
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced photocatalytic activity of a B12-based catalyst co-photosensitized by TiO2 and Ru(II) towards dechlorination†
Ying Suna,
Wei Zhang*a,
Tian-Yi Mab,
Yu Zhanga,
Hisashi Shimakoshi c,
Yoshio Hisaedac and
Xi-Ming Song*a
c,
Yoshio Hisaedac and
Xi-Ming Song*a
aLiaoning Key Laboratory for Green Synthesis and Preparative Chemistry of Advanced Materials, College of Chemistry, Liaoning University, Shenyang 110036, P. R. China. E-mail: weizhanghx@lnu.edu.cn; songlab@lnu.edu.cn; Fax: +86-24-62207922; Tel: +86-24-62207792
bSchool of Chemical Engineering, University of Adelaide, Adelaide, SA 5005, Australia
cDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University, Motooka, Fukuoka 819-0395, Japan
First published on 2nd January 2018
Abstract
A novel hybrid photocatalyst denoted as B12–TiO2–Ru(II) was prepared by co-immobilizing a B12 derivative and trisbipyridine ruthenium (Ru(bpy)32+) on the surface of a mesoporous anatase TiO2 microspheres and was characterized by DRS, XRD, SEM and BET et al. By using the hybrid photocatalyst, DDT was completely didechlorinated and a small part of tridechlorinated product was also detected in the presence of TEOA only after 30 min of visible light irradiation. Under simulated sunlight, the hybrid exhibited a significantly enhanced photocatalytic activity for dechlorination compared with B12–TiO2 under the same condition or itself under visible light irradiation due to the additivity in the contribution of UV and visible part of the sunlight to the electron transfer. In addition, this hybrid catalyst can be easily reused without loss of catalytic efficiency. This is the first report on a B12-based photocatalyst co-sensitized by two photosensitizers with wide spectral response.
Introduction
Photocatalysis has played a special role in both pollutant degradation and organic synthesis from the viewpoint of eco-friendliness in recent decades.1–5 As photocatalysts, vitamin B12 and its derivatives have exhibited excellent photocatalytic activity for decomposing halogenated organic compounds due to the supernucleophilic reactivity of Co(I) species, which could be produced from the Co(III) or Co(II) center of B12 and its derivatives under ultraviolet light irradiation in the presence of electron donor precursors.6–11 In 2004, Hisaeda et al.12 firstly reported the photocatalytic activity of a heptamethyl cobyrinate perchlorate, [Co(II)7C1ester]ClO4, in methanol for DDT dechlorination under visible light irradiation in the presence of Ru(bpy)32+ as photosensitizer and proved that the Co(I) species could be formed through accepting electrons from the photosensitizer under visible light irradiation. In order to establish a more efficient, economical and environmental friendly visible-light photocatalytic system, heterogenous catalysis has been performed through immobilizing the B12 catalyst and a photosensitizer onto catalyst supports and some recycled hybrid catalysts based on B12 were obtained.13,14 In 2011, Hisaeda group14 immobilized a B12 derivative and trisbipyridyl ruthenium proportionally on the same polymer backbone through radical polymerization reaction, and the obtained copolymer catalyst showed high catalytic activity for dehalogenation reaction. They proved that the electron transfer rate from the photosensitizer to the Co center of the B12 catalyst was significantly accelerated attributing to the short distance between the catalyst and the photosensitizer in the copolymer.TiO2 as a semiconductor has been widely applied in various fields such as dye-sensitized solar cell, organic pollution degradation, organic synthesis and so on for its strong oxidation and reduction abilities generated by UV light irradiation.15–18 Hiseada group has successfully immobilized B12 on P25 TiO2 or AMT600 TiO2 nanoparticles and the obtained hybrid catalysts showed high catalytic activity for dehalogenation reactions under UV light irradiation.19–22 In the hybrid systems, TiO2 served not only as a catalyst support but also an effective photosensitizer. In 2011, Reisner group23 reported a self-assembled system in which a cobalt catalyst (Et3NH)[CoIIICl(dmgH)2 (pyridyl-4-hydrophosphonate)] was attached on a ruthenium dye [Ru(bpy)3]2+-sensitized P25 TiO2 nanoparticles. It was proved that TiO2 played an important role as an electron mediator in the hybrid and promoted the electron transfer from the excited [Ru(bpy)3]2+ moieties to the cobalt catalyst under visible light irradiation. In addition, in recent years, many efforts have been focused on the photocatalytic applications of mesoporous TiO2 microspheres for their high surface areas, suitable pore structures, and uniform pore size distributions, which could supply more active sites for adsorption and photocatalytic reactions.24–27 Aiming to develop more efficient photocatalyst for dechlorination, in this paper, a novel B12-based hybrid catalyst co-sensitized by TiO2 and Ru(bpy)32+, B12–TiO2–Ru(II), was prepared through immobilizing a vitamin B12 derivative, cobyrinic acid, and a derivative of trisbipyridine ruthenium, Ru(dcb)(bpy)2(PF6)2, on the surface of mesoporous anatase TiO2 microspheres as shown in Fig. 1. The catalytic properties of this hybrid catalyst for 1,1-bis(4-chlorophenyl)-2,2,2-trichloroethane (DDT) dechlorination under visible light irradiation or simulated sunlight irradiation were reported. This is the first report on B12-based photocatalyst co-sensitized by two photosensitizers with wide spectral response.
 | ||
| Fig. 1 Illustration of the preparation of the hybrid B12–TiO2–Ru(II). | ||
Experimental
Materials
1,1-Bis(4-chlorophenyl)-2,2,2-trichloroethane (DDT), Ru(bpy)2Cl2, Tetrabutyl titanate, and Ru(bpy)3Cl2 was purchased from J & K Scientific Ltd. Cyanoaqua cobyrinic acid [(CN)(H2O)Cob(III)7COOH]Cl,28 [(CN)(H2O)Cob(III)7C1ester]Cl,19 mesoporous anatase TiO2 microspheres,25 4,4′-diethyl ester-2,2′-bipyridine (deep) and Ru(dcb)(bpy)2(PF6)2 (ref. 29) were synthesized according to the literatures. Other reagents were purchased from Sinopharm Chemical Reagent Beijing Co. (SCRC). All the reagents were of analytical grade and were used as received without further purification.Characterization
The SEM images and EDS analysis were acquired using a Hitachi SU-8010 equipped with an EDX analyzer operated at an accelerating voltage of 5 kV. The TEM images were obtained on a JEM 2100 operating at 200 kV. The UV-vis diffuse reflectance spectroscopy (DRS) measurements were obtained on a UV-vis spectrometer (Shimadzu UV-2550) using BaSO4 as a reference standard. The specific surface area of the samples was measured by the Brunauer–Emmett–Teller (BET) method using nitrogen adsorption and desorption isotherms on a Micrometrics ASAP 2020 system. The UV-vis spectra were obtained on a Perkin Elmer Lamda 25 spectrophotometer. The NMR spectra were recorded on a Mercury Vx-300 MHz NMR spectrometer. XRD patterns were obtained on a Bruker D8-Advance. The luminescence spectra were measured using a Hitachi F-4500 spectrophotometer in MeOH at room temperature. A 500 W xenon lamp (CHFXQ 500 W, Global xenon lamp power) with a λ ≥ 420 nm optical filter, AM 1.5 optical filter and a heat cut-off filter provided visible light or simulated sunlight illumination.Synthesis of Ru(dcb)(bpy)2(PF6)2
Ru(dcb)(bpy)2(PF6)2 was synthesized according to the literature29 with slight modification. Ru(bpy)2Cl2·2H2O (75 mg, 0.15 mmol) and 4,4′-diethyl ester-2,2′-bipyridine (44 mg, 0.15 mmol) were dissolved in ethanol (15 mL) and the mixture refluxed for 8 h. The solution was allowed to cool, filtered, and taken to dryness. The residue was dissolved in NaOH solution (0.3 mol L−1, 15 mL) and refluxed for 1 h and cooled to room temperature. A large excess of NH4PF6 was added to the solution under vigorously stirring and then HCl (5 mol L−1) was added to the mixture dropwise until much precipitate appeared. Then the precipitate was filtered and washed with a minimal amount of water, CH2Cl2, Et2O respectively. After being dried in vacuum for 6 h, a dark red crystal was obtained. 1H NMR (CD3CN), δ: 7.39 (q, 4H), 7.68 (dd, 4H), 7.80 (d, 2H), 7.87 (d, 2H), 8.06 (q, 4H), 8.50 (dd, 4H), 9.19 (s, 2H).Preparation of B12–TiO2–Ru(II)
Mesoporous anatase TiO2 microspheres (30 mg) was added into the methanol solution (6 mL) containing [(CN)(H2O)Cob(III)7COOH]Cl (8 × 10−4 mol L−1) and Ru(dcb)(bpy)2(PF6)2 (1.6 × 10−3 mol L−1). After the mixture was stirred at room temperature for 4 h, the hybrid B12–TiO2–Ru(II) was obtained by centrifugation and washed with methanol for three times.General catalytic procedure
A methanol solution (5 mL) containing B12–TiO2–Ru(II) (3 mg), DDT (2.4 × 10−3 mol L−1) and triethanolamine (0.2 mol L−1) was degassed after three of freeze–pump–thaw circles. After the solution was irradiated for 0.5 h using a xenon lamp with a λ ≥ 420 nm optical filter and a heat cut-off filter or a AM 1.5 optical filter, the catalyst was separated by centrifugation. Methanol was evaporated under reduced pressure and the residue was dissolved in CHCl3 (5 mL). The CHCl3 layer was extracted with water (10 mL) to remove TEOA, and then the solvent was evaporated under reduced pressure to dryness. The residues were analyzed by 1H NMR using 1,4-dioxane as the internal standard. The hybrid catalyst B12–TiO2–Ru(II) was washed with methanol and then reused after adding fresh triethanolamine and the substrate.Results and discussion
Fig. 1 illustrates the preparation procedure of the hybrid B12–TiO2–Ru(II). The mesoporous anatase TiO2 microspheres were firstly synthesized by hydrothermal reaction and then the catalyst [(CN)(H2O)Cob(III)7COOH]Cl and the photosensitizer Ru(dcb)(bpy)2(PF6)2 were grafted on the surface of mesoporous anatase TiO2 microspheres through their carboxylic groups only at room temperature. After the decoration, the colour of TiO2 microspheres changed from white to saffron yellow as shown in Fig. 1. The Co and Ru contents in the hybrid B12–TiO2–Ru(II) were 6.83 × 10−5 mol g−1 and 1.5 × 10−4 mol g−1, which were determined through detecting the absorbance change of the characteristic peaks of [(CN)(H2O)Cob(III)7COOH]Cl and Ru(dcb)(bpy)2(PF6)2 at 523 nm and 480 nm respectively in the supernatant by UV-vis spectra. The Co content in this hybrid was obviously increased and was 5 times higher than that of the hybrid B12–TiO2 (AMT600) reported in the literature,22 which should be attributed to the large surface area and pores of the mesoporous anatase TiO2 microspheres. In addition, it is worth to mention that the ratio of Co and Ru in the hybrid was about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.2 which was similar to their ratio 1:2 in the raw solution before adding TiO2. This result indicated that the proportional co-immobilization of these two different molecules, [(CN)(H2O)Cob(III)7COOH]Cl and Ru(dcb)(bpy)2(PF6)2, on the surface of TiO2 microspheres could be easily realized by one pot reaction. In this hybrid, the Ru(II) complexes and the TiO2 microspheres as photosensitizer are expected to be electron donors for B12 activation to form reactive Co(I) species.
:2.2 which was similar to their ratio 1:2 in the raw solution before adding TiO2. This result indicated that the proportional co-immobilization of these two different molecules, [(CN)(H2O)Cob(III)7COOH]Cl and Ru(dcb)(bpy)2(PF6)2, on the surface of TiO2 microspheres could be easily realized by one pot reaction. In this hybrid, the Ru(II) complexes and the TiO2 microspheres as photosensitizer are expected to be electron donors for B12 activation to form reactive Co(I) species.
Fig. 2 shows the UV-vis diffuse reflectance spectra of the hybrid B12–TiO2–Ru(II) and mesoporous anatase TiO2 microsphere and the UV-vis spectra of [(CN)(H2O)Cob(III)7COOH]Cl and Ru(dcb)(bpy)2(PF6)2 in methanol. The mesoporous anatase TiO2 microspheres strongly absorbed ultraviolet light and had very weak absorption after 400 nm.25 The catalyst [(CN)(H2O)Cob(III)7COOH]Cl exhibited a dominant absorption peak located at λmax = 490 nm accompanied by a prominent shoulder located at 523 nm and a small absorption peak at 404 nm.28 Ru(dcb)(bpy)2(PF6)2 exhibits a broad characteristic absorptions at λmax = 480 nm accompanied a small shoulder at 423 nm, which coincided with the literature.29 Comparing with the UV-vis spectra of the above reference compounds, the broad absorption from 400 nm to 600 nm shown by the hybrid B12–TiO2–Ru(II) fully indicated that the B12 catalyst and the Ru(II) photosensitizer had been co-immobilized on the surface of mesoporous anatase TiO2 microspheres successfully. The hybrid could absorb not merely ultraviolet but also visible light.
 | ||
| Fig. 2 UV-vis diffuse reflectance spectra of B12–TiO2–Ru(II) and TiO2, and UV-vis spectra of [(CN)(H2O)Cob(III)7COOH]Cl and Ru(dcb)(bpy)2(PF6)2 in methanol. | ||
Fig. 3 gives the XRD patterns of the hybrid B12–TiO2–Ru(II) and the raw material mesoporous anatase TiO2 microspheres. The as-prepared TiO2 exhibited seven obviously diffraction peaks at 2θ = 25.4°, 37.9°, 48.1°, 55.0°, 62.8°, 68.8° and 75.0°, which corresponding to the (101), (004), (200), (211), (204), (116) and (215) crystal faces of anatase TiO2 according to the JCPDS No. 21-1272.25 The sharp diffraction peaks indicated that the crystallinity of the anatase TiO2 was relatively high. After the modification with the two functional molecules, the shape and the intensity of the diffraction peaks did not change significantly, confirming that the crystallinity of the mesoporous anatase TiO2 microspheres was well kept.
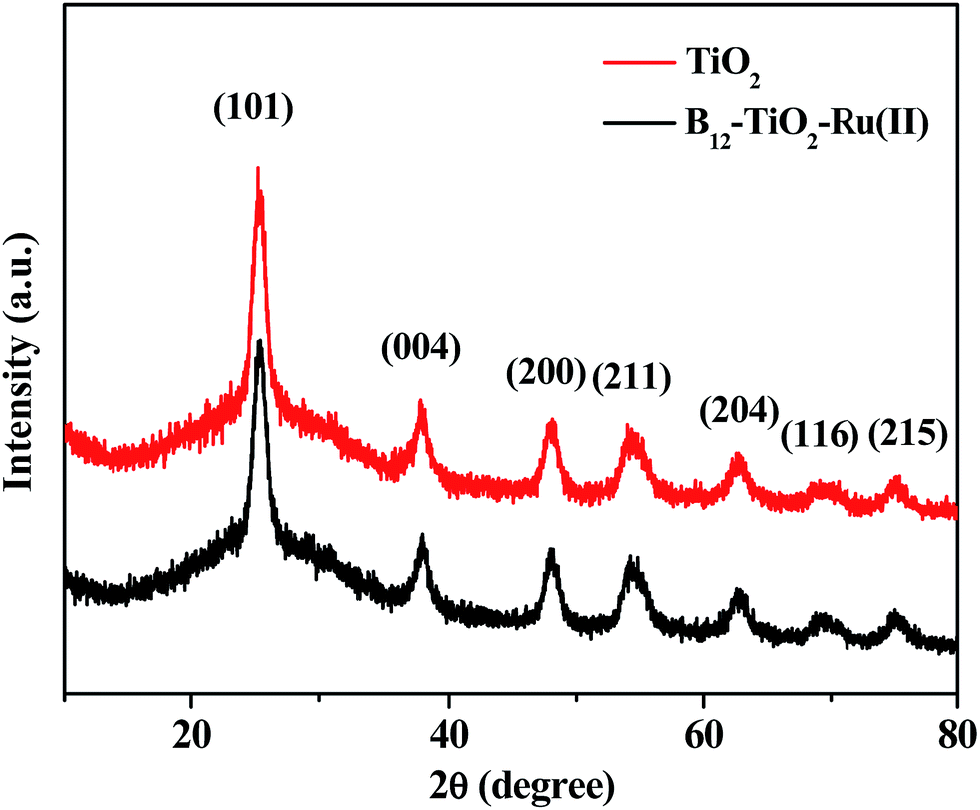 | ||
| Fig. 3 XRD patterns of the hybrid B12–TiO2–Ru(II) and the raw material mesoporous anatase TiO2 microspheres. | ||
The morphologies and structures of the mesoporous anatase TiO2 microspheres and the hybrid B12–TiO2–Ru(II) were characterized by SEM and TEM as shown in Fig. 4. Before or after the modification, the support TiO2 exhibited good spherical morphology with an uniform size of about 500 nm in diameter and each TiO2 microsphere was stacked by innumerable interconnected nanocrystals. Compared with TiO2 nanocrystals (P25 or AMT600), the larger particle diameter could make them more easily separated from the dispersion and recycled.
 | ||
| Fig. 4 SEM images of TiO2 (A) and B12–TiO2–Ru(II) (B); TEM images of TiO2 (C) and B12–TiO2–Ru(II) (D). | ||
The chemical composition of the hybrid B12–TiO2–Ru(II) was also determined by EDS as shown in Fig. 5. The element Ti was derived from TiO2, while the Ru, P and F peaks arose from the photosensitizer Ru(dcb)(bpy)2(PF6)2, Co peak originated from [(CN)(H2O)Cob(III)7COOH]Cl and the C and N peaks originated from both Ru(dcb)(bpy)2(PF6)2 and [(CN)(H2O)Cob(III)7COOH]Cl. The element O was originated from Ru(dcb)(bpy)2(PF6)2, TiO2 and [(CN)(H2O)Cob(III)7COOH]Cl.
 | ||
| Fig. 5 EDS spectrum of the hybrid B12–TiO2–Ru(II). | ||
High dispersity of a heterocatalyst in the reaction medium could significantly increase the effective collision probability between the catalyst and the substrate and thus promote its catalytic activity in a heterogeneous catalytic system. Therefore, the dispersity of the mesoporous TiO2 microspheres and the hybrid B12–TiO2–Ru(II) in methanol was evaluated and the pictures were taken after standing for 1 h at room temperature. As shown in Fig. 6, both the mesoporous anatase TiO2 microspheres and the hybrid B12–TiO2–Ru(II) dispersed well in methanol though their diameter were larger than that of the P25 or AMT600.
 | ||
| Fig. 6 Dispersions of TiO2 (a) and B12–TiO2–Ru(II) (b) in methanol after standing for 1 h at room temperature. | ||
Nitrogen adsorption–desorption isotherms and the Barrett–Joyner–Halenda (BJH) pore-size distribution analysis are usually employed to detect structural information for porous materials. The nitrogen adsorption–desorption isotherms at 77 K of the mesoporous anatase TiO2 microspheres and the hybrid B12–TiO2–Ru(II) are shown in Fig. 7. The as-prepared TiO2 microspheres showed an apparent capillary condensation step at a relative pressure (P/P0) of 0.5–0.85, which was corresponded to a pore size of 8 nm obtained by the BJH method. Their BET specific surface area was 172 m2 g−1, which is three times that of P25 (ref. 19) and AMT600,21 and the total pore volume was 0.64 cm3 g−1. From the isotherms of the hybrid B12–TiO2–Ru(II), it can be confirmed that the pore size of the hybrid reduced to 7 nm and the BET specific surface area and the total pore volume decreased to 159 m2 g−1 and 0.55 cm3 g−1 respectively, which should be attributed to the modification of the B12 catalyst [(CN)(H2O)Cob(III)7COOH]Cl and the photosensitizer Ru(dcb)(bpy)2(PF6)2. These data indicated that the two functional molecules were immobilized not only onto the surface but also inside the pore of the TiO2 microspheres.
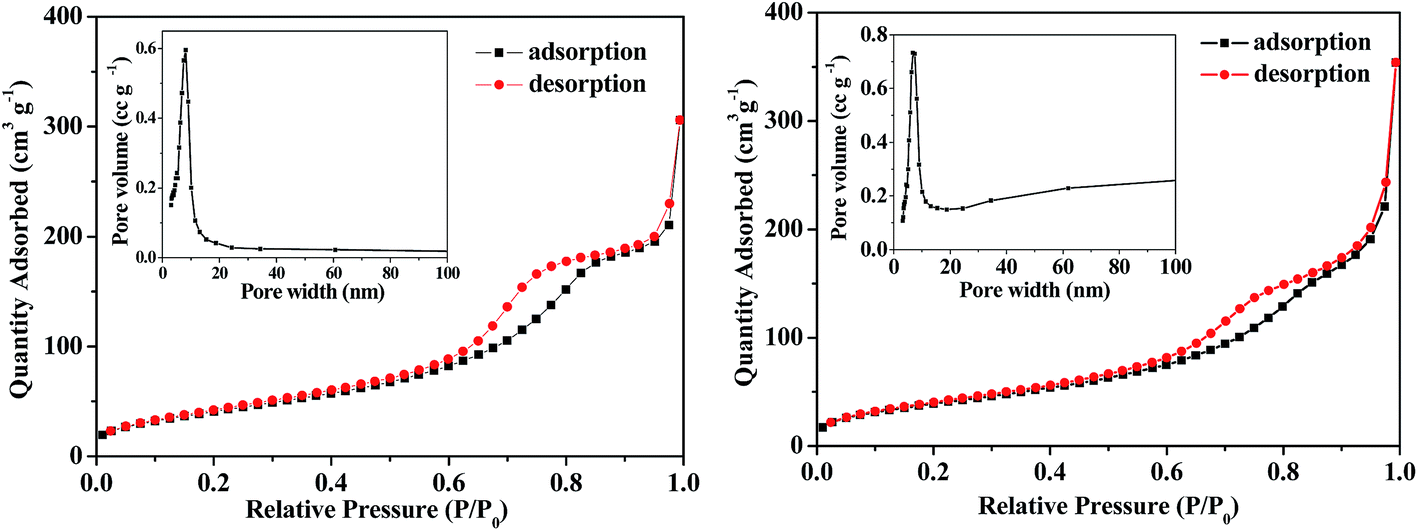 | ||
| Fig. 7 Nitrogen adsorption–desorption isotherms measured at 77 K and the pore-size distribution (inset) of the mesoporous anatase TiO2 microspheres (left) and the hybrid B12–TiO2–Ru(II) (right). | ||
Formation of the Co(I) species of the B12 complex in the presence of TiO2 was monitored by the UV-vis spectral change in methanol containing a sacrificial reductant triethanolamine (TEOA) under visible light irradiation (λ ≥ 420 nm). In order to avoid that the UV-vis spectral change of B12 in solution can't be detected sensitively after it was fixed on the TiO2 microspheres, the compounds [(CN)(H2O)Cob(III)7C1ester]Cl and Ru(bpy)3Cl2 without carboxylic groups (see Fig. S1, ESI†), were used instead of [(CN)(H2O)Cob(III)7COOH]Cl and Ru(dcb)(bpy)2(PF6)2 respectively. As shown in Fig. 8a, the mixture of [(CN)(H2O)Cob(III)7C1ester]Cl, Ru(bpy)3Cl2 and TiO2 produced three characteristic peaks at 352 nm, 463 nm and 542 nm respectively. Comparing to the UV-vis absorption spectra of [(CN)(H2O)Cob(III)7C1ester]Cl and Ru(bpy)3Cl2 (see Fig. S2, ESI†), the absorbance at 352 nm and 542 nm was assigned to the Co(III) species of the B12 complex and the peak at 463 nm was originated from both Co(III) and Ru(II) before irradiation. After irradiation for 25 minutes, one new peak at 392 nm was clearly observed, which was typical for the Co(I) state of the B12 complex,13 and simultaneously the peaks at 352 nm and 542 nm disappeared and the absorbance at 463 nm decreased. In contrast, a slow Co(I) formation was observed in the mixture of the B12 catalyst and the photosensitizer without TiO2 (Fig. 8b). Through the comparison, it was proved that the introduction of TiO2 can efficiently promote the formation of Co(I) species, attributing to the semiconductor property of TiO2 as an electron mediator, not only accelerating the electron transfer from the Ru(II) photosensitizer to the Co center of the catalyst, but also effectively suppressing the back electron-transfer process. When both the B12 catalyst and the Ru(II) photosensitizer were grafted on the TiO2 microspheres to give a new hybrid catalyst, the distance among B12, TiO2 and Ru(II) would be sharply shortened and the electron transfer from the photosensitizer to the Co center of the B12 catalyst via TiO2 is expected to be further accelerated under visible light irradiation.
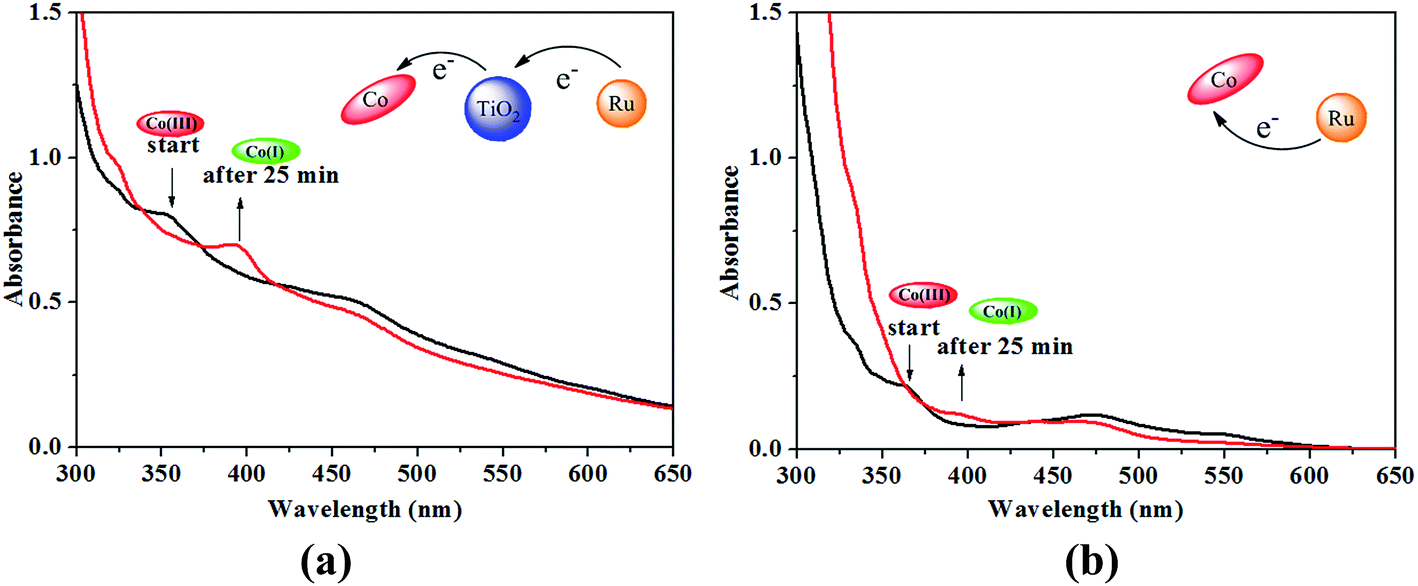 | ||
| Fig. 8 UV-vis spectral change of [(CN)(H2O)Cob(III)7C1ester]Cl (5 × 10−6 mol L−1) with TiO2 (a) and without TiO2 (b) in the presence of Ru(bpy)3Cl2 (5 × 10−6 mol L−1) and TEOA (0.2 mol L−1) by visible light irradiation (λ ≥ 420 nm) under N2 atmosphere in methanol. | ||
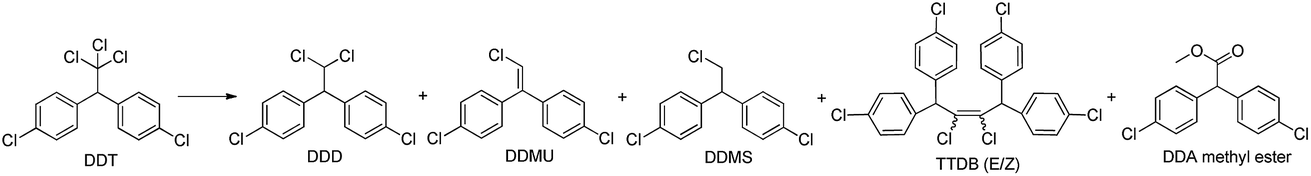
The visible light photocatalytic activity of the hybrid B12–TiO2–Ru(II) for DDT dechlorination was investigated firstly using triethanolamine as the electron sacrificer in methanol. The results are summarized in Table 1. When using the hybrid B12–TiO2–Ru(II) as catalyst, DDT conversion was 65% only after 10 min of visible light irradiation and four kinds of dechlorinated products were obtained including one mono-dechlorinated product 1,1-bis(4-chlorophenyl)-2,2-dichloroethane (DDD, 35% yield), three di-dechlorinated products 1,1-bis(4-chlorophenyl)-2-chloroethylene (DDMU, 4% yield) and 1,1,4,4-tetrakis(4-chlorophenyl)-2,3-dichloro-2-butene (TTDB (E/Z), 9% yield), one tri-dechlorinated product methyl 2,2-bis(4-chlorophenyl) acetate (DDA methyl ester, 3% yield) (entry 1 in Table 1). When the irradiation time was increased to 30 min, not only DDT was totally converted, but also the mono-dechlorinated product DDD disappeared completely. Accordingly, the yields of the di- and tri-dechlorinated products rose and one more di-dechlorinated product 1,1-bis(4-chlorophenyl)-2-chloroethane (DDMS) was also found (entry 2 in Table 1). TTDB (E/Z) were the products of coupling reaction between two didechlorinated carbene intermediates,12 therefore di-dechlorinated products obtained with a high yield of 93% after 30 min of irradiation, which was the highest among the B12-based photocatalytic systems reported in the literatures. In addition, from the data it can be concluded that DDD could be further degraded as soon as it was produced in such a hybrid catalytic system. The reaction did not proceed under dark conditions or using TiO2 instead of the hybrid B12–TiO2–Ru(II) (entries 3 and 4 in Table 1). When we used the hybrid B12–TiO2 or Ru(II)–TiO2 (see Fig. S3–S5, ESI†) alone (entries 5 and 6 in Table 1), the DDT conversion decreased to 24% and 13% respectively and DDD was detected as the single product after 30 min of irradiation. In contrast to the result in the hybrid system (entry 2 in Table 1), the DDT conversion was only 11% with DDD as the single product when a 1:2.2 mixture of the B12 catalyst [(CN)(H2O)Cob(III)7C1ester]Cl and the photosensitizer Ru(bpy)32+ was used under the same experimental conditions (entry 7 in Table 1). While adding a certain amount of mesoporous TiO2 microspheres into the simple mixture system (entry 8 in Table 1), the DDT conversion was increased to 42% and the di-dechlorinated products TTDB (E/Z) were found with 10% yield except DDD. The results shown here indicated that after the introduction of TiO2 to the simple mixture, especially after the co-immobilization of the B12 catalyst and the Ru(II) photosensitizer on TiO2 microspheres, reaction efficiency was significantly enhanced compared to that of the simple mixture system, which should be attributed to the very important role of the mesoporous anatase TiO2 microspheres not only as an adsorption reagent for the B12 catalyst, the photosensitizer and the substrate DDT or DDD etc. but also as an electron mediator.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalytic system | Irradiation time(min) | Conversion (%) | Product yieldb (%) | TONc | TOFc (h−1) | ||||
| DDD | DDMU | DDMS | TTDB (E/Z) | DDA | ||||||
| a B12–TiO2–Ru(II) = 3 mg, [DDT] = 2.4 × 10−3 mol L−1, [TEOA] = 0.2 mol L−1, MeOH: 5 mL, irradiation: λ ≥ 420 nm, 50 mW cm−2, distance: 10 cm.b DDT conversion and the product yields were determined by 1H HMR.c Turnover numbers (TON) and the turnover frequency (TOF) were based on the concentration of B12 and the product yields.d In the dark.e TiO2 = 3 mg.f B12–TiO2 = 3 mg.g Ru(II)–TiO2 = 3 mg.h [Ru(bpy)3Cl2] = 1.1 × 10−4 mol L−1, [(CN)(H2O)Cob(III)7C1ester]Cl = 4.9 × 10−5 mol L−1. | ||||||||||
| 1 | B12–TiO2–Ru(II) | 10 | 65 | 35 | 4 | Trace | 9 | 3 | 52 | 312 |
| 2 | B12–TiO2–Ru(II) | 30 | 100 | 0 | 40 | 13 | 20 | 6 | 119 | 238 |
| 3d | B12–TiO2–Ru(II) | 30 | Trace | — | — | — | — | — | 0 | 0 |
| 4e | TiO2 | 30 | 0 | — | — | — | — | — | 0 | 0 |
| 5f | B12–TiO2 | 30 | 24 | 16 | — | — | — | — | 9 | 18 |
| 6g | Ru(II)–TiO2 | 30 | 13 | 7 | — | — | — | — | — | — |
| 7h | B12, Ru(bpy)32+ | 30 | 11 | 10 | — | — | — | — | 5 | 10 |
| 8e,h | B12, Ru(bpy)32+, TiO2 | 30 | 42 | 21 | Trace | Trace | 10 | Trace | 30 | 60 |
TiO2 and the Ru(II) moieties in the hybrid could be excited at the same time under sunlight irradiation because they absorb light in UV and visible region respectively. Therefore, the hybrid B12–TiO2–Ru(II) should exhibited enhanced photocatalytic activity for dechlorination under sunlight irradiation. To evaluate the co-photosensitized effect of TiO2 and the Ru(II) complex on the B12 catalyst, DDT dechlorination was subsequently carried out under simulated sunlight (in Table 2). After 5 min of simulated sunlight irradiation, the DDT conversion was 49% and five dechlorinated products including DDD, DDMU, TTDB (E/Z) and DDA were found when using the hybrid B12–TiO2 alone as catalyst (entry 2 in Table 2). While using the hybrid B12–TiO2–Ru(II) instead, the DDT conversion increased to 70% and one more product DDMS were detected (entry 1 in Table 2). When the irradiation time was prolonged to 10 min, the DDT conversion reached 100% in the hybrid B12–TiO2–Ru(II) system (entry 3 in Table 2), which was still 1.4 times that of the hybrid B12–TiO2 system (entry 4 in Table 2) and 1.5 times that of itself under visible light irradiation (entry 1 in Table 1). Comparing the data, the effect of co-photosensitization was clearly observed. TiO2 was activated by the UV part of simulated sunlight and then transferred the electrons directly to the Co(I) center of the B12 catalyst, while at the same time the Ru(bpy)32+ moieties in the hybrid were excited by the visible part and transferred its electrons to the Co(I) center through TiO2. In other words, the hybrid B12–TiO2–Ru(II) realized the simultaneous utilization of both UV and visible part of the sunlight with high efficiency and the contribution of UV and visible part of the sunlight to the electron transfer has additivity.
| Entry (%) | Catalyst | Irradiation time (min) | Conversion | Product yieldb (%) | TONc | TOFc (h−1) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| DDD | DDMU | DDMS | TTDB (E/Z) | DDA | ||||||
| a B12–TiO2–Ru(II) = 3 mg, B12–TiO2 = 3 mg, [DDT] = 2.4 × 10−3 mol L−1, [TEOA] = 0.2 mol L−1, MeOH: 5 mL, irradiation illuminant: simulated sunlight, 50 mW cm−2, distance: 10 cm.b DDT conversion and the product yields were determined by 1H HMR.c Turnover numbers (TON) and the turnover frequency (TOF) were based on the concentration of B12 and the product yields. | ||||||||||
| 1 | B12–TiO2–Ru(II) | 5 | 70 | 22 | 4 | 2 | 17 | 4 | 67 | 804 |
| 2 | B12–TiO2 | 5 | 49 | 13 | 2 | Trace | 12 | 2 | 41 | 492 |
| 3 | B12–TiO2–Ru(II) | 10 | 100 | 29 | 15 | 5 | 19 | 5 | 173 | 1038 |
| 4 | B12–TiO2 | 10 | 74 | 25 | 2 | Trace | 20 | 4 | 70 | 42 |
Furthermore, the recycled catalysis of this hybrid catalyst B12–TiO2–Ru(II) under visible light irradiation and simulated sunlight irradiation was performed respectively since it can be easily separated from the reaction system by centrifugation and the relevant data were shown in Table 3. According to the data, this hybrid catalyst still kept high catalytic activity after 3 recyclings under 30 min of visible-light irradiation (entry 1 in Table 3). The recyclability of the hybrid B12–TiO2–Ru(II) at lower conversion has also been investigated under 5 min of simulated sunlight irradiation (entry 2 in Table 3). These data indicated that this hybrid catalyst had good stability and recyclability, and kept its catalytic activity after 3 recyclings even at lower conversion. The recovered B12–TiO2–Ru(II) after 3 recyclings irradiated by visible light was characterized by UV-vis spectrum and SEM respectively in Fig. 9, showing that the morphology and the optical property of the hybrid had no distinct change compared with its original.
| Entry | Irradiation illuminant (irradiation time) | Cycle | Conversion (%) | Product yield (%)b | ||||
|---|---|---|---|---|---|---|---|---|
| DDD | DDMU | DDMS | TTDB (E/Z) | DDA | ||||
| a B12–TiO2–Ru(II) = 3 mg, [DDT] = 2.4 × 10−3 mol L−1, [TEOA] = 0.2 mol L−1, MeOH: 5 mL, irradiation conditions: 50 W cm−2, distance: 10 cm.b DDT conversion and the product yields were determined by 1H HMR. | ||||||||
| 1 | Visible light (30 min) | 1 | 100 | 0 | 40 | 13 | 20 | 6 |
| 2 | 100 | 0 | 38 | 14 | 19 | 7 | ||
| 3 | 100 | 0 | 39 | 12 | 19 | 6 | ||
| 2 | Simulated sunlight (5 min) | 1 | 70 | 22 | 4 | 2 | 17 | 4 |
| 2 | 69 | 23 | 3 | 1 | 17 | 3 | ||
| 3 | 68 | 23 | 3 | 1 | 16 | 3 | ||
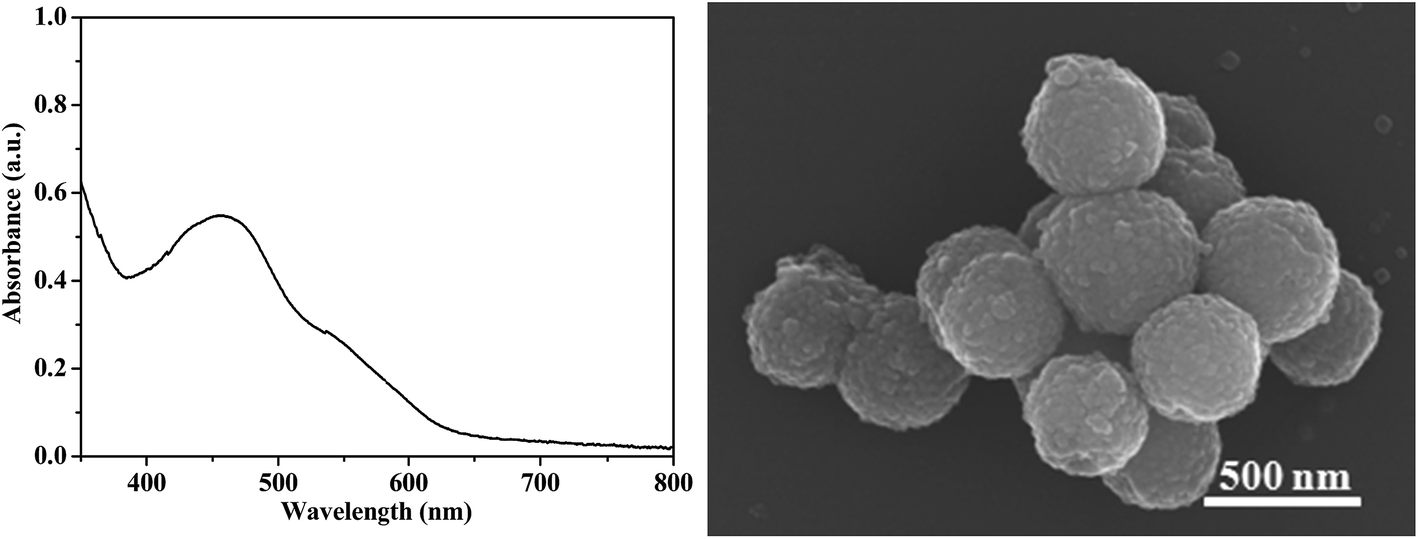 | ||
| Fig. 9 UV-vis spectrum (left) and SEM (right) of B12–TiO2–Ru(II) after 3 recyclings irradiated by visible light. | ||
Finally, we investigated the photosensitizing mechanism of the Ru(II) complex in the hybrid. In general, reductive and oxidative quenching mechanisms exist in the complex Ru(bpy)32+ when it is activated by visible light irradiation.30 To elucidate the quenching mechanism, the steady-state emission from the excited state of the Ru(II) complex before and after adding the B12 complex and TiO2 was measured, as shown in Fig. 10. After exciting the metal to ligand charge transfer (MLCT) transition band of the Ru(II) complex at 450 nm, a strong emission at 600 nm was observed, while the complex [(CN)(H2O)Cob(III)7C1ester]Cl showed no emission at this position. After adding [(CN)(H2O)Cob(III)7C1ester]Cl to the solution of Ru(bpy)32+, the emission intensity of the Ru(II) complexes was only slightly decreased, but if further adding a small amount of TiO2 microspheres, the excited state of the Ru(II) complex was quenched obviously, which indicated that electron transfer should proceed efficiently between the excited state Ru(bpy)32+* and TiO2 microspheres, which is consistent with the literature.31 Therefore, the so-called oxidative quenching mechanism could be expected for the visible-light-driven catalytic reaction in this hybrid system.
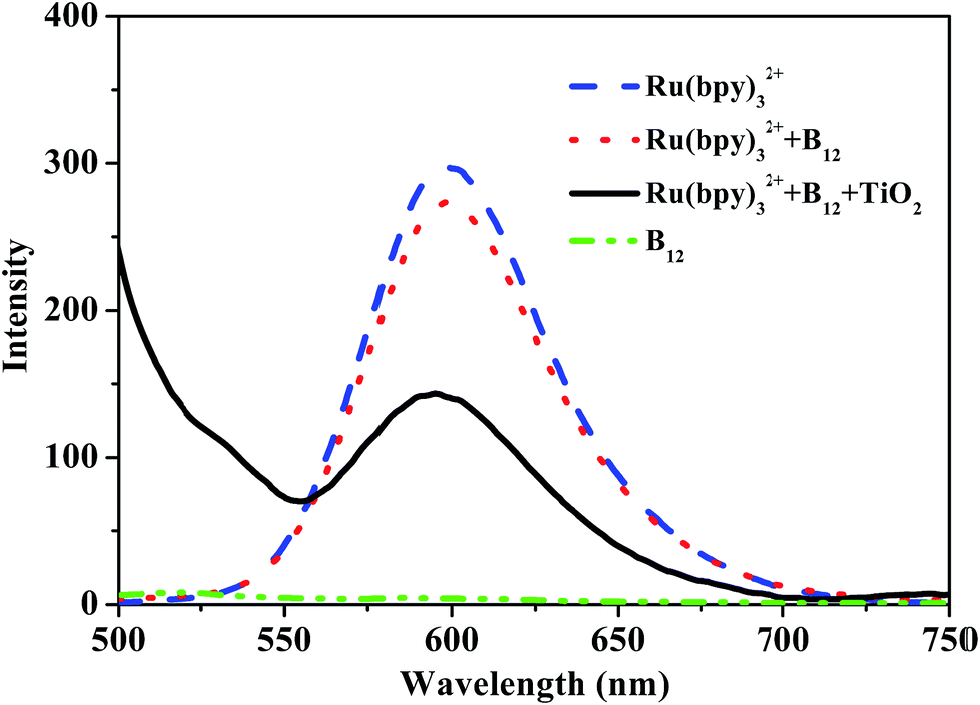 | ||
| Fig. 10 The steady-state emission of Ru(bpy)3Cl2 before and after adding [(CN)(H2O)Cob(III)7C1ester]Cl and TiO2 in methanol. ([Ru(bpy)3Cl2] = [[(CN)(H2O)Cob(III)7C1ester]Cl] = 4.7 × 10−5 mol L−1, TiO2 = 0.2 g L−1, λex = 450 nm). | ||
The photocatalytic mechanism of the hybrid system under sunlight was illustrated in Fig. 11. The photosensitizer Ru(bpy)32+ was excited by visible light irradiation and then injected one electron to the conduction band of the support TiO2 microspheres, and simultaneously, some electrons in the valence band of TiO2 moved up to the conduction band under UV light irradiation. Subsequently, the Co(III) center of the B12 derivative accepted two electrons from the conduction band of TiO2 and was reduced to the Co(I) species. The supernucleophilic Co(I) species had a high reactivity with organic halides to induce the oxidative addition of the alkylating agents to the metal center with dehalogenation.19,32 The obtained Ru(bpy)33+ and the holes in the valence band of TiO2 were rapidly reduced by the sacrificial reagent TEOA.
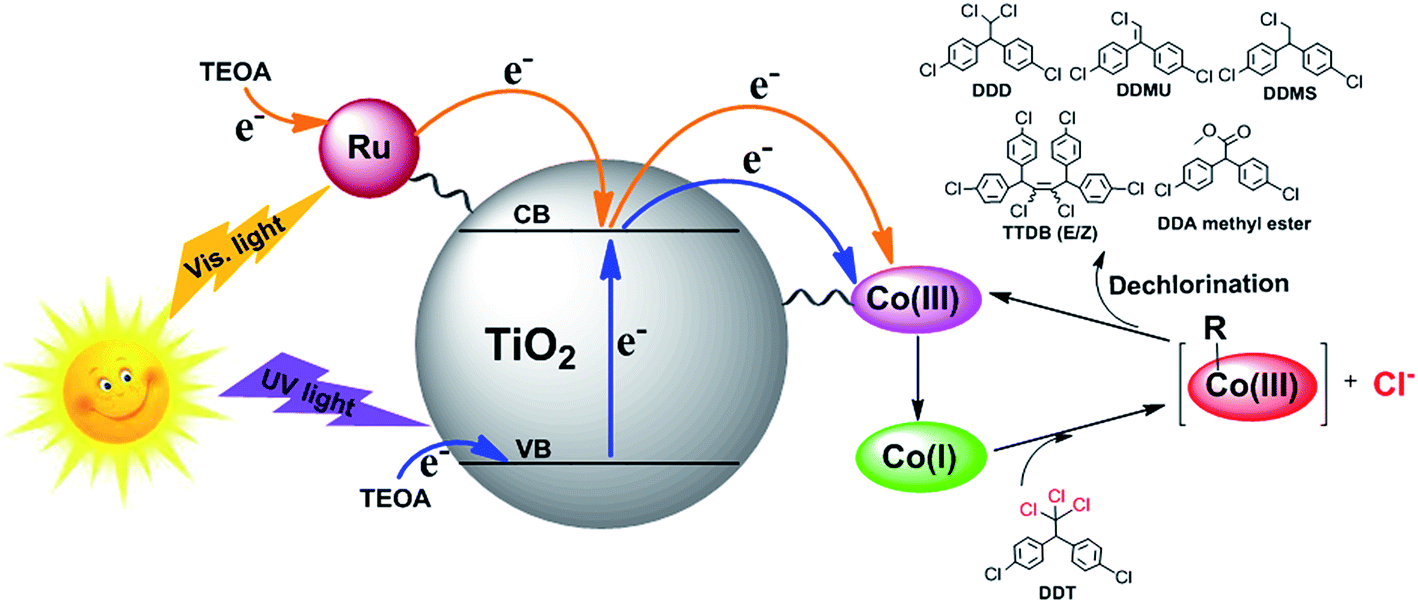 | ||
| Fig. 11 Proposed mechanism for DDT dechlorination catalyzed by B12–TiO2–Ru(II) under simulated sunlight. | ||
Conclusions
In conclusion, a B12-based catalyst co-sensitized by two photosensitizers, TiO2 and a Ru(II) complex, as a UV-vis light-driven photocatalyst was successfully synthesized. In the catalytic system composed of B12–TiO2–Ru(II) and TEOA, DDT could be completely converted into didechlorinated products and a small part was tridechlorinated only after 30 min of visible light irradiation. The catalytic activity was much higher than that of the other reported B12-based photocatalytic systems. Especially, under simulated sunlight, the hybrid exhibited a significantly enhanced photocatalytic activity for dechlorination compared with B12–TiO2 at the same condition or itself under visible light irradiation attributing to the additivity in the contribution of UV and visible part of the sunlight to the electron transfer. In addition, this hybrid catalyst can be easily reused without loss of catalytic efficiency. Therefore, the presented design strategy for photocatalytic system is efficient in full utilization of solar energy and it would be readily applicable to the design of an eco-friendly catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from Science Foundation of Educational Department of Liaoning Province (LYB201604) and the National Natural Science Foundation of China (51773085, 21203082).Notes and references
- D. Friedmann, A. Hakki, H. Kim, W. Choi and D. Bahnemann, Green Chem., 2016, 18, 5391 RSC.
- S. Klementova and M. Zlamal, Photochem. Photobiol. Sci., 2013, 12, 660 CAS.
- M. Y. Ye, Z. H. Zhao, Z. F. Hu, L. Q. Liu, H. M. Ji, Z. R. Shen and T. Y. Ma, Angew. Chem., Int. Ed., 2017, 56, 8047 Search PubMed.
- X. Fan, K. Lai, L. Wang, H. Qiu, J. Yin, P. Zhao, S. Pan, J. Xu and C. Wang, J. Mater. Chem. A, 2015, 3, 12179 CAS.
- Y. Sun, W. Zhang, J. Tong, Y. Zhang, S. Wu, D. Liu, H. Shimakoshi, Y. Hisaeda and X. M. Song, RSC Adv., 2017, 7, 19197 RSC.
- H. Tian, H. Shimakoshi, K. Imamura, Y. Shiota, K. Yoshizawa and Y. Hisaeda, Chem. Commun., 2017, 53, 9478 RSC.
- M. Giedyk, K. Goliszewska and D. Gryko, Chem. Soc. Rev., 2015, 44, 3391 RSC.
- K. Gruber, B. Puffer and B. Kräutler, Chem. Soc. Rev., 2011, 40, 4346 RSC.
- H. Shimakoshi, L. Li, M. Nishi and Y. Hisaeda, Chem. Commun., 2011, 47, 10921 RSC.
- Y. Hisaeda and H. Shimakoshi, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, 2010, vol. 10, pp. 313–370 Search PubMed.
- K. Tahara, H. Shimakoshi, A. Tanaka and Y. Hisaeda, Dalton Trans., 2010, 39, 3035 RSC.
- H. Shimakoshi, M. Tokunaga, T. Baba and Y. Hisaeda, Chem. Commun., 2004, 16, 1806 RSC.
- W. Zhang, H. Shimakoshi, N. Houfuku, X. M. Song and Y. Hisaeda, Dalton Trans., 2014, 43, 13972 RSC.
- H. Shimakoshi, M. Nishi, A. Tanaka, K. Chikama and Y. Hisaeda, Chem. Commun., 2011, 47, 6548 RSC.
- W. Ke, C. C. Stoumpos, J. L. Logsdon, M. R. Wasielewski, Y. Yan, G. Fang and M. G. Kanatzidis, J. Am. Chem. Soc., 2016, 138, 14998 CrossRef CAS PubMed.
- A. Romeiro, D. Freitas, M. Emilia Azenha, M. Canle and H. D. Burrows, Photochem. Photobiol. Sci., 2017, 16, 935 CAS.
- T. Yui, A. Kan, C. Saitoh, K. Koike, T. Ibusuki and O. Ishitani, ACS Appl. Mater. Interfaces, 2011, 3, 2594 CAS.
- N. Hoffmann, Aust. J. Chem., 2015, 68, 1621 CrossRef CAS.
- H. Shimakoshi, E. Sakumori, K. Kaneko and Y. Hisaeda, Chem. Lett., 2009, 38, 468 CrossRef CAS.
- H. Shimakoshi, M. Abiru, S. Izumi and Y. Hisaeda, Chem. Commun., 2009, 14, 6427 RSC.
- H. Shimakoshi and Y. Hisaeda, Chempluschem, 2014, 79, 1250 CrossRef CAS.
- H. Shimakoshi and Y. Hisaeda, Angew. Chem., Int. Ed., 2015, 54, 15439 CrossRef CAS PubMed.
- F. Lakadamyali and E. Reisner, Chem. Commun., 2011, 47, 1695 RSC.
- C. Guo, M. Ge, L. Liu, G. Gao, Y. Feng and Y. Wang, Environ. Sci. Technol., 2010, 44, 419 CrossRef CAS PubMed.
- Y. Zhang, S. Lin, W. Zhang, Y. Zhang, F. Qi, S. Wu, Q. Pei, T. Feng and X. M. Song, Electrochim. Acta, 2014, 150, 167 CrossRef CAS.
- Y. M. Wu, M. Y. Xing, B. Z. Tian, J. L. Zhang and F. Chen, Chem. Eng. J., 2010, 162, 710 CrossRef CAS.
- Y. C. Feng, L. Li, M. Ge, C. S. Guo, J. F. Wang and L. Liu, ACS Appl. Mater. Interfaces, 2010, 2, 3134 CAS.
- K. Tahara, H. Shimakoshi, A. Tanaka and Y. Hisaeda, Dalton Trans., 2010, 39, 3035 RSC.
- C. A. Kelly, F. Farzad, D. W. Thompson, J. M. Stipkala and G. J. Meyer, Langmuir, 1999, 15, 7047 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. V. Zelewsky, Coord. Chem. Rev., 1988, 19, 85 CrossRef.
- E. E. Beauvilliers and G. J. Meyer, Inorg. Chem., 2016, 55, 7517 CrossRef CAS PubMed.
- H. Shimakoshi, Y. Nagami and Y. Hisaeda, Appl. Chem. Eng., 2015, 4, 9 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13037f |
| This journal is © The Royal Society of Chemistry 2018 |