells. The absorbance was measured at a dual wavelength, 540 nm and 690 nm, on a Labsystem Multiskan microplate reader (Merck Eurolab, Switzerland). Each dosed concentration was performed in triplicate wells and repeated twice for the MTT assay.
HeLa cells (3 × 103 per well) were incubated in 96-well plates for 24 hours prior to exposure to drugs. The cells were treated with samples in the dark overnight. Afterwards, the cells were exposure to yellow light (1–16 J cm−2) produced from a 400 W tungsten lamp fitted with a heat-isolation filter and a 500 nm long-pass filter. The fluence rate was 6 mW cm−2. Cell viability was determined by the MTT reduction assay at 24 h post-PDT.21 The cell monolayers were rinsed twice with PBS and then incubated with 50 μL MTT solution (0.5 mg mL−1) at 37 °C for 3 h. Then the media were removed, and 100 μL of DMSO solubilizing reagent was added and shaked for 30 minutes to dissolve the formed formazan crystals in living cells. The absorbance was measured at dual wavelength, 540 nm and 690 nm, on a Labsystem Multiskan microplate reader (Merck Eurolab, Switzerland). Each dosed concentration at individual light exposure was performed in quadruplicate wells for the PDT assay.
HeLa cells (2 × 105 per coverslip in 6-well plate) were treated with compound 2 (8 μM) for 6 h at 37 °C. After that, the supernatant was carefully removed and the cells were washed three times with PBS. Subcellular localization of compound 2 was examined using an Olympus FV1000 confocal microscope. A diode laser line at 514 nm was used for the excitation of compounds. Emission band pass filter setting at 600–750 nm was used for compound 2. Pinhole size of 80–100 μm was selected to exclude fluorescence light emitted from out-of-focus planes above and below the focusing plane. A 40× objective was used for image capturing. Images were processed and analyzed by using the FV10-ASW software (Olympus).
Results and discussions
Effects of the stoichiometric addition of ct-DNA on the compound 1 and 2 were illustrated by absorption spectra respectively (see Fig. 1a and c). The results in Table S1† showed that increasing DNA concentration resulted in moderate hypochromism for compound 1 and 2 (H = 20%–30%). Besides, significant red shift for compound 1 (Δλ = 12 nm) was observed, which was ascribed to the intercalative interaction between methylene violet 3RAX moiety and DNA at high compound-to-base pair ratio,22 and interaction between porphyrins and the bases of DNA.23 Generally, for a given DNA strand, intercalation exhibits a large red shift of the Soret band (>15 nm) and a substantial hypochromicity (>35%), while the small change normally occurs in the groove binding mode.24 In the presence of high concentrations of ct-DNA, the absorbance band of compound 2 exhibited substantial hypochromicity (>20%) without significant spectral shift, suggesting that compounds 2 may interact with DNA by groove binding.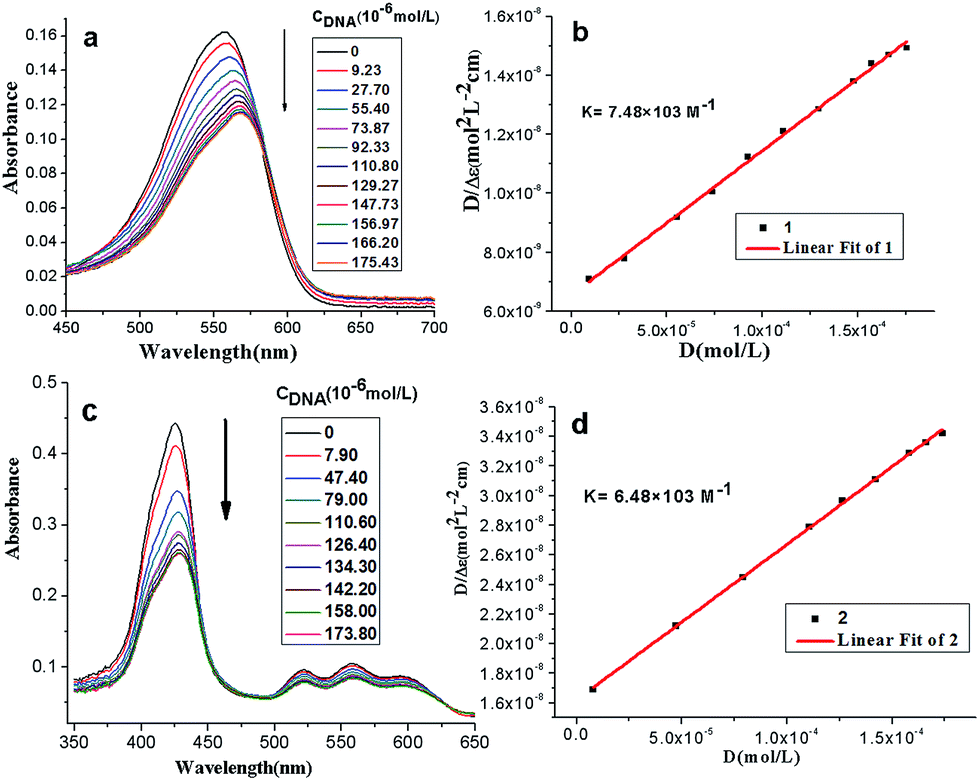 | ||
| Fig. 1 UV-vis absorption spectra of compound 1 (a) and compound 2 (c) with the titration of ct-DNA; maximal absorption intensity of compound 1 (b) and 2 (d) with the titration to ct-DNA. The concentration of compound 1 and 2 is 4 μmol L−1. Buffer solution (pH = 7.4, 0.05 mol L−1 Tris–HCl, 0.1 mol L−1 NaCl). | ||
The intercalative binding mode was characterized and the binding constants K of compound 1 and 2 were calculated respectively according to eqn (1):
| [DNA]total/|εA − εF| = [DNA]total/|εB − εF| + 1/(|εB − εF|K) | (1) |
Fluorescent titration was performed for further investigation on the binding mode between compound 1, 2 and ct-DNA. As shown in Fig. 2a and b, the fluorescence intensities of compound 1 and 2 increased with the addition of ct-DNA. The emission enhancement may be attributed to the insertion of compounds 1 with π-conjugated planar structure into the compact double helix groove space of CT-DNA. Subsequently, the compound 1 in the excited state can be effectively protected by the strong hydrophobic core of DNA, avoiding the quenching effect of ambient solvents.27 Besides, there were electrostatic interactions between the compound 1, 2 (with positive charges) and phosphate groups of DNA (with negative charges), which enhance the electron transfer from porphyrin to methylene violet. To this end, the emission from porphyrin part is suppressed relative to that of methylene violet 3RAX part. The bathochromic shift and enhanced fluorescence of compound 2 after interaction with DNA suggests it may adopt an end-to-end interaction with DNA (J aggregate).28
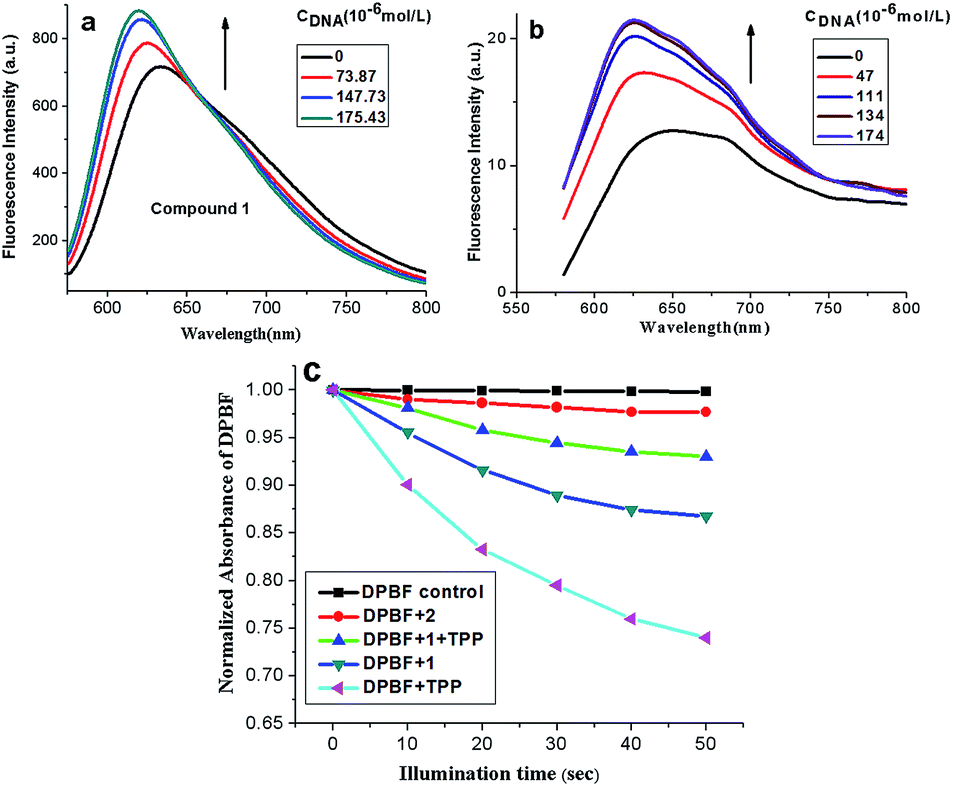 | ||
| Fig. 2 Fluorescence spectra of compound 1 (a) and 2 (b) with the titration to ct-DNA. Each spectrum was recorded in pH = 7.4, 0.05 mol L−1 Tris–HCl, 0.1 mol L−1 NaCl buffer solution. (c) Singlet oxygen generation of 1 and 2 (50 μM). The change of absorption of DPBF at 418 nm (A/A0) upon the irradiation time. The spectrum was recorded in DMSO. | ||
1,3-Diphenylisobenzofuran (DPBF) was used as a fluorescent probe for detection of singlet oxygen (1O2) generated from compound 1 and 2 upon irradiation24 with 5,10,15,20-tetraphenylporphyrin (TPP) as control groups. The relationship between absorbance ratio of DPBF (A/A0) and illumination time could indirectly reflect the 1O2 yield of the compounds. As shown in Fig. 2c, the absorbance of DPBF at 418 nm decreased in the presence of either compound 1 or 2 with the increase of illumination time. According to the slopes of the curves, the relative rates of 1O2 generation for compound 1 and 2 could be compared. The larger slope in values indicated the higher 1O2 quantum yield. Thus, the order of 1O2 quantum yield for compounds was Φ(TPP) > Φ(1) > Φ(2), indicating the introduction of porphyrin ring into compound 1 leads to the reduction of 1O2 yields. Moreover, we used TPP mixing with compound 1 to investigate the DPBF influence. Also in Fig. 2c, the generation of 1O2 decreased with the addition of compound 1 (see DPBF + 1 + TPP system), it might be because the compound 1 absorbed or quenched singlet oxygen 1O2 produced by TPP to some extent.
DNA photocleavage activities of compound 1 and 2 were measured by monitoring the conversion of a supercoiled form (form I) to the nicked circular form (form II),29 with the radiation of light at 400–500 nm for 60 minutes. As shown in Fig. 3, compound 2 at 400 μM had only 20% DNA photocleavage, while compound 1 at 300 μM showed over 60% photocleavage activity towards DNA. Generally, the DNA photocleavage activity of organic molecules was mainly depended on their singlet oxygen quantum yield and the electrostatic interaction with DNA. Thus, the significantly enhanced cleavage of DNA for compound 1 could be due to its relatively stronger electrostatic interaction with DNA (higher density of positive charge) and higher 1O2 yield of compound 1 comparing with that of compound 2.30
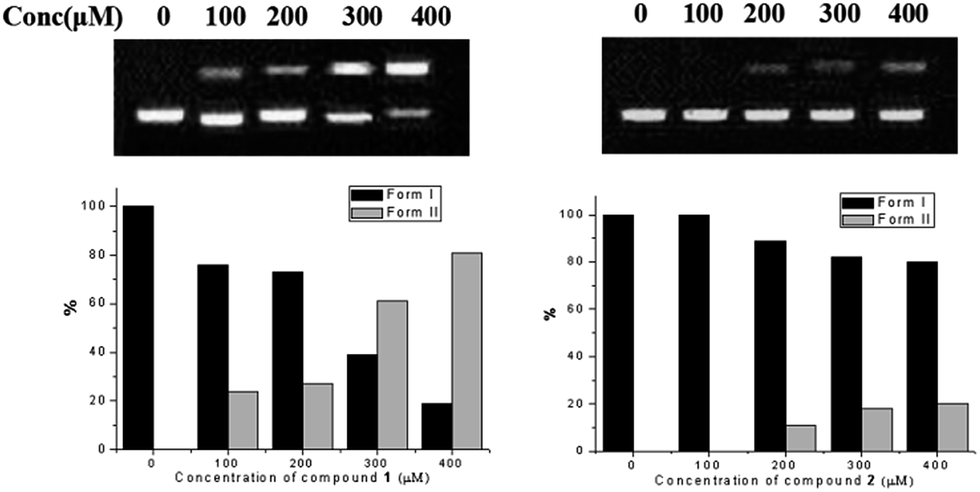 | ||
| Fig. 3 DNA binding profiles of compound 1 and 2 in the one-phor-all plus buffer (10 mM Tris–acetate, 10 mM magnesium acetate, 50 mM potassium acetate, pH = 7.5). | ||
The cell inhibition ability is one of crucial features for the conjugated porphyrins serving as anticancer drugs. Therefore, MTT and PDT assays were performed to determine the cytotoxicity of compound 2 to HeLa cells. As shown in Fig. 4d, there was no obvious decrease in cell viability even the incubating concentration of the compound 2 reached 100 μM after 24 h incubation, so it proves compound 2 with low dark toxicity, which means compound 2 results in few adverse effects as an anticancer drug. With the light excitation, compound 2 showed obvious photocytotoxicity as in Fig. 4d. With the increase of concentrations and light dose, the porphyrins exerted high cytotoxicity because of the increasing 1O2 generation through PDT mechanism. The light dose required to kill 60% of HK-1 cells was approximately 1 J cm−2 at 10 μM for compound 2. The dramatic difference between toxicity with/without light indicates that compound 2 has great PDT efficiency against HeLa cells.
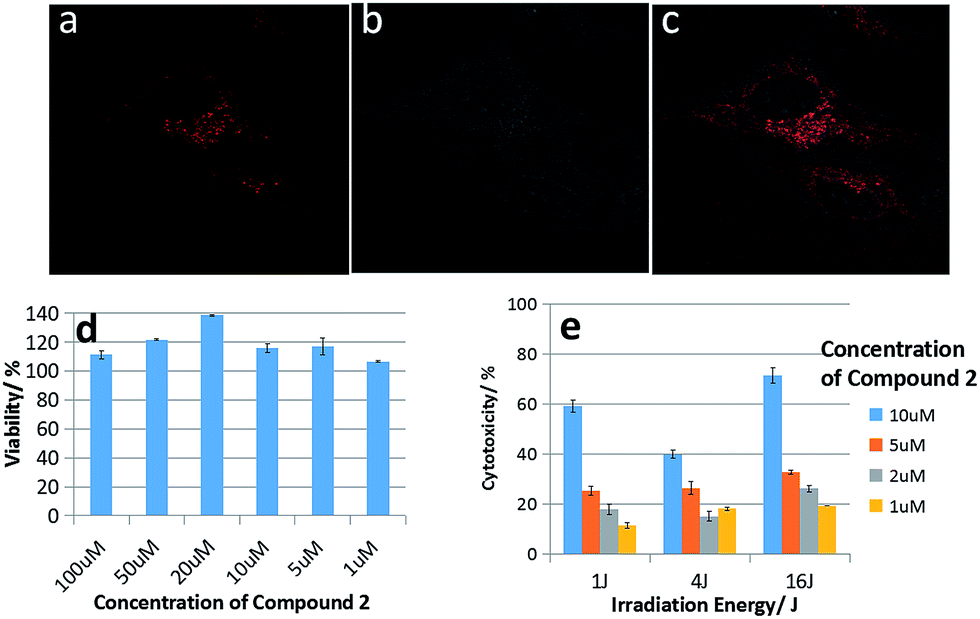 | ||
| Fig. 4 (a) Confocal laser scanning microscopy (CLSM) in vitro images; (b) bright-field images; (c) overlapping images. (6 h incubation time; 5 μM of compound 2); (d) MTT dark and (e) photo-cytotoxicity of compound 2 after incubation for 24 h and then irradiation with light (yellow light, 6 mW cm−2). | ||
Confocal microscopy was used to further investigate the subcellular localization of compound 2 in HeLa cells. As shown in Fig. 4a–c, the red fluorescence of compound 2 was mainly localized in the mitochondria of HeLa cells, because methylene violet 3RAX is a key dye to stain the mitochondria of cells, such as Janus green B.31 This result suggests porphyrins tailed with methylene violet 3RAX could be effective in promoting the targeting on mitochondria for antitumor activity. Besides, the hydrophilic part (methylene violet 3RAX) and lipophilic part (porphyrin ring) in the structure of compound 2 are important moieties for good photocytotoxicity in PDT.32
Conclusions
In summary, we first time designed and synthesized compound 2 by conjugating porphyrin with synthesized methylene violet 3RAX (1). The structures of both compounds were confirmed by 1H NMR, MS, UV-vis, IR, and elemental analysis. Meanwhile, UV-vis absorption, fluorescence evidence indicated that compounds 1 had intercalative interaction with DNA, while compounds 2 interacted strongly with DNA through groove binding and electrostatic modes. The quantum yield of 1O2 generated by TPP decreased with the addition of compound 1. At 400 μM, the DNA photocleavage activities of compound 1 and 2 were 80% and 20%, respectively. In addition, compound 2 with low dark toxicity but high photocytotoxicity against HeLa cells was mainly localized in mitochondria. The results from photophysical studies and cell experiments suggest porphyrin–methylene violet 3RAX conjugate could be developed as potential photosensitizers for mitochondrial targeting action in PDT.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China [grant no. 21601142, 20902071]; Wuhan Science and Technology Talent Training Program of Chenguang Project [grant no. 2015070404010190]; Key Project of Scientific Research Project of Hubei Provincial Department of Education [grant no. D20141506]; and Natural Science Foundation of Hubei Province [grant no. 2017CFB689] and Changshu HKBU Technology Company Limited.References
- B. W. Henderson and V. H. Fingar, Cancer Res., 1987, 47, 3110–3114 CAS.
- Z. B. Xu, F. Q. Yu, F. S. Wu, H. Zhang, K. Wang and X. L. Zhang, J. Porphyrins Phthalocyanines, 2015, 19, 1046–1052 CrossRef CAS.
- Y. L. Zhao, D. Yin, Z. Gao and C. S. Xu, Laser J., 2008, 29, 81–82 Search PubMed.
- P. Juzenas and J. Moan, J. Environ. Pathol., Toxicol. Oncol., 2006, 25, 29–50 CrossRef CAS.
- M. L. Lavaggi, M. Nieves, M. Cabrera, C. O. Azar, A. L. Cerain, A. Monge, H. Cerecetto and M. Gonzalez, Eur. J. Med. Chem., 2010, 45, 5362–5369 CrossRef CAS PubMed.
- M. L. Lavaggi, G. Aguirre, L. Boiani, L. Orelli, B. Garci, H. Cerecetto and M. Gonzalez, Eur. J. Med. Chem., 2008, 43, 1737–1741 CrossRef CAS PubMed.
- H. Cerecetto, M. Gonzalez, M. L. Lavaggi, A. Azqueta, A. L. Cerain and A. Monge, J. Med. Chem., 2005, 48, 21–23 CrossRef CAS PubMed.
- E. M. Lene, N. Gyrid, G. Gro, S. Havard, T. Knut, F. D. Kristin, Z. Kolbjorn, B. Anders, B. Oystein, O. D. Stein, S. Frode and H. Lars, Mar. Drugs, 2013, 11, 332–349 CrossRef PubMed.
- B. S. Gary, P. S. Steven and G. D. Brian, J. Phys. Chem., 1980, 84, 1841–1843 CrossRef.
- L. Yang, X. Zhang, W. S. Qu and J. L. Hua, Imaging Sci. Photochem., 2014, 32, 113–120 CAS.
- J. B. Laursen and J. Nielsen, Chem. Rev., 2004, 104, 1663–1686 CrossRef CAS PubMed.
- A. Cimmino, A. Evidente, V. Mathieu, A. Andolfi, F. Lefranc, A. Kornienko and R. Kiss, Nat. Prod. Rep., 2012, 29, 487–501 RSC.
- (a) K. Wang, X. Lin, X. Wan, T. Jia, X. L. Zhang, H. Zhang and X. L. Ju, Chem. J. Chin. Univ., 2012, 33, 2663–2669 CAS; (b) Y. Zheng, S. Zhu, L. Jiang, F. Wu, C. Huang, Z. Li, K. L. Wong, Z. Xu and K. Wang, J. Porphyrins Phthalocyanines, 2017, 21, 122–127 CrossRef CAS.
- (a) J. P. Kamat and T. P. A. Devasagaya, Toxicology, 2000, 155, 73–82 CrossRef CAS PubMed; (b) S. Zhu, S. Yao, F. Wu, L. Jiang, K. L. Wong, J. Zhou and K. Wang, Org. Biomol. Chem., 2017, 15, 5764–5771 RSC.
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- I. Chambrier, C. Banerjee, S. Remiro-Buenamanana, Y. M. Chao, A. N. Cammidge and M. Bochmann, Inorg. Chem., 2015, 54, 7368–7380 CrossRef CAS PubMed.
- J. Y. Chen, X. X. Ren, Q. Gu and X. Y. Yue, Chem. Res. Appl., 2011, 23, 35–40 CAS.
- Y. Y. Gao, L. Cao, Z. Z. Ou, C. Chen, Y. Li and X. S. Wang, Acta Phys.-Chim. Sin., 2013, 29, 2162–2172 CAS.
- K. Wang, C. T. Poon, W. K. Wong, W. Y. Wong, D. W. J. Kwong, H. Zhang and Z. Y. Li, Eur. J. Inorg. Chem., 2009, 922–928 CrossRef CAS.
- A. P. Wilson, in Cytotoxicity and Viability Assays in Animal Cell Culture: A Practical Approach, ed. J. R. W. Masters, Oxford University Press, Oxford, 3rd edn, 2000, vol. 1, pp. 175–219 Search PubMed.
- N. K. Mak, K. M. Li, W. N. Leung, R. N. S. Wong, D. P. Huang, M. L. Lung, Y. K. Lau and C. K. Chang, Biochem. Pharmacol., 2004, 68, 2387–2396 CrossRef CAS PubMed.
- S. Ishita and S. K. Gopinatha, Dyes Pigm., 2013, 96, 81–91 CrossRef.
- U. Sehlstedt, S. K. Kim, P. Carter, J. Goodisman, J. F. Vollano, B. Norden and J. C. Dabrowiak, Biochemistry, 1994, 33(2), 417–426 CrossRef CAS PubMed.
- E. I. Kochevar and W. R. Redmond, Method. Enzymol., 2000, 319, 20–28 CrossRef PubMed.
- Z. Xue, A. X. Hou, D. W. J. Kwong and W. K. Wong, Bioorg. Med. Chem. Lett., 2007, 17, 4266–4270 CrossRef CAS PubMed.
- R. F. Pasternack, E. J. Gibbs and J. J. Villafranca, J. Biochem., 1983, 22, 2406 CrossRef CAS.
- H. T. Wang, T. T. Hu, Q. L. Zhang, J. H. Liu, X. Z. Ren, C. H. Li, F. Wang and P. X. Zhang, Acta Chimica Sinica, 2008, 66, 1565–1571 CAS.
- (a) K. M. Sovenyhazy, J. A. Bordelon and J. T. Petty, Nucleic Acids Res., 2003, 31, 2561–2569 CrossRef CAS PubMed; (b) D. Mobius, Adv. Mater., 1995, 7, 437–444 CrossRef.
- (a) K. Wang, T. P. Chun, Y. C. Chun, K. W. Wong, W. J. K. Daniel, F. Q. Yu, H. Zhang and Z. Y. Li, J. Porphyrins Phthalocyanines, 2012, 16, 85–92 CrossRef CAS; (b) K. Yang, X. Zhang, F. Yang, F. Wu, X. Zhang and K. Wang, Aust. J. Chem., 2017, 70, 830–836 CrossRef CAS.
- (a) Y. M. Zheng, K. Wang, T. Li, X. L. Zhang and Z. Y. Li, Molecules, 2011, 16, 3488–3498 CrossRef CAS PubMed; (b) Y. Ishikawa, N. Yamakawa and T. Uno, Bioorg. Med. Chem., 2007, 15, 5230–5238 CrossRef CAS PubMed.
- (a) S. Brenner, Biochim. Biophys. Acta, 1953, 11, 480–486 CrossRef CAS PubMed; (b) S. J. Cooperstein, A. Lazarow and J. W. Patterson, Exp. Cell Res., 1953, 5, 69–82 CrossRef CAS PubMed.
- S. Z. Zu, F. S. Wu, K. Wang, Y. M. Zheng, Z. Y. Li, X. L. Zhang and W. K. Wong, Bioorg. Med. Chem. Lett., 2015, 25, 4513–4517 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13176c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |