
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism studies of addition reactions between the pyrimidine type radicals and their 3′/5′ neighboring deoxyguanosines†
Shoushan Wang ab,
Min Zhangb,
Peng Liub,
Shilei Xieb,
Faliang Cheng*b and
Lishi Wang*a
ab,
Min Zhangb,
Peng Liub,
Shilei Xieb,
Faliang Cheng*b and
Lishi Wang*a
aSchool of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510641, People's Republic of China. E-mail: wanglsh@scut.edu.cn
bGuangdong Engineering and Technology Research Center for Advanced Nanomaterials, School of Environment and Civil Engineering, Dongguan University of Technology, Dongguan 523808, People's Republic of China. E-mail: chengfl@dgut.edu.cn
First published on 12th January 2018
Abstract
To clarify the biologically significant sequence effect existing in the formation of the pyrimidine-type radicals induced DNA intrastrand cross-links, addition mechanisms between the uridine-5-methyl (˙UCH2), 6-hydroxy-5,6-dihydrothymidine-5-yl (˙T6OH), and 6-hydroxy-5,6-dihydrocytidine-5-yl (˙C6OH) radicals and their 3′/5′ neighboring deoxyguanosines (dG) are explored in the present study employing the model 5′-G(˙UCH2)-3′, 5′-(˙UCH2)G-3′, 5′-G(˙T6OH)-3′, 5′-(˙T6OH)G-3′, 5′-G(˙C6OH)-3′, and 5′-(˙C6OH)G-3′ sequences. It is found that the 5′ G/C8 additions of the three radicals are all simple direct one-step reactions inducing only relatively small structural changes, while a conformational adjustment involving orientation transitions of both nucleobase moieties and twisting of the DNA backbone is indispensable for each 3′ G/C8 addition. Furthermore, markedly positive reaction free energy requirements are estimated for these conformational transformations making the 3′ G/C8 additions of the three radicals thermodynamically much more unfavorable than the corresponding 5′ G/C8 additions. Such essential conformational adjustments along the 3′ G/C8 addition paths that structurally greatly influence the local DNA structures and thermodynamically substantially reduce the addition efficiencies may be the reasons responsible for the differences in the formation yields and biological consequences of the pyrimidine-type radicals induced DNA intrastrand cross-link lesions.
Introduction
Hydroxyl radicals (OH˙), which can be generated endogenously via transition metal ion mediated Fenton-type reactions1 and exogenously by ionizing radiation,2 are able to cause numerous types of DNA lesions, including single-base modifications, strand-breaks, abasic sites, DNA intra- and interstrand cross-links.3–5 DNA lesions that are not repaired in time are mutagenic, genotoxic, and cytotoxic, which are capable of giving rise to cell apoptotic death, and thus are believed to be associated with aging6 and a variety of human diseases, such as neurological disorders,7 cardiovascular diseases,8 and cancers.9,10DNA intrastrand cross-links, in which two neighboring nucleobases in the same strand are linked together by a single covalent bond, have attracted great concern and have been continuously investigated in the past two decades.11–33 Since the seminal finding of the G(8-5m)T intrastrand cross-link (where the C8 atom of guanine is covalently tethered to the 5-methyl carbon atom of its 3′ neighboring thymine, partial atomic numberings of related species are shown in Scheme 1) in X-irradiated deoxygenated aqueous solution of d(CpGpTpA) oligomer,11 more than 15 DNA intrastrand cross-links have now been isolated.12–23 Formation of DNA intrastrand cross-links having experimentally definitely identified structures is proposed as a single-radical event.5 The OH˙ radical produced via endogenous or exogenous path can either add to the C5![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C6 double bond or abstract a hydrogen from the 5-methyl group of pyrimidine bases forming the corresponding secondary radicals, which can then attack their surrounding purine nucleosides (mainly the deoxyguanosine) eventually leading to diverse DNA intrastrand cross-links.34 UV-light irradiation experiments of DNA fragments modified by related photolabile precursors confirmed the reasonability of this reaction mechanism, and many other DNA intrastrand cross-links were further detected.24–33,35,36 One point to be noted is that an obvious sequence effect is observed in the formation yields of these pyrimidine-type radicals induced DNA intrastrand cross-links, namely the yields of the 5′-purine-pyrimidine-3′ cross-links are considerably higher than those of the corresponding 5′-pyrimidine-purine-3′ cross-links.15–17,20,26,28,29,31 This sequence effect was only simply and tentatively attributed to the difference in the distances between the two bonding atoms, the shorter the distance is, the higher the yield is,15,16,28 and convincing explanations are still unclear and lacking. Exploring the addition paths of the pyrimidine-type radicals to their 3′/5′ neighboring purine nucleosides may offer some reasonable and valuable insights into this sequence effect.
C6 double bond or abstract a hydrogen from the 5-methyl group of pyrimidine bases forming the corresponding secondary radicals, which can then attack their surrounding purine nucleosides (mainly the deoxyguanosine) eventually leading to diverse DNA intrastrand cross-links.34 UV-light irradiation experiments of DNA fragments modified by related photolabile precursors confirmed the reasonability of this reaction mechanism, and many other DNA intrastrand cross-links were further detected.24–33,35,36 One point to be noted is that an obvious sequence effect is observed in the formation yields of these pyrimidine-type radicals induced DNA intrastrand cross-links, namely the yields of the 5′-purine-pyrimidine-3′ cross-links are considerably higher than those of the corresponding 5′-pyrimidine-purine-3′ cross-links.15–17,20,26,28,29,31 This sequence effect was only simply and tentatively attributed to the difference in the distances between the two bonding atoms, the shorter the distance is, the higher the yield is,15,16,28 and convincing explanations are still unclear and lacking. Exploring the addition paths of the pyrimidine-type radicals to their 3′/5′ neighboring purine nucleosides may offer some reasonable and valuable insights into this sequence effect.
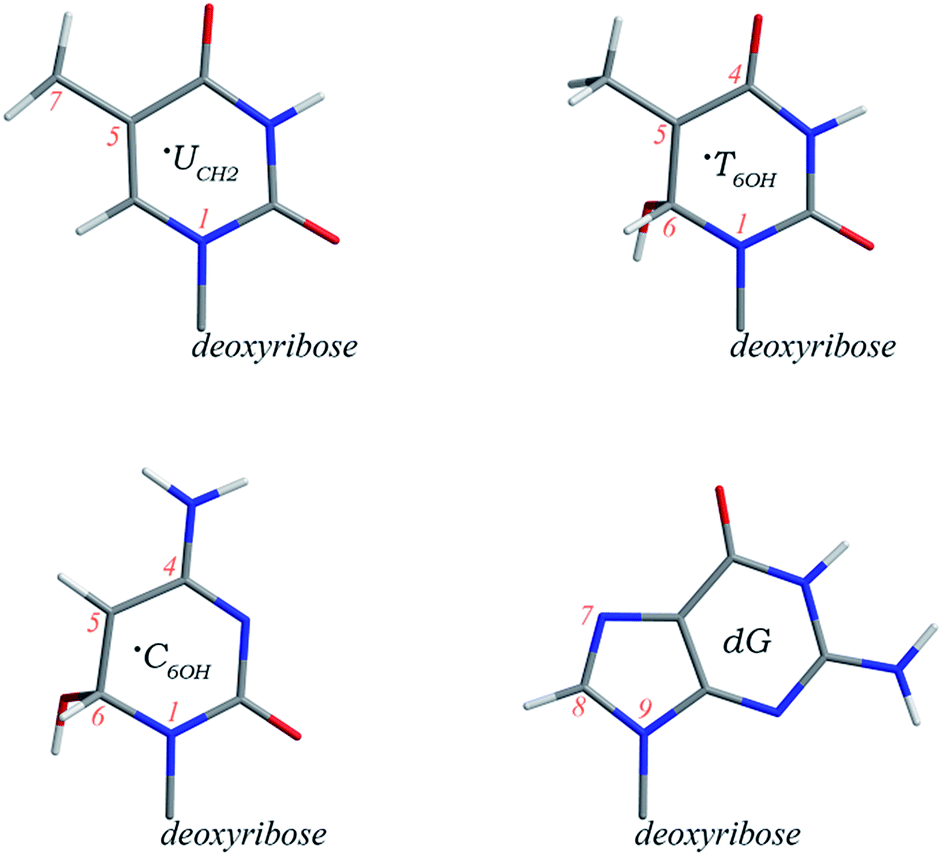 | ||
| Scheme 1 Schematic structures and partial atomic numberings of the ˙UCH2, ˙T6OH, and ˙C6OH radicals as well as deoxyguanosine. | ||
Apart from markedly interrupting DNA replication and significantly decreasing the efficiency and fidelity of nucleotide incorporation,16,18,19,35,37,38 DNA intrastrand cross-links are also able to lead to substantial targeted and semitargeted base transition and transversion mutations,18,19,21,38 with the mutational frequencies induced by the 5′-pyrimidine-purine-3′ cross-links greatly higher than those induced by the 5′-purine-pyrimidine-3′ cross-links,21 indicating that a sequence effect may also exist in the biological consequences of DNA intrastrand cross-links. The unique structures of DNA intrastrand cross-links of different types are tentatively speculated to be responsible for these observations.16,18,19 On the other hand, DNA intrastrand cross-links can resist, to some extent, the excision repair of DNA nucleotide excision repair system.39,40 Again, the repair efficiencies were proposed to be correlated with the different degrees of structural distortions of DNA double helixes induced by different types of DNA intrastrand cross-links.40–42 Thus, exploring the probable structures and formation mechanisms of different types of DNA intrastrand cross-links are much helpful for better understanding of these biological consequences. However, recent theoretical efforts were mainly focused on the studies of formation paths of 5′-purine-pyrimidine-3′ type cross-links and their influences on DNA double helical structures.41–48 To the best of our knowledge, probable formation mechanisms of the corresponding 5′-pyrimidine-purine-3′ type cross-links are scarcely investigated.
On the basis of the above discussions, reaction paths of the ˙UCH2, ˙T6OH, and ˙C6OH radicals adding to the C8 site of their 3′/5′ neighboring deoxyguanosines are studied here. For each radical, different from the simple direct one-step 5′ G/C8 addition, a conformational transformation not only structurally significant but also thermodynamically markedly unfavourable is firstly required for the 3′ G/C8 addition, which may be the possible reasons leading to different formation yields and biological consequences of the corresponding DNA intrastrand cross-links.
Computational details
Dinucleoside monophosphates are demonstrated to be relevant models for investigations of DNA intrastrand cross-links,47 and thus the models 5′-G(˙UCH2)-3′, 5′-(˙UCH2)G-3′, 5′-G(˙T6OH)-3′, 5′-(˙T6OH)G-3′, 5′-G(˙C6OH)-3′, and 5′-(˙C6OH)G-3′ sequences were employed here, which were constructed based on the canonical 5′-GT-3′, 5′-TG-3′, 5′-GC-3′, and 5′-CG-3′ sequences extracted from an experimental X-ray crystal structure of a B-DNA (PDB code: 5FMP). One hydrogen atom was deleted from the 5-methyl group of the T moiety or a hydroxyl group was added on the C6 site of the T or C moiety, the two phosphodiester bonds were capped with methyl groups to prevent the formation of parasitic hydrogen bonds,49 and a sodium ion was inserted between the two phosphate oxygens to mimic the physiological conditions.50 The M06-2X functional combined with the standard 6-31G(d,p) basis set was used for all geometry optimizations and energy calculations. The M06-2X/6-31G(d,p) level of theory has been proved to be reliable for studies of DNA intrastrand cross-links,44,51 and actually is recommended to explore the structure and/or reactivity of a dinucleoside monophosphate.50 All structures obtained were shown to be minimum or first-order saddle points on potential energy surfaces by frequency analyses. Intrinsic reaction coordinate calculations were performed to confirm the connections of each transition state with its corresponding reactant and product. The IEF-PCM formalism52 with a dielectric constant ε = 78.4 was employed to approximate the solvated environment.47,50 Related kinetic and thermodynamic characteristics for each reaction process were estimated on the basis of Gibbs free energies calculated in the rigid-rotor harmonic oscillator approximation at T = 298 K and p = 1 atm. Geometry optimizations and energy calculations were carried out using Gaussian 09 package,53 and structures of all stationary points were analysed employing the X3DNA suite of programs.54Results and discussion
Formation of DNA intrastrand cross-links is generally a three-step reaction,41,45–47 consisting of (step 1) formation of the highly reactive pyrimidine-type radicals, (step 2) additions of these radicals to their 3′/5′ neighboring purine nucleosides (mainly deoxyguanosine), and (step 3) formation of the final closed-shell intrastrand cross-links. The present study is mainly focused on the step 2, namely exploring the probable addition paths of ˙UCH2, ˙T6OH, and ˙C6OH radicals to the C8 site of their 3′/5′ neighboring deoxyguanosines.Additions of the ˙UCH2, ˙T6OH, and ˙C6OH radicals to the C8 site of their 5′ neighboring deoxyguanosines
Among all known DNA intrastrand cross-links, the G(8-5m)T cross-link (Fig. S1 in the ESI†) resulting from the ˙UCH2 radical attacking the C8 site of its 5′ neighboring deoxyguanosine is determined to have the highest yield. As depicted in Fig. 1, addition of the ˙UCH2 radical to the C8 site of its 5′ neighboring dG is unsurprisingly a one-step reaction. The distance (denoted as dG/C8-T/C7) between the radical center (the C7 atom, related spin density distributions are shown in Fig. S2 and S5 in the ESI†) of the ˙UCH2 radical and the C8 site of dG is decreased to 2.09 Å in the located transition state (denoted as TSGU) from a value of 3.60 Å in the canonical 5′-G(˙UCH2)-3′ sequence, and eventually reduced to 1.55 Å forming a standard C–C single bond in the addition product (denoted as 5′-˙G8H(8-5m)T-3′). Meanwhile, evolution of the ˙UCH2/C7 and G/C8 atom hybridizations from sp2 to sp3 makes the C7–C5 bond in the 3′ ˙UCH2 moiety and the C8–N7 and C8–N9 bonds in the 5′ dG moiety gradually elongated to 1.50, 1.45, and 1.48 Å from 1.39, 1.31, and 1.38 Å, respectively, with the other bond length changes no more than 0.04 Å. Notably, along the reaction path, the torsion angle χ5′G (∠O4′C1′N9C4) continuously increases to −77° from −130° while the torsion angle χ3′U (∠O4′C1′N1C2) continuously decreases to −126° from −101°, but the nucleobase moieties of both 3′ ˙UCH2 and 5′ dG always maintain the anti-orientations, with the angle ϕ (defined as the dihedral angle between the planes of the two base moieties in a dinucleoside monophosphate) ranged from 4° to 51°, which are well consistent with the simulation results reported previously.47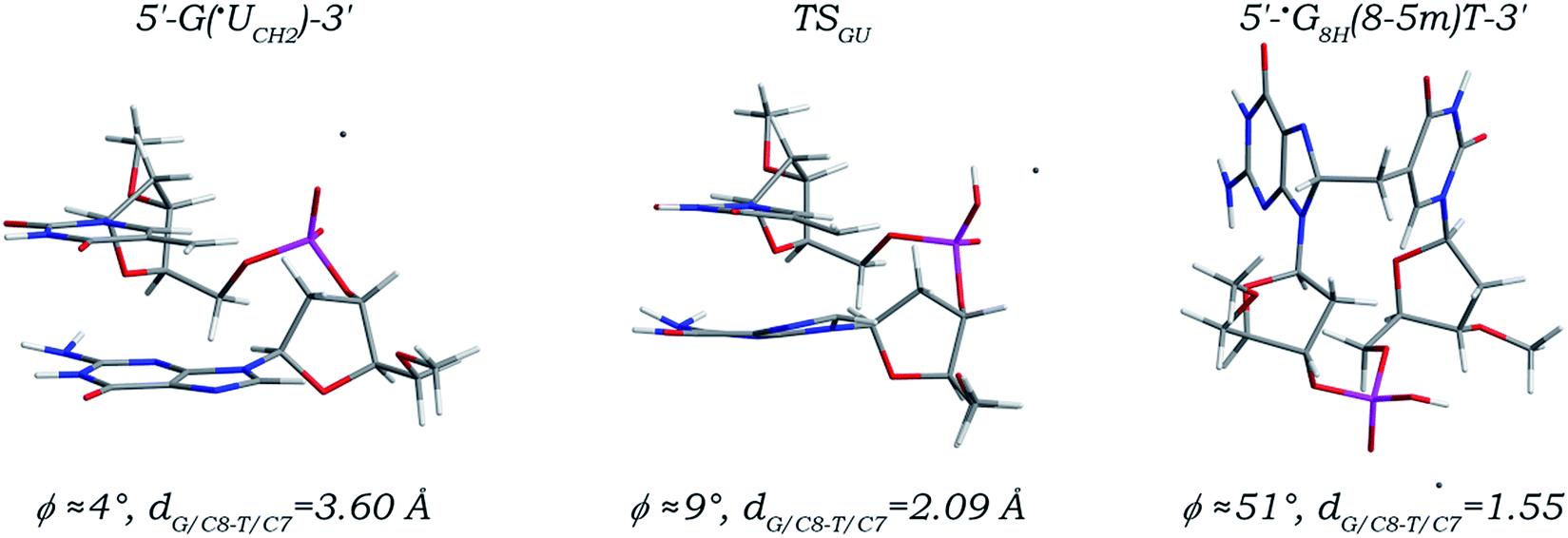 | ||
| Fig. 1 Optimized structures and partial structural parameters for stationary points along the reaction path of the ˙UCH2 radical addition to the C8 site of its 5′ neighboring deoxyguanosine. | ||
As listed in Table 1, an activation free energy of 17.27 kcal mol−1 is calculated, indicating that addition of the ˙UCH2 radical to the C8 site of its 5′ neighboring deoxyguanosine is kinetically feasible. The current free energy barrier is 6–9 kcal mol−1 higher than those reported in the QM/MM simulation studies of an explicitly solvated 5′-G(˙UCH2)-3′ sequence and a solvated dodecameric sequence,41,46,47 which may be attributed to the inherent defect of the BLYP functional, namely always underestimating the transition state barriers by several kilocalories per mole.55 A reaction free energy of 1.41 kcal mol−1 is obtained for the 5′ G/C8 addition of the ˙UCH2 radical, separately comparable to and ca. 5 kcal mol−1 lower than those reported in the above-mentioned two simulation studies.41,46,47 A positive reaction free energy here does not mean that the corresponding reaction step cannot occur. The ˙UCH2 radical addition step is actually coupled with the preceding step of formation of the ˙UCH2 radical and the subsequent step of formation of the closed-shell G(8-5m)T intrastrand cross-link.41,45–47 As shown in Fig. S8 in the ESI,† 28.07 kcal mol−1 reaction free energy is released for the preceding step of formation of the canonical 5′-G(˙UCH2)-3′ sequence, which makes the driving force for formation of the radical adduct 5′-˙G8H(8-5m)T-3′ estimated from the canonical 5′-GT-3′ sequence much negative (−26.66 kcal mol−1). Moreover, the subsequent step of transformation of the radical adduct 5′-˙G8H(8-5m)T-3′ into the closed-shell 5′-G(8-5m)T-3′ cross-link further releases 81.67 kcal mol−1 reaction free energy (Fig. S9 in the ESI†). Thus, besides involving only minor structural changes, the 5′ G/C8 addition of the ˙UCH2 radical is also kinetically and thermodynamically feasible.
| System | ΔG1a | ΔG* | ΔG2d | ΔGTe |
|---|---|---|---|---|
| a ΔG1 = Gintermediate − Gcanonical sequence.b ΔG* = GTS state − Gcanonical sequence.c ΔG* = GTS state − Gintermediate.d ΔG2 = Gaddition product − Gintermediate.e ΔGT = Gaddition product − Gcanonical sequence. | ||||
| 5′-G(˙UCH2)-3′ | — | 17.27b | 1.41 | 1.41 |
| 5′-G(˙T6OH)-3′ | — | 17.45b | 5.81 | 5.81 |
| 5′-G(˙C6OH)-3′ | — | 15.25b | 0.54 | 0.54 |
| 5′-(˙UCH2)G-3′ | 9.12 | 17.37c | 2.48 | 11.60 |
| 5′-(˙T6OH)G-3′ | 18.23 | 14.74c | 2.40 | 20.63 |
| 5′-(˙C6OH)G-3′ | 17.06 | 10.79c | −5.13 | 11.94 |
The ˙T6OH radical, which is produced three times more frequently than the ˙UCH2 radical,36 is shown to be able to attack the C8 site of its 3′/5′ neighboring deoxyguanosine forming the G(8-5)T6OH or T6OH(5-8)G intrastrand cross-link (Fig. S1 in the ESI†), although the yields of both cross-links are much lower than those of the corresponding G(8-5m)T and T(5m-8)G cross-links, respectively.12,13,15,23,56 A complete reaction path of the ˙T6OH radical addition to the C8 site of its 5′ neighboring deoxyguanosine is depicted in Fig. 2, and related kinetic and thermodynamic characteristics are listed in Table 1. The 5′ G/C8 addition of the ˙T6OH radical is also a one-step reaction. In the located transition state (denoted as TSGT), the distance (denoted as dG/C8-T/C5) between the radical center (the C5 atom, related spin density distributions are shown in Fig. S3 and S6 in the ESI†) of the ˙T6OH radical and the C8 site of dG is reduced to 2.13 Å from 3.38 Å in the canonical 5′-G(˙T6OH)-3′ sequence, and finally decreased to 1.60 Å forming a C–C single bond in the addition product (denoted as 5′-˙G8H(8-5)T6OH-3′). All bond length changes are found to be no more than 0.04 Å except that the C8–N7 and C8–N9 bonds in the 5′ dG moiety and the C5–C4, C5–C6, and C5–C7 bonds in the 3′ ˙T6OH moiety are separately elongated to 1.46, 1.47, 1.53, 1.54, and 1.53 Å from values of 1.31, 1.38, 1.45, 1.49, and 1.48 Å due to hybridization transitions of the G/C8 and ˙T6OH/C5 atoms from sp2 to sp3. The anti-orientations of both nucleobase moieties are always maintained along the reaction path, with the two torsion angles χ5′G (∠O4′C1′N9C4) and χ3′T (∠O4′C1′N1C2) merely continuously increased to −80° and −119° from −111° and −146°, respectively. Thus, just like the 5′ G/C8 addition of the ˙UCH2 radical, addition of the ˙T6OH radical to the C8 site of its 5′ neighboring deoxyguanosine also gives rise to only small effects on the local DNA structure.
 | ||
| Fig. 2 Optimized structures and partial structural parameters for stationary points along the reaction path of the ˙T6OH radical addition to the C8 site of its 5′ neighboring deoxyguanosine. | ||
As listed in Table 1, the activation free energy for the 5′ G/C8 addition of the ˙T6OH radical is calculated to be 17.45 kcal mol−1, kinetically comparable to that needed for the 5′ G/C8 addition of the ˙UCH2 radical. Starting from the canonical 5′-G(˙T6OH)-3′ sequence, the reaction free energy for formation of the 5′-˙G8H(8-5)T6OH-3′ adduct is estimated to be 5.81 kcal mol−1, 4.4 kcal mol−1 larger than that for formation of the 5′-˙G8H(8-5m)T-3′ adduct. The 5′ G/C8 addition of the ˙T6OH radical is thus thermodynamically less efficient than the 5′ G/C8 addition of the ˙UCH2 radical, providing a plausible explanation for the experimental observation that the yield of the G(8-5)T6OH cross-link is much lower than that of the G(8-5m)T cross-link. The driving force for formation of the 5′-˙G8H(8-5)T6OH-3′ adduct is reduced to ca. −16 kcal mol−1 when taking into account the preceding step of formation of the canonical 5′-G(˙T6OH)-3′ sequence (Fig. S8 in the ESI,† −21.83 kcal mol−1 reaction free energy is calculated). Furthermore, 81.33 kcal mol−1 reaction free energy is released for subsequent transformation of the radical adduct 5′-˙G8H(8-5)T6OH-3′ into the 5′-G(8-5)T6OH-3′ cross-link (Fig. S9 in the ESI†). Therefore, as in the case of the 5′ G/C8 addition of the ˙UCH2 radical, the 5′ G/C8 addition of the ˙T6OH radical is also thermodynamically feasible.
G(8-5)C and C(5-8)G intrastrand cross-links (Fig. S1 in the ESI†) have been definitely detected in DNA oligomer, duplex DNA, and mammalian cells irradiated by γ-/X-ray13,16,19,23 or treated with Fenton-type reagents.20 Formation of both intrastrand cross-links is believed to be a single-radical event, and be associated with the ˙C6OH radical.13 Reaction path of the ˙C6OH radical addition to the C8 site of its 5′ neighboring deoxyguanosine and related kinetic and thermodynamic characteristics are shown in Fig. 3 and Table 1, respectively. Obviously, as in the cases of the 5′ G/C8 additions of the ˙UCH2 and ˙T6OH radicals, the 5′ G/C8 addition of the ˙C6OH radical is also a simple direct one-step reaction. The distance (denoted as dG/C8-C/C5) between the radical center (the C5 atom, related spin density distributions are shown in Fig. S4 and S7 in the ESI†) of the ˙C6OH radical and the C8 site of dG is lowered to 2.15 Å in the located transition state (denoted as TSGC) from 3.41 Å in the canonical 5′-G(˙C6OH)-3′ sequence, and ultimately reduced to 1.57 Å forming a standard C–C single bond in the addition product (denoted as 5′-˙G8H(8-5)C6OH-3′). Meanwhile, the C8–N7 and C8–N9 bonds in the 5′ dG moiety and the C5–C4 and C5–C6 bonds in the 3′ ˙C6OH moiety are separately stretched to 1.45, 1.47, 1.51, and 1.53 Å from values of 1.31, 1.38, 1.44, and 1.49 Å due to hybridization transitions of the G/C8 and ˙C6OH/C5 atoms from sp2 to sp3, with the other bond length changes no more than 0.04 Å. Once again, the anti-orientations of both nucleobase moieties are maintained along the reaction path with the angle ϕ increased from 18° to 35°, implying that formation of the 5′-˙G8H(8-5)C6OH-3′ adduct has only relatively small influences on the local DNA structure. Addition of the ˙C6OH radical to the C8 site of its 5′ neighboring deoxyguanosine is kinetically feasible, with 15.25 kcal mol−1 activation free energy being calculated (Table 1). On the other hand, the reaction free energy is estimated to be 0.54 kcal mol−1, indicating that the 5′ G/C8 addition step of the ˙C6OH radical is thermodynamically slightly unfavourable. But as shown in Fig. S8 and S9 in the ESI,† the preceding step of formation of the canonical 5′-G(˙C6OH)-3′ sequence and the subsequent step of formation of the closed-shell 5′-G(8-5)C-3′ cross-link are both thermodynamically favourable. Hence, when coupled with these reaction steps, the ˙C6OH radical addition to the C8 site of its 5′ neighboring deoxyguanosine forming the 5′-˙G8H(8-5)C6OH-3′ adduct is thermodynamically feasible.
 | ||
| Fig. 3 Optimized structures and partial structural parameters for stationary points along the reaction path of the ˙C6OH radical addition to the C8 site of its 5′ neighboring deoxyguanosine. | ||
In short, additions of the ˙UCH2, ˙T6OH, and ˙C6OH radicals to the C8 site of their 5′ neighboring deoxyguanosines are all simple direct one-step reactions, imposing only minor influences on the local DNA structure. On the other hand, all these three addition reactions are kinetically and thermodynamically feasible, and the more unfavourable thermodynamic requirement for the 5′ G/C8 addition of the ˙T6OH radical than that for the 5′ G/C8 addition of the ˙UCH2 radical may be the reasons leading to the fact that the yield of the G(8-5)T6OH intrastrand cross-link is much lower than that of the G(8-5m)T intrastrand cross-link.
Additions of the ˙UCH2, ˙T6OH, and ˙C6OH radicals to the C8 site of their 3′ neighboring deoxyguanosines
Different from the simple one-step 5′ G/C8 addition, as depicted in Fig. 4, the reaction path of the ˙UCH2 radical addition to the C8 site of its 3′ neighboring deoxyguanosine is rather complex. Before the 3′ G/C8 addition step proceeds, a large structural adjustment from the initial canonical 5′-(˙UCH2)G-3′ sequence to an intermediate (denoted as INTUG) is firstly required. The torsion angles χ5′U (∠O4′C1′N1C2) and χ3′G (∠O4′C1′N9C4) are increased to 25° and −39° from values of −107° and −87° (Table 2), eventually making the nucleobase moieties of 5′ ˙UCH2 and 3′ dG adopt a syn-orientation and a near T-shaped orientation, respectively. Meanwhile, the DNA backbone rotates primarily around the C3′–O3′, O3′–P, and P–O5′ bonds, with the torsion angles α (∠C4′C3′O3′P), β (∠C3′O3′PO5′), and γ (∠O3′PO5′C5′) changed from 179°, −112°, and −62° to 155°, −86°, and −80°, respectively. Rotation of both nucleobase moieties about their corresponding glycosidic bonds and twist of the DNA backbone collectively lead to the two nucleobase moieties parallel with the angle ϕ being 4° in the intermediate INTUG, and further the distance dG/C8-T/C7 is obviously decreased from 7.01 Å to 3.38 Å. Starting from the INTUG intermediate, the 3′ G/C8 addition step proceeds (Fig. 4). The distance dG/C8-T/C7 is lowered to 2.11 Å in the located transition state (denoted as TSUG), and ultimately reduced to 1.57 Å forming a standard C–C single bond in the addition product (denoted as 5′-T(5m-8)(˙G8H)-3′). Hybridization transitions of both ˙UCH2/C7 and G/C8 atoms from sp2 to sp3 make the bond length changes of the related C7–C5 bond in the 5′ ˙UCH2 moiety and the C8–N7 and C8–N9 bonds in the 3′ dG moiety nearly the same as those observed in the case of the 5′ G/C8 addition, and the other bonds are relatively stable with their bond length changes no more than 0.04 Å. Along the 3′ G/C8 addition path, the angle ϕ only fluctuates within the range of 4–27°, but the prerequisite requirement of a large conformational transformation from the initial canonical 5′-(˙UCH2)G-3′ sequence to an intermediate INTUG implies that addition of the ˙UCH2 radical to the C8 site of its 3′ neighboring deoxyguanosine is obviously structurally disadvantageous, and may greatly influence the local DNA structure. | ||
| Fig. 4 Optimized structures and partial structural parameters for stationary points along the reaction path of the ˙UCH2 radical addition to the C8 site of its 3′ neighboring deoxyguanosine. | ||
| Structure | χ5′ | χ3′ | α | β | γ | δ |
|---|---|---|---|---|---|---|
| 5′-(˙UCH2)G-3′ | −107° | −87° | 179° | −112° | −62° | −175° |
| INTUG | 25° | −39° | 155° | −86° | −80° | 173° |
| 5′-(˙T6OH)G-3′ | −78° | −103° | 161° | −103° | −56° | 178° |
| INTTG | 51° | −52° | 144° | −95° | −80° | 153° |
| 5′-(˙C6OH)G-3′ | −77° | −102° | 162° | −101° | −57° | −179° |
| INTCG | 54° | −48° | 149° | −94° | −84° | 150° |
For the 3′ G/C8 addition step, an activation free energy of 17.37 kcal mol−1 (Table 1), which is nearly equal to that required in the 5′ G/C8 addition, and a reaction free energy of 2.48 kcal mol−1 are calculated. One point to be noted is that, besides structurally disadvantageous, the prerequisite conformational adjustment from the canonical 5′-(˙UCH2)G-3′ sequence to the intermediate INTUG is also thermodynamically unfavourable, with 9.12 kcal mol−1 reaction free energy being absorbed. As a result, the reaction free energy for the 3′ G/C8 addition estimated from the canonical 5′-(˙UCH2)G-3′ sequence is increased to as positive as 11.60 kcal mol−1, ca. 10 kcal mol−1 higher than that for the simple 5′ G/C8 addition. Hence, from the thermodynamic point of view, addition of the ˙UCH2 radical to the C8 site of its 3′ neighboring deoxyguanosine forming the 5′-T(5m-8)(˙G8H)-3′ adduct is much more inefficient than that to the C8 site of its 5′ neighboring deoxyguanosine forming the 5′-˙G8H(8-5m)T-3′ adduct. However, the 11.60 kcal mol−1 reaction free energy does also not mean that the 3′ G/C8 addition of the ˙UCH2 radical cannot occur. When coupled with the thermodynamically favourable preceding step of formation of the canonical 5′-(˙UCH2)G-3′ sequence and the subsequent step of formation of the closed-shell 5′-T(5m-8)G-3′ cross-link (Fig. S8 and S9 in the ESI,† −26.03 and −88.06 kcal mol−1 reaction free energies are separately estimated), addition of the ˙UCH2 radical to the C8 site of its 3′ neighboring deoxyguanosine is in fact thermodynamically feasible.
A reaction path (Fig. 5) much similar to that of the 3′ G/C8 addition of the ˙UCH2 radical is found for the ˙T6OH radical addition to the C8 site of its 3′ neighboring deoxyguanosine. The initial canonical 5′-(˙T6OH)G-3′ sequence must be firstly transformed into an intermediate (denoted as INTTG) before the 3′ G/C8 addition step proceeds. As reflected by the changes of the related torsion angles listed in Table 2, the two nucleobase moieties are separately converted to syn- and T-shaped orientations from anti- and anti-orientations, and the DNA backbone twists around the C3′–O3′, O3′–P, P–O5′, and C5′–O5′ bonds. Synergy of orientation transitions of both nucleobase moieties and twist of DNA backbone makes the distance dG/C8-T/C5 lowered from 4.84 Å in the canonical 5′-(˙T6OH)G-3′ sequence to 3.08 Å in the INTTG intermediate, which is further reduced to 2.16 Å in the located transition state (denoted as TSTG) for the 3′ G/C8 addition step, and ultimately decreased to 1.59 Å forming a C–C single bond in the addition product (denoted as 5′-T6OH(5-8)(˙G8H)-3′). Since the ˙T6OH/C5 and G/C8 atom hybridizations are also changed from sp2 to sp3, bond length alterations of the C5–C4, C5–C6, and C5–C7 bonds in the 5′ ˙T6OH moiety and the C8–N7 and C8–N9 bonds in the 3′ dG moiety as well as the other bonds are shown to be nearly the same as those observed in the 5′ G/C8 addition of the ˙T6OH radical. Although the increment of the angle ϕ (28°) along the 3′ G/C8 addition path is comparable to that (29°) along the 5′ G/C8 addition path, different from its sequence isomer 5′-˙G8H(8-5)T6OH-3′, formation of the 5′-T6OH(5-8)(˙G8H)-3′ adduct may greatly influence the local DNA structure due to the essential conformational adjustment.
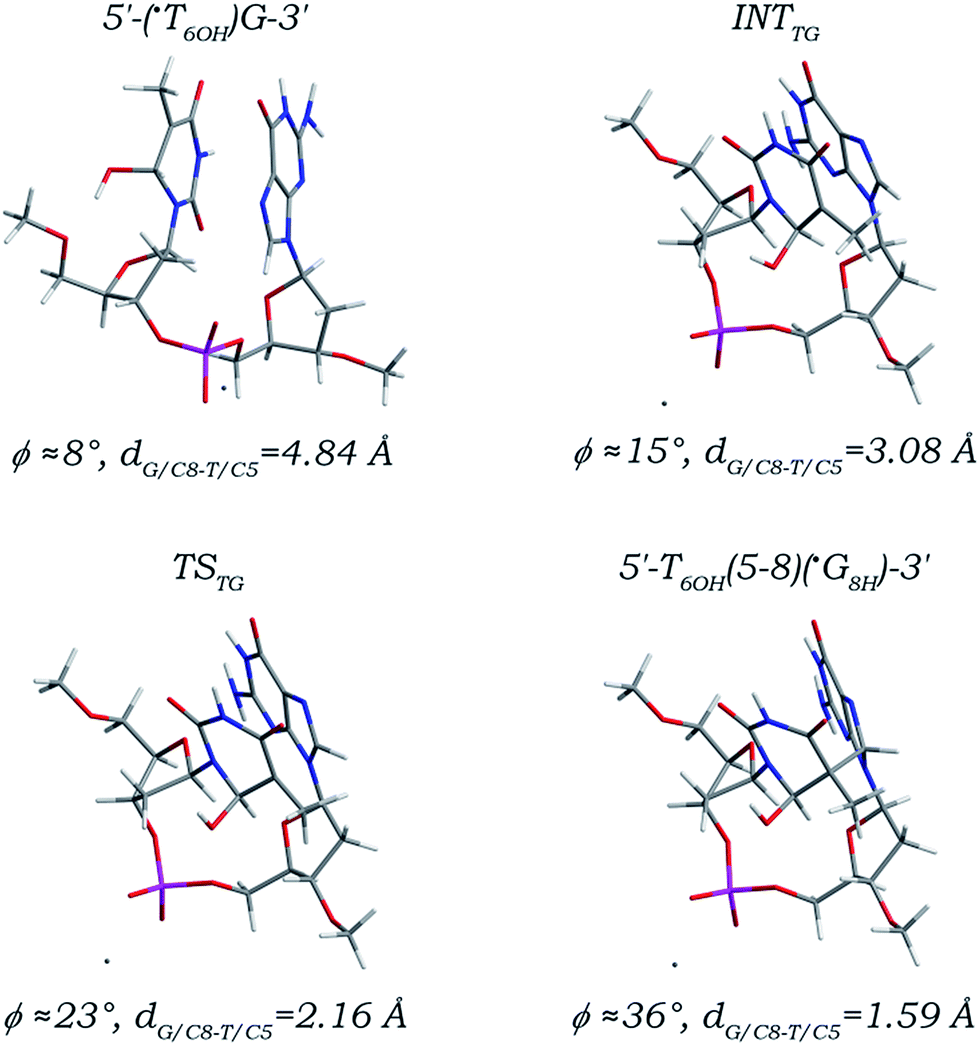 | ||
| Fig. 5 Optimized structures and partial structural parameters for stationary points along the reaction path of the ˙T6OH radical addition to the C8 site of its 3′ neighboring deoxyguanosine. | ||
Among the five addition steps investigated above, the 3′ G/C8 addition of the ˙T6OH radical has the relatively lowest free energy barrier (14.74 kcal mol−1, Table 1), and the reaction free energy is calculated to be 2.40 kcal mol−1, comparable to and 3.41 kcal mol−1 smaller than those for the 3′ G/C8 addition step of the ˙UCH2 radical and the 5′ G/C8 addition of the ˙T6OH radical, respectively. But it does not suggest that the yield of the T6OH(5-8)G intrastrand cross-link is larger than those of the T(5m-8)G and G(8-5)T6OH intrastrand cross-links. As listed in Table 1, a thermodynamic free energy requirement as high as 18.23 kcal mol−1 is estimated for the essential conformational adjustment, making the reaction free energy for the 3′ G/C8 addition of the ˙T6OH radical increased to 20.63 kcal mol−1, separately ca. 9 kcal mol−1 and 15 kcal mol−1 higher than those for the 3′ G/C8 addition of the ˙UCH2 radical and the 5′ G/C8 addition of the ˙T6OH radical. Thus, although formation of the 5′-T6OH(5-8)(˙G8H)-3′ adduct is thermodynamically feasible when taking into account the preceding step of formation of the canonical 5′-(˙T6OH)G-3′ sequence and the subsequent step of formation of the closed-shell 5′-T6OH(5-8)G-3′ cross-link (Fig. S8 and S9 in the ESI,† the driving forces are separately estimated to be −29.13 and −82.68 kcal mol−1), the thermodynamically more unfavourable conformational adjustment indicates that the formation efficiency of the 5′-T6OH(5-8)(˙G8H)-3′ adduct is much lower than those of the 5′-˙G8H(8-5)T6OH-3′ and 5′-T(5m-8)(˙G8H)-3′ adducts, which may explain why the yield of the corresponding T6OH(5-8)G intrastrand cross-link is much less than those of the G(8-5)T6OH and T(5m-8)G intrastrand cross-links.15,23
The reaction path of the ˙C6OH radical addition to the C8 site of its 3′ neighboring deoxyguanosine is found to be nearly the same as that of the 3′ G/C8 addition of the ˙T6OH radical (Fig. 6). An essential conformational adjustment involving orientation transitions of both nucleobase moieties and twist of DNA backbone with alterations of the related torsion angles (Table 2) comparable to those observed in the 3′ G/C8 addition of the ˙T6OH radical is firstly required, which makes the configuration of thus formed intermediate (denoted as INTCG) much analogous to that of the intermediate INTTG. The distance dG/C8-C/C5 that is lowered to 3.12 Å in the intermediate INTCG is decreased to 2.20 Å in the located transition state (denoted as TSCG), and eventually reduced to 1.58 Å forming a standard C–C single bond in the addition product (denoted as 5′-C6OH(5-8)(˙G8H)-3′). Changes of the C5–C4 and C5–C6 bonds in the 5′ ˙C6OH moiety and the C8–N7 and C8–N9 bonds in the 3′ dG moiety as well as the other bonds are demonstrated to be nearly the same as those observed in the corresponding 5′ G/C8 addition. The increment of the angle ϕ along the reaction path is relatively small (31°), but as in the cases of the 3′ G/C8 additions of the ˙UCH2 and ˙T6OH radicals, the prerequisite conformational transformation also makes formation of the adduct 5′-C6OH(5-8)(˙G8H)-3′ give rise to substantial effects on the local DNA structure. Kinetically, the activation free energy (10.79 kcal mol−1, Table 1) for the step of the ˙C6OH radical addition to the C8 site of its 3′ neighboring deoxyguanosine is ca. 4.5 kcal mol−1 smaller than that for the 5′ G/C8 addition. Thermodynamically, a negative reaction free energy (−5.13 kcal mol−1) is estimated for the 3′ G/C8 addition step, but the sizably positive free energy requirement for the essential conformational adjustment (17.06 kcal mol−1) makes the reaction free energy estimated from the canonical 5′-(˙C6OH)G-3′ sequence increased to 11.94 kcal mol−1, which is ca.11 kcal mol−1 larger than that for the 5′ G/C8 addition. Thus, as in the case of the 3′ G/C8 addition of the ˙T6OH radical, although formation of the 5′-C6OH(5-8)(˙G8H)-3′ adduct is thermodynamically feasible when coupled with the preceding step of formation of the canonical 5′-(˙C6OH)G-3′ sequence and the subsequent step of formation of the closed-shell 5′-C(5-8)G-3′ cross-link (Fig. S8 and S9 in the ESI,† −21.26 and −102.61 kcal mol−1 reaction free energies are calculated, respectively), the formation efficiency of which is much lower than that of its sequence isomer 5′-˙G8H(8-5)C6OH-3′.
 | ||
| Fig. 6 Optimized structures and partial structural parameters for stationary points along the reaction path of the ˙C6OH radical addition to the C8 site of its 3′ neighboring deoxyguanosine. | ||
In short, different from the simple direct one-step 5′ G/C8 additions, structurally significant and thermodynamically markedly unfavourable conformational adjustments involving orientation transitions of nucleobase moieties and twist of DNA backbone are essential for the 3′ G/C8 additions of the ˙UCH2, ˙T6OH, and ˙C6OH radicals. Notably, in double helical DNA, the stabilizing interactions (including hydrogen bonds and base-stacking arrangements) may penalize, to a larger extent, these conformational transformations, making the 3′ G/C8 additions of the ˙UCH2, ˙T6OH, and ˙C6OH radicals even more unpreferred. Such essential conformational transformations may thus be the reasons leading to the differences in the formation yields and biological significance of the G(8-5m)T/G(8-5)T6OH/G(8-5)C and T(5m-8)G/T6OH(5-8)G/C(5-8)G intrastrand cross-links. In addition, the more unfavourable thermodynamic requirement for the prerequisite conformational transformation along the 3′ G/C8 addition path of the ˙T6OH radical may provide a plausible explanation for the observation that the yield of the T6OH(5-8)G intrastrand cross-link is much lower than that of the T(5m-8)G intrastrand cross-link.
Conclusions
Reaction mechanisms of the ˙UCH2, ˙T6OH, and ˙C6OH radicals separately adding to the C8 site of their 3′ and 5′ neighboring deoxyguanosines are investigated in the present work. It is found that additions of all these three radicals to the C8 site of their 5′ neighboring deoxyguanosines are straightforward one-step reactions, and along each addition path only relatively minor structural alterations are induced with both nucleobase moieties always maintaining the anti-orientations. In contrast, additions of the ˙UCH2, ˙T6OH, and ˙C6OH radicals to the C8 site of their 3′ neighboring deoxyguanosines are rather complex. The canonical 5′-(˙UCH2)G-3′, 5′-(˙T6OH)G-3′, and 5′-(˙C6OH)G-3′ sequences must be firstly transformed into certain intermediates, during which the anti-orientations of the two nucleobase moieties are translated to syn-(for the 5′ nucleobase) and (near) T-shaped orientations (for the 3′ dG) and the DNA backbone are twisted around the C3′–O3′, O3′–P, P–O5′, and C5′–O5′ bonds, respectively. Compared to the simple direct 5′ G/C8 additions, such significant conformational adjustments along the 3′ G/C8 addition paths may greatly influence the local DNA structure, and thus may give rise to distinctively different biological significance of the corresponding intrastrand cross-links. On the other hand, greatly positive reaction free energies are estimated for these prerequisite conformational transformations, making the total driving forces for 3′ G/C8 additions 10–15 kcal mol−1 larger than those for 5′ G/C8 additions. Furthermore, such essential conformational adjustments may be disfavoured, to a larger extent, in double-stranded DNA due to existence of the stabilizing interactions from hydrogen bonds and base-stacking arrangements. Combined, the 3′ G/C8 additions of the ˙UCH2, ˙T6OH, and ˙C6OH radicals are thus both structurally and thermodynamically significantly more unfavourable than the corresponding 5′ G/C8 additions, which may be the reasons why the yields of the 5′-pyrimidine-purine-3′ cross-links are obviously lower than those of the 5′-purine-pyrimidine-3′ cross-links. All results reported here provide a plausible explanation for the sequence effect observed both in the formation yields and biological significance of pyrimidine-type radicals induced DNA intrastrand cross-links.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by China Postdoctoral Science Foundation (No. 2016M602455), National Natural Science Foundation of China (No. 21375016, 21475022, and 21505019), Natural Science Foundation of Guangdong Province (No. 2015A030310272), Technology Planning Project of Guangdong Province (No. 2015B090927007), and Guangdong Provincial Key Platform and Major Scientific Research Projects for Colleges and Universities (No. 2014KZDXM073, 2015KCXTD029, 2016KTSCX136, 2016KTSCX135, and 2016KQNCX166), and Research Start-up Funds of DGUT (GC300501-04).Notes and references
- J. Prousek, Pure Appl. Chem., 2007, 79, 2325 CrossRef CAS.
- B. G. Ershov and A. V. Gordeev, Radiat. Phys. Chem., 2008, 77, 928 CrossRef CAS.
- S. Steenken, Chem. Rev., 1989, 89, 503 CrossRef CAS.
- M. D. Evans, M. Dizdaroglu and M. S. Cooke, Mutat. Res., 2004, 567, 1 CAS.
- Y. Wang, Chem. Res. Toxicol., 2008, 21, 276 CrossRef CAS PubMed.
- T. Finkel and N. J. Holbrook, Nature, 2000, 408, 239 CrossRef CAS PubMed.
- M. A. Lovell, S. Soman and M. A. Bradley, Mech. Ageing Dev., 2011, 132, 443 CrossRef CAS PubMed.
- K. Satoh, B. C. Berk and H. Shimokawa, Nitric Oxide, 2011, 25, 211 CrossRef CAS PubMed.
- T. B. Kryston, A. B. Georgiev, P. Pissis and A. G. Georgakilas, Mutat. Res., 2011, 711, 193 CrossRef CAS PubMed.
- A. Kotsinas, V. Aggarwal, E. J. Tan, B. Levy and V. G. Gorgoulis, Cancer Lett., 2012, 327, 97 CrossRef CAS PubMed.
- H. C. Box, E. E. Budzinski, J. D. Dawidzik, J. C. Wallace, M. S. Evans and J. S. Gobey, Radiat. Res., 1996, 145, 641 CrossRef CAS PubMed.
- H. C. Box, E. E. Budzinski, J. B. Dawidzik, J. S. Gobey and H. G. Freund, Free Radical Biol. Med., 1997, 23, 1021 CrossRef CAS PubMed.
- H. C. Box, E. E. Budzinski, J. B. Dawidzik, J. C. Wallace and H. Iijima, Radiat. Res., 1998, 149, 433 CrossRef CAS PubMed.
- H. C. Box, H. B. Patrzyc, J. B. Dawidzik, J. C. Wallace, H. G. Freund, H. Iijima and E. E. Budzinski, Radiat. Res., 2000, 153, 442 CrossRef CAS PubMed.
- S. Bellon, J. L. Ravanat, D. Gasparutto and J. Cadet, Chem. Res. Toxicol., 2002, 15, 598 CrossRef CAS PubMed.
- C. Gu and Y. Wang, Biochemistry, 2004, 43, 6745 CrossRef CAS PubMed.
- H. Hong, H. Cao, Y. Wang and Y. Wang, Chem. Res. Toxicol., 2006, 19, 614 CrossRef CAS PubMed.
- Y. Jiang, H. Hong, H. Cao and Y. Wang, Biochemistry, 2007, 46, 12757 CrossRef CAS PubMed.
- H. Hong, H. Cao and Y. Wang, Nucleic Acids Res., 2007, 35, 7118 CrossRef CAS PubMed.
- H. Cao and Y. Wang, Nucleic Acids Res., 2007, 35, 4833 CrossRef CAS PubMed.
- L. C. Colis, P. Raychaudhury and A. K. Basu, Biochemistry, 2008, 47, 8070 CrossRef CAS PubMed.
- J. Wang, H. Cao, C. You, B. Yuan, R. Bahde, S. Gupta, C. Nishigori, L. J. Niedernhofer, P. J. Brooks and Y. Wang, Nucleic Acids Res., 2012, 40, 7368 CrossRef CAS PubMed.
- H. B. Patrzyc, J. B. Dawidzik, E. E. Budzinski, H. G. Freund, J. H. Wilton and H. C. Box, Radiat. Res., 2012, 178, 538 CrossRef CAS PubMed.
- A. Romieu, S. Bellon, D. Gasparutto and J. Cadet, Org. Lett., 2000, 2, 1085 CrossRef CAS PubMed.
- S. Bellon, D. Gasparutto, A. Romieu and J. Cadet, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 967 CAS.
- Q. Zhang and Y. Wang, J. Am. Chem. Soc., 2003, 125, 12795 CrossRef CAS PubMed.
- Q. Zhang and Y. Wang, J. Am. Chem. Soc., 2004, 126, 13287 CrossRef CAS PubMed.
- Y. Zeng and Y. Wang, J. Am. Chem. Soc., 2004, 126, 6552 CrossRef CAS PubMed.
- Q. Zhang and Y. Wang, Nucleic Acids Res., 2005, 33, 1593 CrossRef CAS PubMed.
- H. Hong and Y. Wang, J. Am. Chem. Soc., 2005, 127, 13969 CrossRef CAS PubMed.
- Y. Zeng and Y. Wang, Nucleic Acids Res., 2006, 34, 6521 CrossRef CAS PubMed.
- Y. Zeng and Y. Wang, Biochemistry, 2007, 46, 8189 CrossRef CAS PubMed.
- G. Lin, J. Zhang, Y. Zeng, H. Luo and Y. Wang, Biochemistry, 2010, 49, 2346 CrossRef CAS PubMed.
- M. Dizdaroglu and P. Jaruga, Free Radical Res., 2012, 46, 382 CrossRef CAS PubMed.
- S. Bellon, D. Gasparutto, C. Saint-Pierre and J. Cadet, Org. Biomol. Chem., 2006, 4, 3831 CAS.
- Q. Zhang and Y. Wang, Chem. Res. Toxicol., 2005, 18, 1897 CrossRef CAS PubMed.
- C. Gu and Y. Wang, Biochemistry, 2005, 44, 8883 CrossRef CAS PubMed.
- P. Raychaudhury and A. K. Basu, Biochemistry, 2011, 50, 2330 CrossRef CAS PubMed.
- Z. Yang, L. C. Colis, A. K. Basu and Y. Zou, Chem. Res. Toxicol., 2005, 18, 1339 CrossRef CAS PubMed.
- C. Gu, Q. Zhang, Z. Yang, Y. Wang, Y. Zou and Y. Wang, Biochemistry, 2006, 45, 10739 CrossRef CAS PubMed.
- J. Garrec, C. Patel, U. Rothlisberger and E. Dumont, J. Am. Chem. Soc., 2012, 134, 2111 CrossRef CAS PubMed.
- E. Dumont, T. Dršata, C. F. Guerra and F. Lankaš, Biochemistry, 2015, 54, 1259 CrossRef CAS PubMed.
- V. Labet, C. Morell, A. Grand, J. Cadet, P. Cimino and V. Barone, Org. Biomol. Chem., 2008, 6, 3300 CAS.
- C. D. M. Churchill, L. A. Eriksson and S. D. Wetmore, Chem. Res. Toxicol., 2011, 24, 2189 CrossRef CAS PubMed.
- C. Dupont, C. Patel and E. Dumont, J. Phys. Chem. B, 2011, 115, 15138 CrossRef CAS PubMed.
- C. Patel, J. Garrec, C. Dupont and E. Dumont, Biochemistry, 2013, 52, 425 CrossRef CAS PubMed.
- J. Garrec and E. Dumont, Chem. Res. Toxicol., 2014, 27, 1133 CrossRef CAS PubMed.
- C. D. M. Churchill, L. A. Eriksson and S. D. Wetmore, J. Phys. Chem. B, 2016, 120, 1195 CrossRef CAS PubMed.
- J. Gu, J. Wang and J. Leszczynski, Nucleic Acids Res., 2010, 38, 5280 CrossRef CAS PubMed.
- C. D. M. Churchill and S. D. Wetmore, Phys. Chem. Chem. Phys., 2011, 13, 16373 RSC.
- J. P. Cerón-Carrasco, D. Jacquemin and E. Dumont, J. Phys. Chem. B, 2013, 117, 16397 CrossRef PubMed.
- G. Scalmania and M. J. Frisch, J. Phys. Chem. B, 2010, 132, 114110 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- X. J. Lu, H. J. Bussemaker and W. K. Olson, Nucleic Acids Res., 2015, 43, e142 CrossRef PubMed.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2012, 112, 289 CrossRef CAS PubMed.
- H. C. Box, J. B. Dawidzik and E. E. Budzinski, Free Radical Biol. Med., 2001, 31, 856 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12713h |
| This journal is © The Royal Society of Chemistry 2018 |