
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of Re promoter on the structure and catalytic performance of Ni–Re/Al2O3 catalysts for the reductive amination of monoethanolamine†
Lei Maabd,
Li Yan*b,
An-Hui Lu a and
Yunjie Ding*bc
a and
Yunjie Ding*bc
aState Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, No. 2 Linggong Road, Dalian 116024, P. R. China
bDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian, 116023, China. E-mail: dyj@dicp.ac.cn; Fax: +86 411 84379143; Tel: +86 411 84379143
cState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian, 116023, China
dUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, China
First published on 20th February 2018
Abstract
In this paper, Ni/Al2O3 catalysts (15 wt% Ni) with different Re loadings were prepared to investigate the effect of Re on the structure and catalytic performance of Ni–Re/Al2O3 catalysts for the reductive amination of monoethanolamine. Reaction results reveal that the conversion and ethylenediamine selectivity increase significantly with increasing Re loading up to 2 wt%. Ni–Re/Al2O3 catalysts show excellent stability during the reductive amination reaction. The characterization of XRD, DR UV-Vis spectroscopy, H2-TPR, and acidity–basicity measurements indicates that addition of Re improves the Ni dispersion, proportion of octahedral Ni2+ species, reducibility, and acid strength for Ni–Re/Al2O3 catalysts. The Ni15 and Ni15–Re2 catalysts were chosen for in-depth study. The results from SEM-BSE, TEM, and CO-TPD indicate that smaller Ni0 particle size and higher Ni0 surface area are obtained in the reduced Ni–Re/Al2O3 catalysts. Results from in situ XPS and STEM-EDX line scan suggest that Re species show a mixture of various valances and have a tendency to aggregate on the surface of Ni0 particles. During reaction, the Ni0 particles on the Al2O3 support are stabilized and the sintering process is effectively suppressed by the incorporation of Re. It could be concluded that sufficient Ni0 sites, the collaborative effect of Ni–Re, and brilliant stability contribute to the excellent catalytic performance of Ni–Re/Al2O3 catalysts for the reductive amination of monoethanolamine.
Introduction
Ethylene amines is the collective name for ethylenediamine (EDA), polyethylene polyamines, and the cyclic amines, including piperazine (PIP), aminoethyl piperazine (AEP), and hydroxyethyl piperazine (HEP) etc. As an important intermediate, ethylene amines have wide applications in the production of pharmaceuticals, lube oil additives, chelating agents, fungicides, oil field chemicals, paper wet-strength resins, surfactants, and fabric softeners. Reductive amination of monoethanolamine (MEA) to ethylene amines is a common synthesis route which has been applied in industrial production process since the 1960s.1–4 The reductive amination method has gained increasing interest since it constitutes a green economy route for the synthesis of ethylene amines.5–14In the reductive amination process, ethylene amines are synthesized by contacting MEA and ammonia under hydrogen atmosphere in the presence of hydrogenation–dehydrogenation metal catalysts. The reaction pathways for the transformation of MEA to ethylene amines on a metal catalyst are postulated as follows: firstly, dehydrogenation of MEA to 2-amino-acetaldehyde; secondly, condensation with ammonia or an amine to form imine; lastly, hydrogenation of imine to amine.7,8,11,15 The hydrogenation and dehydrogenation processes are catalyzed by the transition metal, while the condensation process could be accelerated by acid or base sites.16–19 The presence of small amount of hydrogen is found to be necessary to maintain the activity of catalyst during the reductive amination process.8,20,21 The dashed box in Fig. 1 indicates the main reaction path for the generation of EDA. Moreover, the product amines could continue to take part in the reductive amination process as reactants. The presence of continuous reaction makes the pathways more complicated and decreases the selectivity of the desired amine. As a result, the production of various by-products increases the cost of separation process. For this reason, it's important for the catalyst to have both high activity and selectivity of the desired product, such as EDA.
 | ||
| Fig. 1 Reductive amination pathways of MEA. | ||
Different catalytic metal systems such as Ni,17–19,21–24 Ni–Re,25–29 Co,20,30,31 Cu,32–35 Fe,36–38 Rh,39,40 Ru,12,13,41,42 Pt,43–45 and Pd46 etc. have been tested in the reductive amination of amino and aliphatic alcohols. Ni-based catalytic system has proved to be cost-effective and highly active for the amination reaction and has been extensive applied in industry. In one of the representative research, Shimizu and his group have studied the influence of acid–base property and metal particle size of Ni/Al2O3 catalysts on N-alkylation of amines with alcohols.18,19 They clarified that the coexistence of acidic and basic sites on Al2O3 is indispensable for the reductive amination reaction. Moreover, surface metallic Ni sites are proved to be active sites and the small Ni particles with low-coordinated Ni0 atoms show higher turnover number during the reaction. The effect of acidity or metallic surface area on the catalytic performance of supported Ni catalyst in other reductive amination reactions has also been reported by researchers.17,21 Furthermore, for the reductive amination of MEA, many patents have reported that the introduction of Re into the Ni-based catalysts could not only improve the MEA conversion but enhance the selectivity of EDA.25,26 Although the Ni–Re/Al2O3 catalysts have been applied in industry for decades, studies on the Ni–Re/Al2O3 catalytic system for reductive amination are scarce and presented mainly in the patent literatures. The role of Re promoter on the Ni–Re catalysts is still ambiguous. In order to comprehend the catalytic processes and design novel catalyst with better performance, it is of great significance to study the effect of Re in the Ni–Re catalysts on the behavior for the reductive amination process.
We report herein that Al2O3 supported Ni–Re catalysts act as heterogeneous catalysts for the reductive amination of MEA with ammonia under hydrogen atmosphere in a trickle bed reactor. A series of Ni–Re/Al2O3 catalysts with different loading of Re were prepared by co-impregnating method. Combined with various catalyst characterizations, the physicochemical properties and catalytic performance of Ni–Re/Al2O3 were discussed in detail so as to investigate the role of Re in the catalytic system.
Experimental
Materials and catalysts
All the catalysts were prepared by incipient-wetness impregnation method. The Ni/Al2O3 catalyst with 15 wt% Ni (the mass ratio of Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al2O3 was 15:100) was synthesized by impregnation of Al2O3 (Shandong Aluminum Corp., A-AS-04) with an aqueous solution of Ni(NO3)2·6H2O (99.9% purity). After impregnation, the catalyst was initially dried at room temperature. The dried catalyst was then kept in an oven at 120 °C for 4 h and in a muffle at 500 °C for another 4 h. The calcined catalyst was denoted as Ni15. The monometallic Re/Al2O3 catalyst with 2 wt% Re (the mass ratio of Re:Al2O3 was 2:100) was synthesized by impregnation of Al2O3 with an aqueous solution of NH4ReO4 (99.99% purity). The drying and roasting processes were the same as before and the obtained catalyst was denoted as Re2. Ni–Re/Al2O3 catalysts were prepared by co-impregnation of the mixture of Ni(NO3)2·6H2O and NH4ReO4 aqueous solution on Al2O3. The Ni loading was fixed as 15 wt% and the Re loadings were 0.5, 1, 2, and 3 wt%, respectively. The drying and roasting processes were the same as before. The calcined Ni–Re/Al2O3 catalysts were denoted as Ni15–Rex, where x represented the loading of Re. Before reaction, the calcined catalysts were reduced in situ for 4 h at 390 or 500 °C in H2 flowing at 100 mL min−1. The reduced catalysts henceforth was designated Ni15–Rex–yR, where y was the reduction temperature.
:Al2O3 was 15:100) was synthesized by impregnation of Al2O3 (Shandong Aluminum Corp., A-AS-04) with an aqueous solution of Ni(NO3)2·6H2O (99.9% purity). After impregnation, the catalyst was initially dried at room temperature. The dried catalyst was then kept in an oven at 120 °C for 4 h and in a muffle at 500 °C for another 4 h. The calcined catalyst was denoted as Ni15. The monometallic Re/Al2O3 catalyst with 2 wt% Re (the mass ratio of Re:Al2O3 was 2:100) was synthesized by impregnation of Al2O3 with an aqueous solution of NH4ReO4 (99.99% purity). The drying and roasting processes were the same as before and the obtained catalyst was denoted as Re2. Ni–Re/Al2O3 catalysts were prepared by co-impregnation of the mixture of Ni(NO3)2·6H2O and NH4ReO4 aqueous solution on Al2O3. The Ni loading was fixed as 15 wt% and the Re loadings were 0.5, 1, 2, and 3 wt%, respectively. The drying and roasting processes were the same as before. The calcined Ni–Re/Al2O3 catalysts were denoted as Ni15–Rex, where x represented the loading of Re. Before reaction, the calcined catalysts were reduced in situ for 4 h at 390 or 500 °C in H2 flowing at 100 mL min−1. The reduced catalysts henceforth was designated Ni15–Rex–yR, where y was the reduction temperature.
Catalyst characterization
N2 physisorption was measured at Quantachrome Autosorb-1 instrument. Before measurement, each catalyst was outgassed at 300 °C for 3 h under vacuum. The BET surface area was calculated from the range P/P0 = 0.05–0.30 in the adsorption branch. The total pore volume was determined by the amount of N2 adsorbed at a P/P0 of 0.99.Total Ni or Re metal contents in calcined catalysts were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES). Perkin Elmer Optima 7300 DV was applied for elemental detection.
The X-ray diffraction (XRD) analysis of catalysts was performed on a PANalytical X'Pert PRO diffractometer equipped with Cu Kα1 radiation source. The data were recorded with a scan speed of 0.04° s−1 and a step size of 0.013°.
Diffuse reflectance UV-vis (DR UV-vis) analysis of the calcined catalysts was performed on a Varian Cary spectrophotometer with BaSO4 used as a reference.
H2-temperature programmed reduction (H2-TPR) was performed on an Altamira Instruments AMI-300 equipped with a thermal conductivity detection (TCD) device. Typically, 100 mg of catalyst was placed in a quartz reactor. Firstly, the sample was pre-treated at 400 °C for 30 min in an Ar flow, followed by cooling to 50 °C. Next, the temperature was raised from 50 °C to 900 °C with a heating rate of 10 °C min−1 in a 10% H2/Ar flow (30 mL min−1). The TCD was used to detect the amount of H2 consumption.
Temperature programmed desorption (TPD) experiments of NH3 were conducted using an Altamira Instruments AMI-300 equipped with a thermal conductivity detection (TCD) device. Approximately 100 mg of catalyst was placed in a quartz reactor.
Firstly, the catalyst was pre-reduced at 390 °C for 1 h under 10% H2/Ar flow. Next, the catalyst was heated under stream of Ar at 600 °C for 30 min then cooled to 50 °C. Further, catalyst was exposed to a flow (30 mL min−1) of 5% NH3/Ar for 2 h. After being purged in Ar for 1 h, the catalyst was heated linearly at 10 °C min−1 to 600 °C in an Ar flow (30 mL min−1).
CO chemisorption experiments were performed at 50 °C using an Altamira Instruments AMI-300 equipped with thermal conductivity detection (TCD) device. Typically, about 100 mg of catalyst was placed in a quartz reactor. Firstly, the catalyst was heated at 400 °C for 30 min in an Ar flow, followed by in situ reduction at 390 or 500 °C for 4 h in a H2 flow. The H2 consumption was detected by TCD. The catalyst was then purged with a He flow of 30 mL min−1, at 400 or 510 °C for 1 h, respectively. The catalyst was cooled to 50 °C and 10% CO/He pulse chemisorption was initiated at 50 °C. The catalyst was then flushed with a He flow of 30 mL min−1 for 30 min at 50 °C. Finally, CO-TPD pattern was recorded in the range of 50–800 °C at a heating rate of 10 °C min−1 in a He flow (30 mL min−1). The area under the TCD curve below the reduction temperature was used for the calculation of chemisorbed CO volume.47,48 The Ni metal surface area, dispersion, and Ni metal crystal sizes were calculated using the equations described by Velu et al.49 As a note, the reduction degrees were determined by the ratios of the amounts of H2 consumption during H2-TPR to the theoretical values calculated by ICP-OES measurements. When calculating the reduction degree of Ni in Ni15–Re2 catalyst, the H2 consumption of Re in the Ni15–Re2 catalyst was supposed to be equal to that in Re2 catalyst at the same reduction condition.
The in situ X-ray photoelectron spectroscopy (XPS) spectra were recorded using Thermo ESCALAB 250Xi X-ray photoelectron spectrometer. A monochromatic X-ray source of Al Kα of 15 kV was applied. Special regions were Al 2p, Ni 2p and Re 4f. The charging effect was adjusted according to the standard Al 2p line at 74.5 eV.50 The peak fitting of Ni 2p3/2, Re 4f7/2, and Al 2p primary peaks were selected for the quantification of surface components. The samples were transferred to the XPS chamber and in situ reduced at 500 °C for 2 h under H2 flow. After each reduction, the XPS chamber was evacuated to about 7.1 × 10−5 Pa and the XPS measurements were sequentially conducted.
The morphology of the catalysts was investigated by a JSM-7800F scanning electron microscopy (SEM) instrument. The segregation of Ni species was characterized by SEM in backscattered electron mode.
Transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), and energy dispersive X-ray (EDX) line scan were acquired with a Tecnai G2 F30 S-Twin electron microscope, operating at 300 kV. The particle size distribution of sample was measured from more than 20 TEM images which obtained at different selected regions.
Materials and catalysts
The details of the reaction method was described in the ESI.† In short, the reductive amination of MEA was carried out in a trickle bed reactor (9 mm inner diameter). About 3.4 mL catalyst (about 2.4 g) was placed in the middle of the reactor and the rest space was filled with about 22 mL silica sand. The raw material consisted of MEA and liquid ammonia. The mole ratio between MEA:NH3 was 1:10. The liquid hourly space velocity (LHSV) of MEA in the reaction was about 0.5 h−1. The H2 flow rate was controlled by mass flowmeter, corresponding to a space velocity of about 60 h−1. Before reaction, the catalysts were in situ reduced by pure H2 for 4 h at either 390 or 500 °C. The reductive amination reactions were conducted at 170 °C and 8 MPa. The liquid phase products were collected at 5–11 h reaction intervals. Then, the samples were quantified by gas chromatograph (Agilent 7890) fitted with a flame ionization detector and a DB-35 capillary column, and N,N-dimethylformamide as an internal standard.
The conversion of MEA and the selectivity of the products were calculated by the equations as follows:

where nMEA,in was the moles of MEA initially added, nMEA,out was the moles of MEA that remained in the products, and ni was the moles amount of compound i in the products.

Results and discussion
N2 adsorption–desorption
The BET equation was conducted to inspect the porous nature of Al2O3 and Ni-based catalysts. Fig. 2 shows the N2 adsorption–desorption isotherms of calcined Al2O3 support and catalysts. All the isotherms were identified as type IV, which was characteristic of mesoporous materials. The isotherms exhibited type H4 hysteresis loops, indicating the existence of irregular channel systems. The structural properties of support and catalysts, such as surface area, pore volume, and average pore diameter, are shown in Table 1. The BET specific surface area of Al2O3 support was calculated from N2 isotherms at −196.6 °C, and was measured as 388.1 m2 g−1. After modified with Ni or Re, obvious decrease in surface area and pore volume was observed. For Ni–Re doped catalysts, surface area values were in the range of 231–268 m2 g−1. The reduction of surface area and pore volume after impregnation may be caused by the partial blockage of mesoporous structure by metal species.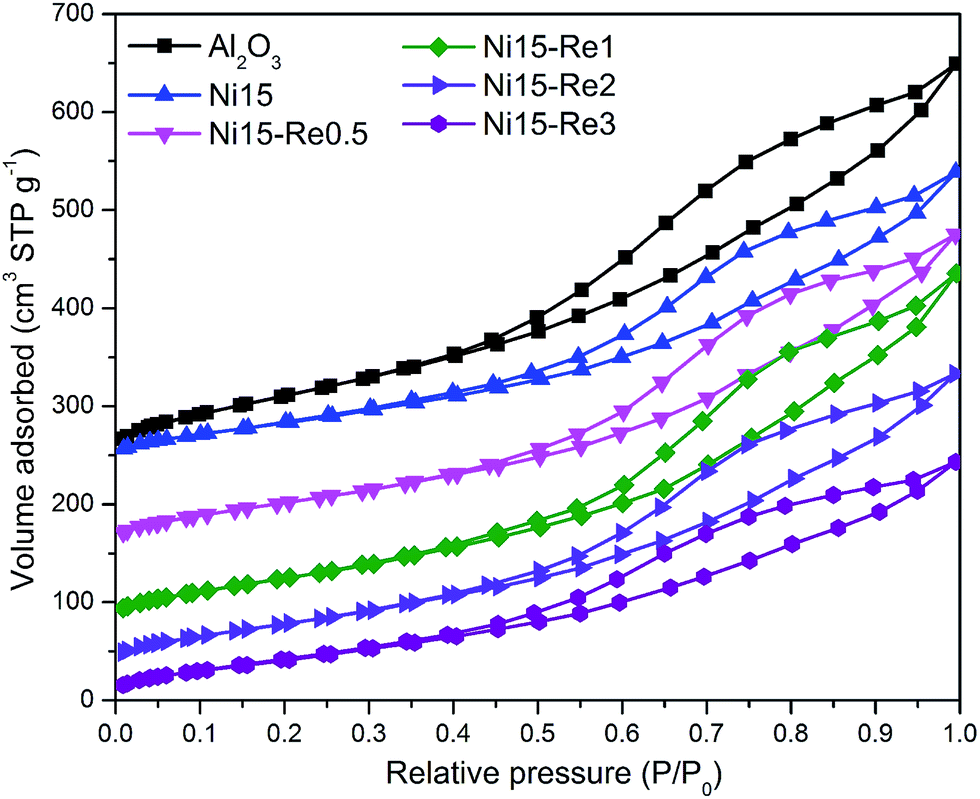 | ||
| Fig. 2 N2 adsorption–desorption isotherms of Al2O3 support, Ni15, Ni15–Re0.5, Ni15–Re1, Ni15–Re2, and Ni15–Re3 catalysts. | ||
| Catalyst | Surface areaa (m2 g−1) | Pore volumeb (mL g−1) | Pore diameterc (nm) | Ni loadingd (wt%) | Re loadingd (wt%) |
|---|---|---|---|---|---|
| a Calculated by BET equation.b BJH desorption pore volume.c BJH desorption average pore volume.d Obtained from ICP-OES results. | |||||
| Al2O3 | 388.1 | 0.690 | 5.6 | — | — |
| Ni15 | 229.7 | 0.482 | 6.5 | 12.5 | — |
| Ni15–Re0.5 | 268.1 | 0.536 | 5.6 | 12.3 | 0.3 |
| Ni15–Re1 | 256.4 | 0.572 | 5.7 | 12.2 | 0.7 |
| Ni15–Re2 | 238.3 | 0.481 | 6.5 | 11.7 | 1.4 |
| Ni15–Re3 | 231.6 | 0.408 | 5.5 | 11.5 | 2.2 |
XRD
X-ray diffraction experiments (XRD) analysis technique was performed to identify the crystal structure of Al2O3 supported Ni and Ni–Re catalysts. As Ni–Re catalysts exhibited similar spectra (Fi. S1†) after calcination, only Ni15 and Ni15–Re2 samples (Fig. 3) were chosen to analyze the crystal structure. Meanwhile, the spectrum of Al2O3 support was also shown for comparison. For Al2O3 support, the XRD pattern showed the diffraction peaks at 2θ = 37.6°, 39.5°, 45.9°, and 67.0°, corresponding to (311), (222), (400), and (440) crystal planes of γ-Al2O3 with low crystallinity (JCPDS 29-0063), respectively. For Ni15 catalyst, only weak diffraction line at 2θ = 42.3° originating from bulk NiO (200) crystal plane (JCPDS 44-1159) was detected. In contrast, the NiO diffraction peak at 2θ value of 42.3° was not observed on Ni15–Re2 sample, implying the good dispersion of Ni species on the Al2O3 surface. Moreover, for Ni15 and Ni15–Re2 catalysts, the broad peak at 66.5°, which was between the values of 2θ = 67.0° for γ-Al2O3 and 2θ = 65.5° for stoichiometric NiAl2O4 spinel (JCPDS 10-0339), can be indexed to non-stoichiometric NiAl2O4 phase on the catalysts. It has been reported that Ni species have a tendency to preserve monolayer dispersion and form surface NiAl2O4-like species on the Al2O3 support until the content of Ni is higher than the threshold coverage.51–54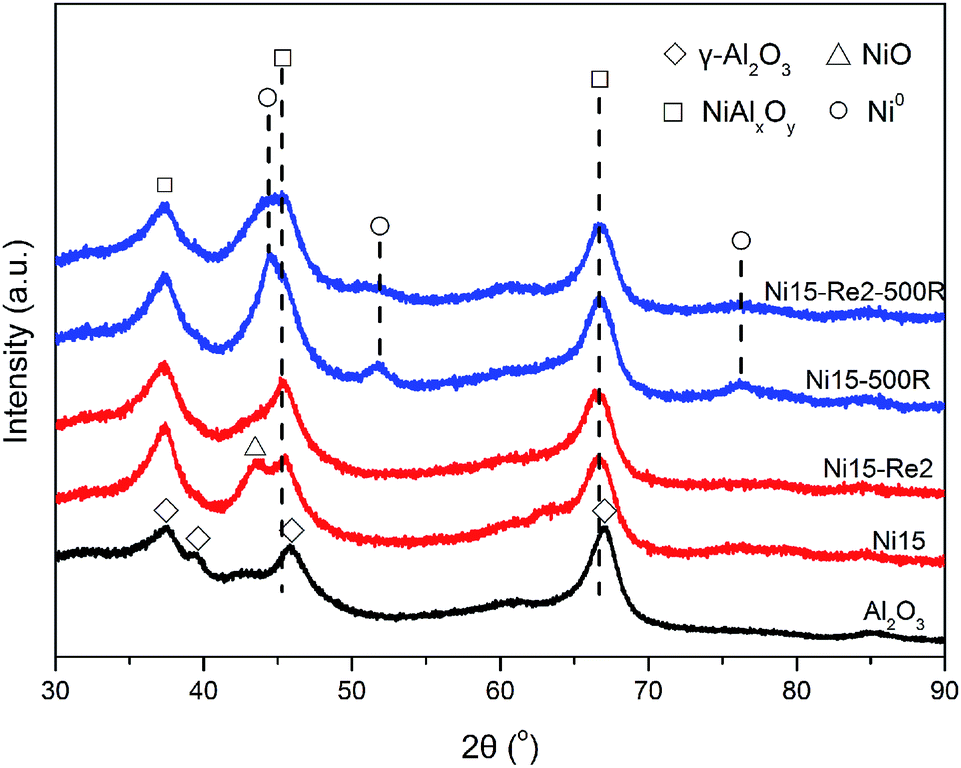 | ||
| Fig. 3 XRD patterns of Al2O3 support, calcined and 500 °C reduced Ni15, Ni15–Re2 catalysts. | ||
The XRD patterns of 390 °C reduced Ni and Ni–Re catalysts are shown in Fig. S1.† There were no significant changes in the diffraction patterns as compared with the calcined samples. The metallic Ni diffraction peaks were not observed, ascribed to the low crystallinity, low reduction degree, or/and small particle size of metallic Ni on the support. For 500 °C reduced Ni15 and Ni15–Re2 catalysts (Fig. 3), diffraction peaks were observed at 2θ = 44.3°, 51.7°, and 76.1°, corresponding to (111), (200), and (220) crystal planes of metallic Ni (JCPDS 04-0850), respectively. With the use of Scherer's formula, average metallic Ni size of 6.2 nm was found for the Ni15–500R catalyst. However, for Ni15–Re2–500R sample, the intensity of the metallic Ni peaks was too weak to calculate the metal size, suggesting the high dispersion of metallic Ni phase. Moreover, the diffraction peaks of Re were absent in all XRD patterns of Ni–Re catalysts, because of its lower loading content and weak crystallization, on the other hand, also implying the good dispersion of Re species. On the basis of the analysis results, it could be concluded that the introduction of Re effectively improves the dispersion of Ni species on the γ-Al2O3 surface after calcination and reduction treatments.
DR UV-Vis spectroscopy
Diffuse reflectance ultraviolet-visible (DR UV-Vis) spectroscopy was applied to research the symmetry and coordination of Ni species in the catalysts. The reflectance spectra are shown in Fig. 4. It can be observed that the Al2O3 and Re2 samples showed no absorption bands in the range of 350–800 nm, while five bands can be observed from the spectrum of the Ni15 catalyst. The band appearing around 377 nm, ascribed to the d–d electron transitions in octahedral Ni2+ (Ni[O]), was a fingerprint for NiO lattice.51 The band at 410 nm was attributed to Ni[O] in surface NiAl2O4-like spinel.55 The adsorption bands in the region 550–650 nm as a doublet with maxima at 592 and 638 nm were assigned to the electron transitions of tetrahedral Ni2+ (Ni[T]) in bulk NiAl2O4 lattice. The adsorption band around 713 nm could be assigned to both d–d electron transition of Ni[O] in NiO and Ni[T] in the NiAl2O4 lattice.55 When Re was incorporated to the Ni-based catalysts, an increase in the intensity of adsorption bands around 377 and 410 nm was observed. Meanwhile, the intensity of the bands at 592, 638, and 713 nm decreased with increase of Re content. The DR UV-Vis results indicated that more NiO and surface NiAl2O4-like spinel were generated and the proportion of bulk NiAl2O4 was reduced on the Ni–Re catalysts. Since the NiO diffraction peaks were not observed for the Ni–Re catalysts in the XRD patterns (Fig. 3), it was reasonable to conclude that the NiO species were highly dispersed on the Re promoted Ni–Re catalysts.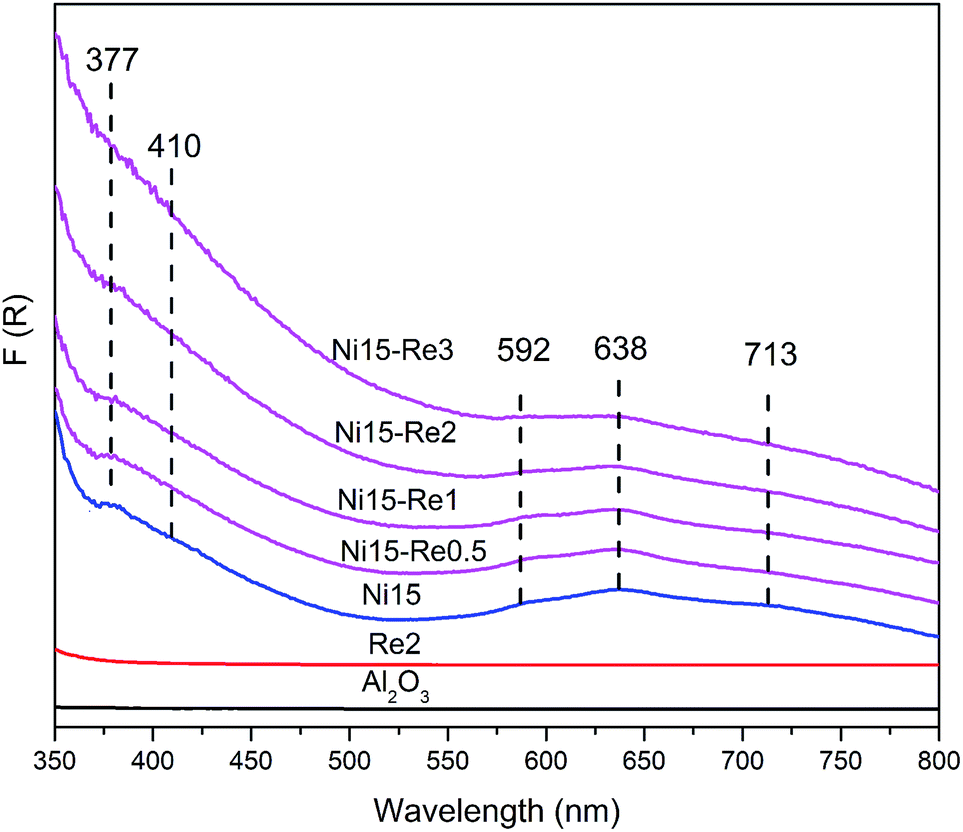 | ||
| Fig. 4 DR UV-Vis spectra of Al2O3 support, Re2, Ni15, Ni15–Re0.5, Ni15–Re1, Ni15–Re2, and Ni15–Re3 catalysts. | ||
To further analyze the proportion of Ni[O] and Ni[T] in the catalysts, the variation of the Kubelka–Munk function ratio of F(R377)/F(R638) and F(R410)/F(R638) with the Re mass content for Ni and Ni–Re catalysts were plotted as a semi-quantitative method. As results shown in Fig. 5, the ratio of F(R377)/F(R638) and F(R410)/F(R638) increased with Re content up to about 1.4 wt%, and started to level off with more Re loading. This result indicates that the addition of Re promoter facilitates the conversion of Ni[T] to Ni[O]. In addition, Ni[O] ions were reported to be reduced more easily than Ni[T].56 The increase of Ni[O] proportion may enhance the reducibility of Ni2+ in Ni–Re catalysts. In conclusion, the addition of Re to Ni catalysts generates more Ni[O] species and improves the proportion of NiO and surface NiAl2O4-like spinel.
 | ||
| Fig. 5 Variation of the F(R) ratio (the ratio calculated from the Kubelka–Munk functions intensities of the band at 377, 410, and 638 nm) with the Re mass content (ICP-OES results) for Ni and Ni–Re catalysts. | ||
H2-TPR
The influence of Re on the reducibility of Ni–Re/Al2O3 catalysts was analyzed by H2-TPR measurements and the profiles are shown in Fig. 6. The corresponding H2 consumptions and reduction degrees are listed in Table S1.† For the Ni15 catalyst, three regions of reduction peaks can be observed. The first range at about 350–470 °C can be assigned to the surface bulk-like NiO species which was weakly interacted with Al2O3 support; the second range around 470–725 °C could be ascribed to the reduction of surface NiAl2O4-like spinel, which was highly dispersed on Al2O3 support; the third range above 725 °C was attributed to the reduction of bulk NiAl2O4 spinel, which had a strong interaction with Al2O3 support and difficult to be reduced.51,57 The reduction degree of Ni15 was 92%. | ||
| Fig. 6 H2-TPR profiles of Re2, Ni15, Ni15–Re0.5, Ni15–Re1, Ni15–Re2, and Ni15–Re3 catalysts. | ||
The reduction profile of Re2 sample displayed two reduction peaks at 367 °C and 482 °C, similar to the results reported by Bare et al.53 The two step reduction of Re2 may be involved in the reduction of different Re valences. The reduction degree of Re2 was calculated to be 85%, indicating rhenium oxide species were incomplete reduced. The average valence of Re was calculated to be about +1. The H2-TPR patterns of Ni–Re catalysts were different from the Ni15 catalyst. The peaks below 470 °C, which could be assigned to reduction of bulk-like NiO or Ni–Re oxide species, shifted to lower temperature and became much stronger as the loading of Re increased from 0.5 to 3 wt%. Moreover, the reduction peaks belonged to surface NiAl2O4-like species (470–725 °C) were shifted toward lower temperature as the increase of Re loading, indicating the addition of Re promoter decreased the reduction temperature of Ni–Re catalysts. Furthermore, as the increase of Re loading, the peaks related to the reduction of bulk NiAl2O4 above 725 °C became much weaker, indicating that the addition of Re can prevent the diffusion of Ni2+ into the Al2O3 lattice to form NiAl2O4 specie, which is hardly reduced and inert for the amination reaction.21 The reduction degrees for Ni–Re/Al2O3 catalysts were around 95%. In conclusion, H2-TPR studies suggest that the introduction of Re significantly improves the reducibility of Ni–Re catalysts and inhibits the formation of bulk NiAl2O4 species.
Acid–base properties
As both the acidic and basic nature of the catalysts were reported to be necessary for the reductive amination reaction,18,19 NH3-TPD, CO2-TPD, and PY-IR experiments were conducted to investigate the acid–base character of catalysts. The NH3-TPD profiles are shown in Fig. 7. The spectrum of Al2O3 support showed a doublet at 120 and 170 °C. The peak at 120 °C could be attributed to desorption of residual physically absorbed NH3. The second peak at 170 °C was assigned to chemisorb NH3 from weak acidic sites. For Al2O3 supported Re sample, desorption peak at 170 °C shifted up to about 225 °C, indicating the presence of Re enhanced the acid strength of Al2O3 support. The NH3-TPD profile of Ni15 catalyst was similar to the Al2O3 support profile. After modified with Re, Ni–Re catalysts exhibited a broad peak in the range of 225–370 °C, indicating the introduction of Re improves the acid strength of Ni–Re catalysts. Moreover, the intensity of desorption peak around 170 °C increases with the increase of Re loading in Ni–Re catalysts. | ||
| Fig. 7 NH3-TPD profiles of Al2O3 support, Ni15, Re2, Ni15–Re0.5, Ni15–Re1, Ni15–Re2, and Ni15–Re3 catalysts. | ||
The pyridine adsorption infrared (IR) spectra (Fig. S2†) demonstrate that Al2O3 support mainly contains surface Lewis acid sites. The impregnation of Ni or Re to the Al2O3 support doesn't affect the type of acidic centers. Furthermore, the basic contents measured from the CO2-TPD results (Fig. S3 and Table S2†) indicate that few basic sites exist on the Al2O3 support and the impregnation of Ni or Re species has little impact on the base character of the catalysts.
In situ XPS
In situ X-ray photoelectron spectroscopy (XPS) was used to analyze the valance state of the elements and the surface composition in the 500 °C reduced Ni15 and Ni15–Re2 catalysts. Fig. 8 presents the XPS spectra of Ni 2p and Re 4f regions from the Ni15–500R and Ni15–Re2–500R samples. The curve fitting results of Ni 2p and Re 4f7/2 are listed in Table 2. The Ni15–500R catalyst had two main peaks at 853.0 and 870.4 eV, corresponding to the Ni0 2p3/2 and Ni0 2p1/2, respectively. Moreover, the presence of peaks at 856.5 and 874.1 eV, respectively corresponding to the BEs of Ni2+ 2p3/2 and Ni2+ 2p1/2, indicating the Ni species in the Ni15 catalyst were partially reduced. The peaks at 862.7 eV and 881.2 eV were ascribed to the shake-up satellite peaks of Ni2+ 2p3/2 and Ni2+ 2p1/2, respectively.50,51,59 It was notable that the BE of Ni2+ 2p3/2 at 856.5 eV was higher than that of published bulk NiO species (around 854.5 eV), indicating the presence of NiAl2O4 species on the catalyst surface.50 For Ni15–Re2–500R catalyst, the peak intensity of Ni0 2p3/2 and Ni0 2p1/2 was higher than the case in Ni15–500R catalyst. According to the calculation results in Table 2, the reduction degree of Ni15–500R catalyst was 29.1%, while that of Ni15–Re2–500R catalyst increased to 39.1%, indicating the presence of Re enhanced the reducibility of Ni species. Furthermore, from curve fitting results, the BE of Ni0 2p3/2 in Ni15–Re2–500R was 0.3 eV higher than that in Ni15–500R catalyst. Chayakul et al. have reported that d-band electron densities of Ni could be reduced by the electron transformation from Ni to Re.58 The shift in BEs of Ni0 2p3/2 when comparing Ni15–500R and Ni15–Re2–500R provided evidence of an interaction between Ni to Re species. In addition, the surface Ni/Al ratios of catalysts were in the range of 0.0678 to 0.0725, which were lower than the theoretical value of 0.122, because of the incorporation of Ni2+ into the Al2O3 lattice by the strong interaction between the surface Ni and Al2O3 support. In conclusion, the XPS results proved that the addition of Re promotes the reduction of Ni2+ compounds and electron interaction existed between Ni and Re. | ||
| Fig. 8 In situ XPS of (1) Ni 2p region for Ni15–500R and Ni15–Re2–500R catalysts, and (2) Re 4f region for Ni15–Re2–500R catalyst. The dash lines in the spectra correspond to the Ni 2P or Re 4f7/2 peaks of different valance states. | ||
| Catalyst | Ni 2P3/2a (eV) | Ni 2P1/2a (eV) | Atomic ratio Ni/Al | Re 4f7/2a (eV) | Atomic ratio Re/Ni | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ni2+ | Ni0 | Ni2+ | Ni0 | Re0 | Re3+ | Re4+ | Re7+ | |||
| a BEs of Ni 2P or Re 4f7/2 species. The percentages of Ni or Re with different valance states are shown in parentheses. | ||||||||||
| Ni15–500R | 856.5 (70.9) | 853.0 (29.1) | 874.1 | 870.4 | 0.0725 | — | — | — | — | — |
| Ni15–Re2–500R | 856.5 (60.9) | 853.3 (39.1) | 874.2 | 870.6 | 0.0678 | 40.5 (4.6) | 41.3 (25.1) | 43.1 (21.0) | 46.5 (49.3) | 0.0836 |
The XPS spectrum of Re 4f for the Ni15–Re2–500R catalyst is shown in Fig. 8(2). The separation of the spin–orbit Re 4f7/2–4f5/2 doublet were fixed for the curve fitting of Re spectra. Four valance states of Re were selected to get the best fitting. The deconvoluted peaks at 40.5, 41.3, 43.1, and 46.5 eV were corresponded to the 4f7/2 BEs of Re0, Re3+, Re4+, and Re7+ components, respectively.60–62 The concentration of Rex+ species was calculated by the intensity of Re 4f7/2 and shown in Table 2. The reduction degree and average valence of Re were calculated to be about 30% and +5, respectively. The results indicated that the major Re species existed as a mixture of various oxidation states after reduction at 500 °C. These multiple oxidation states after reduction might arise from the strong oxophilicity of Re species. Bare et al. have studied the oxidized and reduced states of Re over Re/Al2O3 using a combination of X-ray absorption fine structure (XAFS) and theoretical approaches.53 They concluded that the oxidized Re species still existed even after reduction at 700 °C under dry H2. Furthermore, for Ni15–Re2–500R catalyst, the surface atomic ratio of Re/Ni was found to be 0.0836, which was much higher than the theoretical value calculated by ICP-OES (0.0377). This result indicates the enrichment of Re species on the surface of Ni–Re/Al2O3 catalysts.
BSE-SEM and CO-adsorption
The dispersion of metal particles for the reduced Ni15 and Ni15–Re2 catalysts at different temperature were further examined by SEM in backscattered electron (BSE) image mode. The images are shown in Fig. 9. It should be noted that BSE image was used to detect differences in the atomic number within the catalysts. The contrast between particles and support was mainly linked to the atomic masses or atomic numbers of the corresponding elements. For the Ni15–390R sample (Fig. 9(1)), Ni containing particles with an average size of about 8 nm were observed on the surface of catalyst. When the reduction temperature was increased to 500 °C (Fig. 9(2)), more Ni particles were detected on the sample and the particle sizes slightly increased to about 10 nm, indicating the aggregation of Ni under higher reduction temperature. For Re promoted Ni15–Re2 catalyst (Fig. 9(3 and 4)), the particle sizes were mostly around 4 and 6 nm after reduced at 390 and 500 °C, respectively. The particle sizes of reduced Ni15–Re2 catalysts were much smaller than those of reduced Ni15 catalysts, indicating the addition of Re promoter effectively improved the dispersion of Ni after reduction.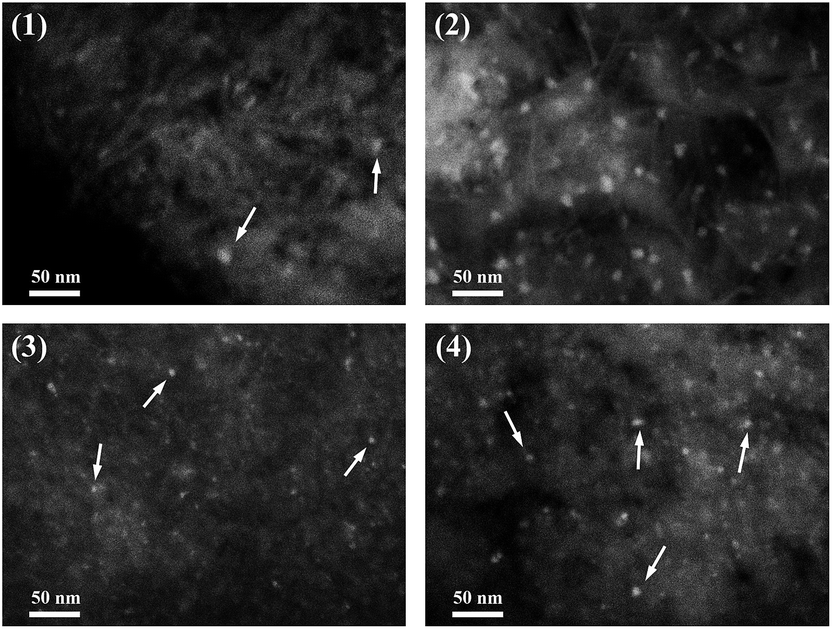 | ||
| Fig. 9 BSE-SEM images of the reduced catalysts: (1) Ni15–390R, (2) Ni15–500R, (3) Ni15–Re2–390R, and (4) Ni15–Re2–500R. | ||
For supported Ni catalysts, metallic Ni sites had proved to be active species for the dehydrogenation–hydrogenation steps. CO-TPD measurement was applied to study the influence of Re promoter on the metallic Ni site, metal dispersion, and average Ni particle size of the Ni based catalyst. Ni15 and Ni15–Re2 catalysts were chosen for comparison. The CO uptakes and analogy computation results, corrected by the reduction degrees, are summarized in Table 3. The CO uptake results indicated that a trace amount CO was adsorbed on the surface of Re2 catalyst. Moreover, since CO adsorption infrared (IR) spectra (Fig. S4†) proved that major CO species were linearly adsorbed on the surface of Ni0 sites, the Ni:CO ratio in chemisorption was taken as 1 to calculate the Ni0 surface area.47 As shown in Table 3, after modified with Re promoter, the Ni0 surface areas of the 390 and 500 °C reduced catalysts increased from 0.7 to 3.0 m2 gcat.−1 and 2.2 to 4.9 m2 gcat.−1, respectively. Furthermore, Re promoted Ni15–Re2 catalyst showed higher dispersion and smaller particle size in comparison with the monometallic Ni15 catalyst after reduction.
| Catalyst | Reduction temperature (°C) | Reduction degree (%) | CO uptake (μmol gcat.−1) | Ni0 surface area (m2 gcat.−1) | Ni0 dispersion (%) | Ni0 crystal size (nm) |
|---|---|---|---|---|---|---|
| Ni15 | 390 | Ni-8.8 | 17.4 | 0.7 | 9.3 | 19.4 |
| 500 | Ni-27.3 | 56.1 | 2.2 | 9.6 | 18.7 | |
| Ni15–Re2 | 390 | Ni-24.8 | 77.6 | 3.0 | 15.7 | 11.5 |
| 500 | Ni-50.9 | 125.3 | 4.9 | 12.4 | 14.5 | |
| Re2 | 390 | Re-47.7 | — | — | — | — |
| 500 | Re-72.8 | — | — | — | — |
It should be noted that the Ni0 particle sizes obtained from previous XRD or BSE-SEM methods were smaller than those calculated from CO uptakes. This difference might arise from the decoration of Ni particles with NiAl2O4 species after reduction. The aluminate shells on the surface of metallic Ni particles might prevent the adsorption of CO gas, resulting in low CO uptake. Salagre et al. have concluded that Ni crystallites surface would be decorated with porous NiAl2O4 species during reduction process.63 A similar model of NiAl2O4 structure after reduction was proposed by Jerzy earlier.64 This speculation was further supported by the XRD, H2-TPR, and XPS analyses which showed that Ni species mainly existed as surface NiAl2O4 spinel in the catalyst. According to the CO-adsorption results, the addition of Re in the Ni based catalysts significantly improves the Ni dispersion and generates more Ni0 sites after reduction.
Reductive amination of MEA
Reductive amination of MEA was carried out in a trickle bed reactor. The amine products collected after reaction about 12 h were analyzed and summarized in Table 4. Interestingly, it was found that Ni15–Rex catalysts could efficiently catalyze the reductive amination of MEA to EAD. The conversion and EDA selectivity increased to 36.6% and 63.0% (entry 5 in Table 4) with Re content up 2 wt% (Ni–15–Re2–390R), and started to level off with more Re loading (entry 6 in Table 4). In comparison, a very low conversion (7.6%) and EDA selectivity (40.0%) were observed on Ni15–390R catalyst (entry 2 in Table 4), and no detectable activity was observed on Re2–390R catalyst under the same reaction conditions (entry 1 in Table 4). The big difference between Ni15–Rex catalysts and Ni15 catalyst revealed that the presence of Re was critical for reductive amination. The addition of Re not only significantly improved the activity of Ni-based catalysts but also enhanced the selectivity of desired amines, such as EDA and PIP. In consideration of many parallel reactions during reductive amination processes of MEA, the enhancement of the EDA yield was essential to avoid the difficulty during products separating.| Entry | Catalyst | Conversion (%) | Selectivity (%) | ||||
|---|---|---|---|---|---|---|---|
| EDAa | AEEAb | PIPc | DETAd | Otherse | |||
| a Ethylenediamine.b Aminoethylethanolamine.c Piperazine.d Diethylenetriamine.e N-(2-Hydroxyethyl)piperazine and N-Aminoethylpiperazine.f Patent (US 5750790):26 170 °C, 12.5 MPa, MEA:NH3 = 1:10.g Patent (US 6534441 B1):27 160 °C, 20.0 MPa, MEA:NH3 = 1:10.h The pilot-scale results of our Ni–Re/Al2O3 catalysts: 175 °C, 17.5 MPa, MEA:NH3 = 1:10. |
|||||||
| 1 | Re2–390R | — | — | — | — | — | — |
| 2 | Ni15–390R | 7.6 | 40.0 | 56.2 | 3.2 | 0.0 | 0.6 |
| 3 | Ni15–Re0.5–390R | 22.3 | 55.6 | 32.2 | 5.8 | 4.3 | 2.1 |
| 4 | Ni15–Re1–390R | 27.6 | 60.3 | 26.2 | 6.4 | 4.6 | 2.5 |
| 5 | Ni15–Re2–390R | 36.6 | 63.0 | 20.4 | 8.1 | 5.7 | 2.8 |
| 6 | Ni15–Re3–390R | 37.6 | 63.6 | 19.2 | 8.9 | 5.2 | 3.1 |
| 7 | Ni15–500R | 14.5 | 46.3 | 37.4 | 8.5 | 5.1 | 2.7 |
| 8 | Ni15–Re2–500R | 43.2 | 67.7 | 16.4 | 8.2 | 5.5 | 2.2 |
| 9 | Ni–Re/Al2O3f | 45.4 | 69.1 | 12.9 | 6.6 | 10.5 | 0.9 |
| 10 | Ni–Re/SiO2–Al2O3g | 53.0 | 82.4 | 9.8 | 1.1 | 5.9 | 0.8 |
| 11 | Ni15–Re/Al2O3h | 50.4 | 82.1 | 4.4 | 5.1 | 7.7 | 0.8 |
The TOF of Ni–Re catalysts (Table S3†) with varied Re loading were estimated for reference according to the method reported by Shimizu.19 The TOF increases from 213 to 251 h−1 with increasing the Re loading from 0.5 to 2 wt% and starts to level off with further increasing Re loading.
In order to compare the activity of Ni15 and Ni15–Re2 catalysts further, we increased the reduction temperature of Ni15 and Ni15–Re2 catalysts from 390 to 500 °C and the results are list in Table 4. The MEA conversion for the Ni15–500R catalyst was still low and product distribution changed little (entry 7 in Table 4). In contrast, the yield of EDA was further improved over the Ni15–Re2–500R catalyst (entry 8 in Table 4). Fig. 10 shows the stability of Ni15–500R and Ni15–Re2–500R catalysts. The results showed that the increase in reduction temperature could increase the initial activity of catalysts. However, for Ni15–500R catalyst, the MEA conversion decreased rapidly with reaction time and nearly deactivated within 23 h. The EDA selectivity declined and the by-product AEEA significantly increased as the activity dropped. For Ni15–Re2–500R catalysts, a slightly decrease of conversion was also observed, but the catalysts performance could reach steady state and maintain a higher activity. The above results indicate that the addition of Re not only improves the yield of EDA but also inhibits the deactivation during the reductive amination process. In combination with the previously CO chemisorption results, the highly Ni0 surface area could correspond to the enhancement in the yield of EDA.
 | ||
| Fig. 10 Stability of (1) Ni15–500R and (2) Ni15–Re2–500R catalysts. | ||
Currently, the reports about MEA reductive amination were presented mainly in the patent literatures.26,27,65 The reaction results of Ni–Re catalysts reported by others were listed in Table 4 (entry 9 and 10). It should be noted that most of patent reported experiments and industrial processes were carried out under high reaction pressure (12–25 MPa). Fischer et al. have reported that ammonia would form a supercritical fluid (critical data of ammonia: Tc = 132.4 °C, Pc = 11.48 MPa) in a fix-bed reactor under high pressure.9,10 The formation of supercritical ammonia is crucial to improve the selectivity of primary diamines. Unfortunately, our reaction pressure should not exceed 8 MPa due to the limitations of experimental conditions. In our subsequent pilot-scale study (entry 11), 50.4 percent conversion could be achieved with a selectivity of 82.1 percent for EDA at 175 °C under 17.5 MPa.
Characterization of the spent catalysts
XRD technique was used to investigate the crystalline structure and morphology of the spent Ni15–500R and Ni15–Re2–500R catalysts. XRD patterns are shown in Fig. 11. Both the spent catalysts exhibited obvious diffraction lines at 2θ = 44.3°, 51.7°, and 76.1°, characteristic of the metallic Ni phase. Average Ni0 particle sizes of 21.3 and 15.8 nm were found for the spent Ni15–500R and Ni15–Re2–500R catalysts with the use of Scherrer's equation, respectively. Combined with the previous XRD patterns in Fig. 3, it was clear that aggregation and growth of Ni0 particles occurred during the reductive amination process. | ||
| Fig. 11 XRD patterns of spent Ni15–500R and Ni15–Re2–500R catalysts. | ||
The TEM micrographs and size distributions of Ni0 particles of spent catalysts are shown in Fig. 12. It was found that Ni particles for the spent Ni15–500R catalyst (Fig. 12(1 and 2)) were heterogeneously dispersed and had a broad distribution of 40 to 550 nm. In addition, large Ni particles with sizes above 1 μm were often observed from the TEM images at some selected regions (Fig. S5†). The TEM results clearly indicated that serious sintering of Ni0 particles happened during reaction for Ni15 catalyst. On the contrary, the Ni0 sintering process was effectively suppressed in the presence of Re promoter. For spent Ni15–Re2–500R catalyst (Fig. 12(3 and 4)), Ni0 particles more uniformly dispersed and the sizes of Ni0 particles densely distributed in a range of 10–60 nm. One should be noted that the difference between the XRD and TEM results was due to the low crystalline of Ni0 or the sizes of metal particles beyond the applicable scope of Scherrer's equation. The elemental distribution of Ni and Re for spent Ni15–Re2–500R catalyst was examined by STEM-EDX line scan. As shown in Fig. 12(5 and 6), similar trend was observed for the Ni and Re elements in the spent catalysts. The EDX line scan results suggest that the surface of Ni0 particles is rich in Re species.
 | ||
| Fig. 12 TEM images and particle size distributions of: (1) and (2) spent Ni15–500R; (3) and (4) spent Ni15–Re2–500R. (5) STEM image of spent Ni15–Re2–500R, and (6) EDX line scans across a large particle as dedicated by the red line. | ||
Discussion
As indicated by characterization results, the addition of Re promoter affects the structure and activity of Ni/Al2O3 catalysts during preparation process and MEA reductive amination. For calcined catalysts, XRD and DR UV-Vis results indicate that most of Ni species existed in the form of non-stoichiometric NiAl2O4 phase in Ni/Al2O3 catalyst. The presence of bulk NiO phase is also detected by XRD. The introduction of Re effectively enhances the NiO dispersion and facilitates the generation of surface Ni[O] species in Ni–Re/Al2O3 catalysts.After reduction, H2-TPR and in situ XPS results show that Re promoter significantly improves the reducibility of Ni. Moreover, as indicated by BSE-SEM and CO-adsorption results, Re promoted Ni/Al2O3 catalysts have smaller Ni0 particle size and more surface Ni0 sites compared with monometallic Ni/Al2O3 catalyst. The enhancement of the Ni reducibility could be explained by two reasons. Firstly, as indicated by DR UV-Vis results, the increase of surface NiO species and Ni[O] proportion for Ni–Re catalysts makes the Ni2+ species more easily to be reduced; secondly, the reduction of Ni2+ may be accelerated by hydrogen spillover effects on Re atoms. It has been reported that Re could promote the reduction of Co2+ or Ce4+ species by hydrogen spillover.59,66 Moreover, both H2-TPR and in situ XPS results indicate that Re species are incomplete reduced and exhibit a mixture of various oxidation states after reduction. Furthermore, the acidity–basicity measurement results suggest that the addition of Re improves the acid strength of catalysts. The generation of new acid sites in bimetallic catalysts comprised of noble metals and oxophilic metal promoters has been reported in recent years.67,68 Some research reported that the acid–base pair sites adjacent to the Ni0 sites assisted important steps such as deprotonation of alcohol or hydrogen transfer.18,19 The enhancement of acidic strength might accelerate the deprotonation of MEA or hydrogen transfer steps.
The characterization results of spent catalysts indicate that Ni/Al2O3 catalyst suffers from serious Ni sintering during reaction. The sintering of Ni particles on Al2O3 support in the presence of NH3 and H2 has been reported by Lif et al.69 They concluded that the presence of NH3 accelerates the rate of sintering by weakening the interaction between Ni particle and Al2O3 support. For Re modified Ni–Re/Al2O3 catalysts, the Ni sintering process could be effectively suppressed. Moreover, monometallic Re2 catalyst has proved to be inactive during reaction. The reductive amination process is mainly catalyzed by Ni0 sites. In combination with the reaction and characterization results of spent catalysts, the decline of activity could be ascribed to the sintering of Ni particles during reaction. As shown in Fig. 10(1) (Ni–500R), the MEA conversion drops by a dramatic 86 percent after reaction for about 30 h, compared to the sampling result at 2 h. Meanwhile, serious Ni sintering is observed from the TEM images of spent Ni–500R sample. For Ni15–Re2–500R catalyst (Fig. 10(2)), the MEA conversion decreases from 54% (2 h) to 38% (28 h) with the growth in Ni particle size. Even more interestingly, for both catalysts, the selectivity of products changes along with the catalyst activity. In the initial period, the selectivities of EDA and PIP are higher and selectivity of by-product AEEA is less than 10%. As the MEA conversion decreases to about 36%, the PIP selectivity decreased and more AEEA generated. Along with further decrease of activity, a sharp rise in AEEA selectivity is detected and EDA selectivity decreases to about only 38%. This phenomenon could be explained by the continuous reaction during MEA reductive amination. As shown in Fig. 1, PIP is mainly generated from the amination of AEEA. For high active catalysts, there are sufficient Ni0 sites on the surface of catalysts and AEEA would proceed to generate PIP. With sintering of Ni particles during reaction, the amount of surface Ni0 sites would decline and AEEA selectivity starts to increase. Moreover, competitive reaction between MEA of NH3 and MEA molecules is in existence during reductive amination and the generation of side product AEEA is inevitable. The reaction results indicate that highly active Ni–Re/Al2O3 catalyst is beneficial to enhance the selectivities of EDA and PIP and reduce the generation of by-product AEEA.
The interaction between Ni and Re has been confirmed in recent years. Liu et al. studied the TiO2 supported Ni–Re catalysts for selective hydrogenation of methyl esters.70 Their XPS results suggested that surface enrichment of metallic Re species in bimetallic Ni–Re catalysts caused the enhancement of activity. Gebrestadik et al. investigated ReOx modified Ni catalysts for the hydrogenolysis of glycidol.71 According to the XPS results, they concluded that Re was decorated by Ni and the collaborative effect between ReOx and Ni particles was beneficial for the production of 1,3-PD. Yang et al. also reported the formation of surface Ni–Re alloy on the Ni–Re/SiO2 catalysts.67 Furthermore, they suggested that oxophilic Re played a crucial role for the adsorption of O atom during reaction. In our research, the XPS and STEM-EDX line scan results indicated the electron transformation from Ni to Re and the enrichment of Re species on the Ni0 particles surface. It was reasonable to speculate that surface of Ni0 particles was decorated with ReOx species. The collaboration effect between Ni–ReOx may has an influence on the mechanism of MEA reductive amination. This speculation will be studied in our further study.
Conclusions
The Al2O3 supported Ni and Ni–Re catalysts were prepared by impregnation method and evaluated for the reductive amination of MEA in a trickle bed reactor. The Ni–Re/Al2O3 catalysts showed high activity, high EDA selectivity, and outstanding stability in comparison with the monometallic Ni/Al2O3 catalyst. The addition of Re promoter improved Ni dispersion and proportion of Ni[O] species on the surface of Ni–Re/Al2O3 catalysts. The reducibility of Ni species was significantly improved by the Re promoter. After reduction, Ni–Re/Al2O3 catalysts exhibited smaller Ni particle size (4–6 nm) and more Ni0 sites as compared to Ni/Al2O3 catalysts. Moreover, the presence of Re improved the acid strength of catalysts. In situ XPS and STEM-EDX line scan results indicated that Re species had a mixture of various oxidation states and tended to aggregate on the surface of Ni0 particles after reduction. The adjacent Ni0–ReOx sites might facilitate the dehydrogenation step of MEA. The analysis results indicated that the deactivation of Ni/Al2O3 catalyst was mainly caused by the aggregation of Ni0 particles during reaction. For Ni–Re/Al2O3 catalysts, the Ni0 particles were stabilized and sintering process was effectively suppressed by the presence of Re promoter. We concluded that the abundant Ni0 sites, which combined with good stability of Ni0 particles and synergic effect of Ni–Re, corresponded to the excellent catalytic performance of Ni–Re/Al2O3 catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21273227) and Strategic Priority Research Program of Chinese Academy of Sciences, Grant No. XDB17000000.Notes and references
- G. F. Mackenzie, US Pat., 2 861 995, The Dow Chemical Company, 1958
.
- F. Weiss and R. Lichtenberger, US Pat., 3 068 290, Societe d'Electro-Chimie d'Electro-Metallurgie et des Acieries Electriques d'Ugine, 1962
- P. H. Moss and N. B. Godfrey, US Pat., 3 151 115, Jefferson Chemical Company, 1964
- J. Johansson and J. Tornquist, US Pat., 3 766 184, Sweden No Drawing Company, 1973
- J. Pašek, P. Kondelik and P. Richter, Ind. Eng. Chem. Prod. Res. Dev., 1972, 11, 333 Search PubMed
- A. Baiker and W. Richarz, Ind. Eng. Chem. Prod. Res. Dev., 1977, 16, 261 CAS
- C. M. Barnes and H. F. Rase, Ind. Eng. Chem. Prod. Res. Dev., 1981, 20, 399 CrossRef CAS
- A. Baiker and J. Kijenski, Catal. Rev., 1985, 27, 653 CAS
- A. Fischer, T. Mallat and A. Baiker, Angew. Chem., Int. Ed., 1999, 38, 351 CrossRef CAS
- A. Fischer, T. Mallat and A. Baiker, J. Catal., 1999, 182, 289 CrossRef CAS
- A. Fischer, M. Maciejewski, T. Bürgi, T. Mallat and A. Baiker, J. Catal., 1999, 183, 373 CrossRef CAS
- S. Imm, S. Bähn, L. Neubert, H. Neumann and P. M. Beller, Angew. Chem., Int. Ed., 2010, 49(8126), 8129 Search PubMed
- K. O. Marichev and J. M. Takacs, ACS Catal., 2016, 6, 2205 CrossRef CAS PubMed
- G. Liang, A. Wang, L. Li, G. Xu, N. Yan and T. Zhang, Angew. Chem., Int. Ed., 2017, 56, 3050 CrossRef CAS PubMed
- H. Kimura, Catal. Rev., 2011, 53, 1 CAS
- Y. Zhang, G. Bai, X. Yan, Y. Li, T. Zeng, J. Wang, H. Wang, J. Xing, D. Luan and X. Tang, Catal. Commun., 2007, 8, 1102 CrossRef CAS
- J. M. Jehng and C. M. Chen, Catal. Lett., 2001, 77, 147 CrossRef CAS
- K. I. Shimizu, K. Kon, W. Onodera, H. Yamazaki and J. N. Kondo, ACS Catal., 2012, 3, 112 CrossRef
- K. I. Shimizu, N. Imaiida, K. Kon, S. M. A. H. Siddiki and A. Satsuma, ACS Catal., 2013, 3, 998 CrossRef CAS
- J. H. Cho, J. H. Park, T. Chang, G. Seo and C. Shin, Appl. Catal., A, 2012, 417, 313 CrossRef
- J. H. Cho, J. Park, T. Chang, J. Kim and C. Shin, Catal. Lett., 2013, 143, 1319 CrossRef CAS
- A. Baiker, D. Monti and S. F. Yuan, J. Catal., 1984, 88, 81 CrossRef CAS
- K. I. Shimizu, S. Kanno, K. Kon, S. Siddiki, H. Tanaka and Y. Sakata, Catal. Today, 2014, 232, 134 CrossRef CAS
- J. H. Cho, S. H. An, T. S. Chang and C. H. Shin, Catal. Lett., 2016, 146, 811 CrossRef CAS
- D. C. Best, US Pat., 4 123 462, Union Carbide Corporation, 1978
- S. W. King, US Pat., 5 750 790, Union Carbide Corporation & Plastics Technology Corporation, 1998
- W. J. Bartley, R. G. Cook, K. E. Curry and S. K. Mierau, US Pat., 6 534 441, Union Carbide Corporation & Plastics Technology Corporation, 2003
- Z. Pan, Y. Ding, D. Jiang, X. Li, G. Jiao and H. Luo, Appl. Catal., A, 2007, 330, 43 CrossRef CAS
- L. Yan, Y. J. Ding, Y. Lv, X. B. Cheng and L. X. Ma, WO Patent 2013152548 A1, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 2013
- G. Jenzer, T. Mallat and A. Baiker, Catal. Lett., 1999, 61, 111 CrossRef CAS
- L. Zhang, C. Yang, Q. Yao, Y. Hou, Q. Lu and J. Xu, Mater. Manuf. Processes, 2014, 29, 738 CrossRef CAS
- J. Runeberg, A. Baiker and J. Kijenski, Appl. Catal., 1985, 17, 309 CrossRef CAS
- W. Hammerschmidt, A. Baiker, A. Wokaun and W. Fluhr, Appl. Catal., 1986, 20, 305 CrossRef CAS
- J. Kijeński, P. J. Niedzielski and A. Baiker, Appl. Catal., 1989, 53, 107 CrossRef
- P. R. Likhar, R. Arundhathi, M. L. Kantam and P. S. Prathima, Eur. J. Org. Chem., 2009, 31, 5383 CrossRef
- T. Yan, B. L. Feringa and K. Barta, ACS Catal., 2015, 6, 381 CrossRef
- H. J. Pan, W. N. Teng and Y. Zhao, Chem. Commun., 2015, 51, 11907 RSC
- B. Emayavaramban, M. Roy and B. Sundararaju, Chem.–Eur. J., 2016, 22, 3952 CrossRef CAS PubMed
- T. Takanashi, Y. Nakagawa and K. Tomishige, Chem. Lett., 2014, 43, 822 CrossRef CAS
- A. Abdukader, H. M. Jin, Y. X. Cheng and C. J. Zhu, Tetrahedron Lett., 2014, 55, 4172 CrossRef CAS
- X. Xie and V. H. Han, ACS Catal., 2015, 5, 4143 CrossRef CAS
- J. G. Donaire, M. Ernst, O. Trapp and T. Schaub, Adv. Synth. Catal., 2016, 358, 358 CrossRef
- K. K. Wu, W. He, C. Sun and Z. Yu, Tetrahedron, 2016, 72, 8516 CrossRef CAS
- Y. Nakamura, K. Kon, A. S. Touchy, K. Shimizu and W. Ueda, ChemCatChem, 2015, 7, 921 CrossRef CAS
- S. Siddiki, K. Kon and K. Shimizu, Chem.–Eur. J., 2013, 19, 14416 CrossRef CAS PubMed
- M. Ousmane, G. Perrussel, Z. Yan, J. M. Clacens, F. De Campo and M. P. Titus, J. Catal., 2014, 309, 439 CrossRef CAS
- A. Tanksale, J. N. Beltramini, J. A. Dumesic and G. Q. Lu, J. Catal., 2008, 258, 366 CrossRef CAS
- S. Abate, K. Barbera, E. Giglio, F. Deorsola, S. Bensaid, S. Perathoner, R. Pirone and G. Centi, Ind. Eng. Chem. Res., 2016, 55, 8299 CrossRef CAS
- S. Velu and S. K. Gangwal, Solid State Ionics, 2006, 177, 803 CrossRef CAS
- C. P. Li, A. Proctor and D. M. Hercules, Appl. Spectrosc., 1984, 38, 880 CrossRef CAS
- E. Heracleous, A. F. Lee, K. Wilson and A. A. Lemonidou, J. Catal., 2005, 231, 159 CrossRef CAS
- G. Li, L. Hu, J. M. Hill, G. Li, L. Hu and J. M. Hill, Appl. Catal., A, 2006, 301, 16 CrossRef CAS
- S. R. Bare, S. D. Kelly, F. D. Vila, E. Boldingh, E. Karapetrova, J. Kas, G. E. Mickelson, F. S. Modica, N. Yang and J. J. Rehr, J. Phys. Chem. C, 2011, 115, 5740 CAS
- B. C. Vicente, R. C. Nelson, A. W. Moses, S. Chattopadhyay and S. L. Scott, J. Phys. Chem. C, 2011, 115, 9012 CAS
- A. Fouskas, M. Kollia, A. Kambolis, C. Papadopoulou and H. Matralis, Appl. Catal., A, 2014, 474, 125 CrossRef CAS
- M. Wu and D. M. Hercules, J. Phys. Chem., 1979, 83, 2003 CrossRef CAS
- J. L. Ewbank, L. Kovarik, F. Z. Diallo and C. Sievers, Appl. Catal., A, 2015, 494, 57 CrossRef CAS
- K. Chayakul, T. Srithanratana and S. Hengrasmee, Catal. Today, 2011, 175, 420 CrossRef CAS
- G. Garbarino, C. Wang, I. Valsamakis, S. Chitsazan, P. Riani, E. Finocchio, M. F. Stephanopoulos and G. Busca, Appl. Catal., B, 2015, 174, 21 CrossRef
- J. Okal, L. K. Piński, L. Krajczyk and W. Tylus, J. Catal., 2003, 219, 362 CrossRef CAS
- J. Okal, W. Tylus and L. Kępiński, J. Catal., 2004, 225, 498 CrossRef CAS
- G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, J. Catal., 2011, 278, 228 CrossRef CAS
- P. Salagre, J. L. G. Fierro, F. Medina and J. E. Sueiras, J. Mol. Catal. A: Chem., 1996, 106, 125 CrossRef CAS
- J. Zieliński, J. Catal., 1982, 76, 157 CrossRef
- D. Chang and F. A. Sherrod, US Pat., 5 817 593, The Dow Chemical Company, 1998
- A. M. Hilmen, D. Schanke and A. Holmen, Catal. Lett., 1996, 38, 143 CrossRef CAS
- F. F. Yang, D. Liu, H. Wang, X. Liu, J. Y. Han, Q. F. Ge and X. L. Zhu, J. Catal., 2017, 349, 84 CrossRef CAS
- D. Hibbitts, Q. Tan and M. Neurock, J. Catal., 2014, 315, 48 CrossRef CAS
- J. Lif, M. Skoglundh and L. Löwendahl, Appl. Catal., A, 2002, 228, 145 CrossRef CAS
- K. Liu, J. Pritchard, L. Lu, R. van Putten, M. W. G. M. Verhoeven, M. Schmitkamp, X. Huang, L. Lefort, C. J. Kiely, E. J. M. Hensen and E. A. Pidko, Chem. Commun., 2017, 53, 9761 RSC
- F. B. Gebretsadik, J. Llorca, Y. Cesteros and P. Salagre, ChemCatChem, 2017, 9, 1 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12891f |
| This journal is © The Royal Society of Chemistry 2018 |