
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical and experimental evidence for rGO-4-PP Nc as a metal-free Fenton-like catalyst by tuning the electron distribution
Guangfei Yuac,
Lai Lyu *bd,
Fagen Zhangb,
Dengbiao Yane,
Wenrui Caobd and
Chun Huabcd
*bd,
Fagen Zhangb,
Dengbiao Yane,
Wenrui Caobd and
Chun Huabcd
aKey Laboratory of Drinking Water Science and Technology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China
bKey Laboratory for Water Quality and Conservation of the Pearl River Delta, Ministry of Education, School of Environmental Science and Engineering, Guangzhou University, Guangzhou, 510006, China. E-mail: lyulai@gzhu.edu.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dResearch Institute of Environmental Studies at Greater Bay, Guangzhou University, Guangzhou, 510006, China
eSchool of Environmental and Chemical Engineering, Tianjin Polytechnic University, Tianjin 300387, China
First published on 17th January 2018
Abstract
The application of the classical Fenton reaction has long been limited by several problems, such as metallic sludge and narrow pH range, which derived from the metal components in the catalyst. Developing a metal-free Fenton catalyst may efficiently address these problems. Here, we firstly perform a density functional theory (DFT) study to explore the possibility of developing the 4-phenoxyphenol molecule-doped reduced graphene oxide nanocomposite (rGO-4-PP Nc) as a metal-free Fenton-like catalyst by tuning the electron distribution. The theoretical calculation results reveal that rGO-4-PP Nc can act as an efficient Fenton-like catalyst for H2O2 activation and pollutant degradation through formation of electron-rich O and electron-deficient C centers on the C–O–C bridge. The actual rGO-4-PP Nc is also prepared via a surface complexation and copolymerization process. The experimental evidence, such as that gained from XRD, FIIR and EPR analysis, confirm the theoretical models and the dual-reaction-center Fenton-like mechanism. This work provides a basis for theoretical calculation to guide the actual synthesis and prediction of catalytic activity of the Fenton-like catalysts, and also offers a creative perspective to develop new generation metal-free Fenton catalysts by tuning the electron distribution using organic polymers.
1. Introduction
The Fenton reaction, in which highly aggressive hydroxyl radicals (˙OH) are generated from H2O2 by reduction with Fe2+, is one of the powerful advanced oxidation processes (AOPs) that can be used for the degradation of organic pollutants.1 Its widespread applications have also been observed in materials synthesis,2 medicine3 and polishing.4 However, several drawbacks hinder the practical application of the classical homogeneous Fenton reaction, including the required strong acidic condition (pH = 2–3.5), the accumulation of Fe-containing sludge and the requirement for a neutralization step of the effluent and sludge disposal.5 To address these problems, the metal-containing heterogeneous Fenton catalysts, such as zero-valent metals, metallic oxides, supported metal composites and metal-doped solids, have been developed as alternatives to the classical Fenton process.Unfortunately, the redox of the metal ions in the single metal site of the metal-containing Fenton catalyst inevitably results in a series of problems, including the narrow available pH range, the rate-limiting step in the reduction of M(n+m)+ to Mn+ by oxidizing H2O2,6,7 the excess consumption of H2O2,8,9 and the secondary pollution owing to the metal leaching.10,11 To avoid these problems caused by metal species thoroughly, development of metal-free Fenton-like catalysts is one of the most effective means of the settlements.
Recently, our study12 shows an efficient Fenton-like degradation of refractory organic pollutants in mild conditions by constructing the electron-rich and electron-deficient areas (the dual reaction centers) on the hydroxylated C-doped g-C3N4/CuCo–Al2O3 nanocomposite (OH–CCN/CuCo–Al2O3). This gives us an inspiration to explore the possibility of developing efficient metal-free Fenton-like catalysts by tuning the electron distribution in theory.
Density functional theory (DFT) calculation, as an effective approach to study the electronic properties of the materials and the oxygen reduction reaction mechanisms, has been widely used during the past few years,13–15 which can be chosen as a credible theoretical research method for the electron distribution analysis and the interaction process analysis of the Fenton-like catalyst and H2O2. Graphene, a two-dimensional single layer of graphite consisting of sp2-hybridized carbon atoms, can be adopted as the substrate material for its unique electrical and mechanical properties.16–18 4-Phenoxyphenol (4-PP) as a molecular material with two benzene rings, a C–O–C bond and a hydroxyl group, shows an unusual flexibility in its moderately hydrogen bonded O–H⋯OR66 (12) rings, which can breathe to accommodate different solvent molecules into the channels,19 which results in that 4-PP can be used as an excellent organic ligand.
Therefore, for the first time, we perform a DFT calculation study to explore the possibility of developing the 4-phenoxyphenol molecule-doped reduced graphene oxide nanocomposite (rGO-4-PP Nc) as an efficient metal-free Fenton-like catalysts by tuning the electron distributions in this work. The theoretical geometric and electronic properties, HOMO/LUMO analysis, electrostatic potential distributions and the computation of the interaction process of active sites with H2O2 reveal that rGO-4-PP Nc can act as an efficient Fenton-like catalyst through formation of the electron-rich O and electron-deficient C centers on the C–O–C bridge. The actual rGO-4-PP Nc is prepared via a surface complexation and copolymerization process. The experimental characterizations and methods successfully verify the theoretical models and the dual-reaction-center Fenton-like mechanism.
2. Computational and experimental methods
2.1. Computational methods
DFT calculations were performed using Gaussian 09 software.20 The structures were optimized at the B3LYP/6-31G (d,p) computational levels. The B3LYP method was chosen for its widely recognized high quality especially in this type of organic chemistry reaction. Subsequent frequency calculations were performed at the same level in order to verify that these structures were the local minimas on the potential energy surface. The SMD is chosen to simulate the effects of water.21For all the models we established and shown in our figures, the C, O and H atoms are represented by the grey, red and white circles, respectively. In addition, the Multiwfn program was used to investigate the valence-electron density.22
To find correct spin multiplicity (SM) values for the structures, single-point energy (SPE) and stable calculations were performed at the beginning of the study. Different SM values were adopted for SPE calculations, and the SM value that had the lowest SPE was chosen as the correct spin multiplicity to perform the subsequent calculations for the corresponding systems. After the SM values had been selected, stable calculations were carried out to verify the wavefunctions were stable.
2.2. Chemicals and reagents
Bisphenol A (BPA, ≥99%) and phenytoin (PHT, 99%) were obtained from Acros (Geel, Belgium). Horseradish peroxidase (POD, 99%), 2-chlorophenol (2-CP, ≥99%) and N,N-diethyl-p-phenylenediamine sulfate (DPD, 98%) were purchased from Sigma-Aldrich (St. Louis, USA). Hydrogen peroxide (H2O2, 30%, w/w) was obtained from Sinopharm Chemical Reagent Co. (Shanghai, China). Deionized water was used throughout the study.2.3. Catalyst preparation
Graphite oxide (GO) was synthesized from natural graphite powder by a modified Hummers method.23 The 4-PP-doped reduced graphene oxide nanocomposite (rGO-4-PP Nc) was synthesized through the following methods: 0.5 g GO powder and 0.2 g 4-phenoxyphenol (4-PP) were dissolved into 25 mL ethanol, then it was stirred for 1 h and got sonication treatment for 30 min to obtain an evenly dispersed solution. This solution was subsequently placed in a water bath and stirred for 4 h at 70 °C, then the temperature was maintained 90 °C before the solution dried out by evaporating to obtain the solid precursors. The precursors were carefully grounded, transferred to an alumina crucible and heated to 350 °C (maintained 1 h) with the rate of 5 °C min−1 by a muffle furnace for the annealing copolymerization process. After it cooled naturally, the black powder we obtained was washed by turns with ethanol and deionized water for several times and then dried out by the oven. Finally the rGO-4-PP Nc sample was prepared.2.4. Characterization
Fourier-transform infrared spectroscopy (FTIR) was obtained using a Nicolet 8700 FTIR spectrophotometer (Thermo Fisher Scientific Inc., USA). X-ray diffraction (XRD) patterns were measured by a Scintag-XDS-2000 diffractometer with Cu Kα radiation (λ = 1.540598 Å) operating at 40 kV and 40 mA. The EPR spectra were recorded on a Bruker A300-10/12 electron paramagnetic resonance (EPR) spectrometer.2.5. Experiment methods
Typically, 50 mL pollutant solution (such as 2-CP, 10 mg L−1) and 20 mg catalyst powder were put into a beaker (pH values were adjusted using HCl and NaOH solution) and mixed with magnetic stirring for 30 min to reach the adsorption/desorption equilibrium. 12 mM H2O2 was then added to the suspension under magnetic stirring, which was kept throughout the experiment. 1 mL aliquots were obtained and filtered through a Millipore filter (pore size 0.22 μm) at the given time intervals, subsequently the pollutants were determined immediately by a 1200 series HPLC (Agilent, U.S.A.) equipped with a UV detector and a ZORBAX Eclipse XDB-C18 column. The mobile phase is composed of a mixture of methanol/water at a flow rate of 1 mL min−1. The H2O2 decomposition experiments were also carried out using the similar procedure. The concentration of H2O2 in filtrate was determined immediately by the DPD-POD method, as previously reported in the literature.243. Results and discussion
3.1. Geometric and electronic properties of catalyst models
A finite-sized graphene sheet was modeled with 6 benzene rings including 22 carbon and 12 hydrogen atoms. Carbon atoms on the edge of the graphene were terminated by hydrogen atoms to avoid the effect of unsaturated boundary. The properties of the finite-sized model may be different to an extent from that in the real system because of the size and edge effects. However, it is expected to be qualitatively reliable for using the results acquired from the current model to predict the local chemical properties. Spin multiplicity (SM) numbers were found to be 1 for the H2O2, 4-PP molecules and graphene sheet; 3 for catalyst model (rGO-4-PP Nc). These selected SM numbers were adopted throughout all calculations.Fig. 1 shows the optimized geometric structures of graphene (C22H12), catalyst model (rGO-4-PP Nc, C34H20O2) and 4-PP molecule (C12H10O2). The optimized graphene has a planar structure with good symmetry, and the 4-PP molecule is connected with graphene by a C–O–C bond in rGO-4-PP Nc. In addition, the C–O bond lengths of C35–O34 and C13–O34 were calculated to be 1.392 and 1.386 Å, respectively, and the C13–O34–C35 bond angle was 119.93° in rGO-4-PP Nc. In contrast, in the 4-PP molecule, the C6–O23 bond length and C6–O23–H24 bond angle in the corresponding position were calculated to be 1.375 Å and 109.03°, respectively.
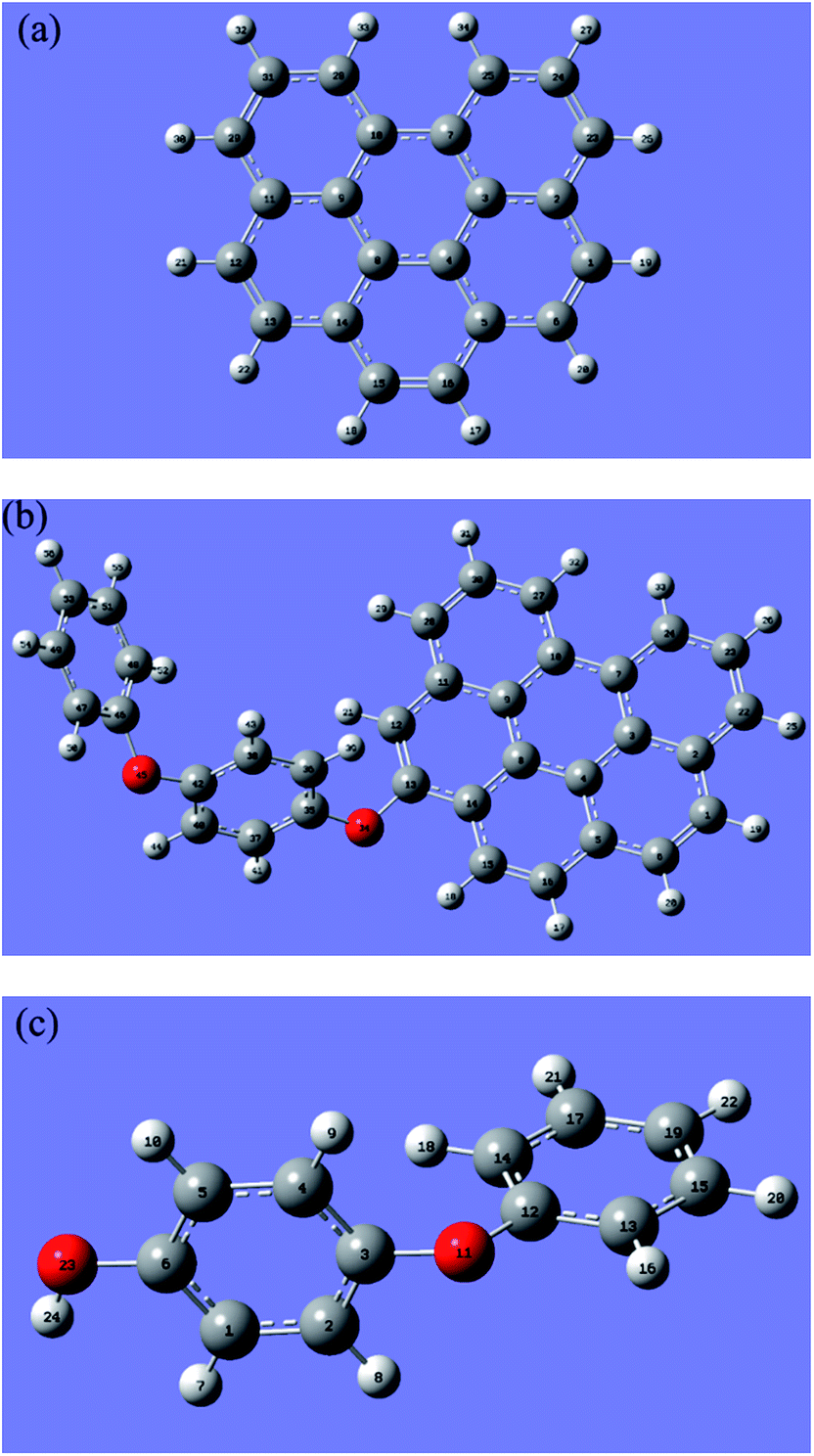 | ||
| Fig. 1 Optimized structures of graphene (a), rGO-4-PP Nc (b), and 4-PP molecule (c) with labeling of all the atoms. Grey, red and white circles represent C, O and H atoms, respectively. | ||
As the electronic properties of the models are of great importance for their chemical activity, here we start to discuss them by two different approaches. Charge distribution of graphene (a) and rGO-4-PP Nc (b) is shown in Fig. 2 with Mulliken charges displaying on the atoms. It is obvious that the atomic charge values in graphene are bilateral symmetry, so either side of this structure can be selected for subsequent analysis. In rGO-4-PP Nc, the oxygen atom O1 has a considerably low atomic charge value −0.605, which is approximately equal to −0.606 of the other oxygen atom. The carbon atoms adjacent to the O1 (C1 and C2) have the positive atomic charge value 0.247 and 0.319, respectively. It is noted that the corresponding carbon atom C3 in graphene has the negative atomic charge −0.163, which is 0.410 lower than the C1 in rGO-4-PP Nc. The reason for this significant change is the larger electronegativity of O compared to the C atom, which makes the charge transfers from neighboring C1 atom to the O1 atom and the carbon atom itself positive in rGO-4-PP Nc.
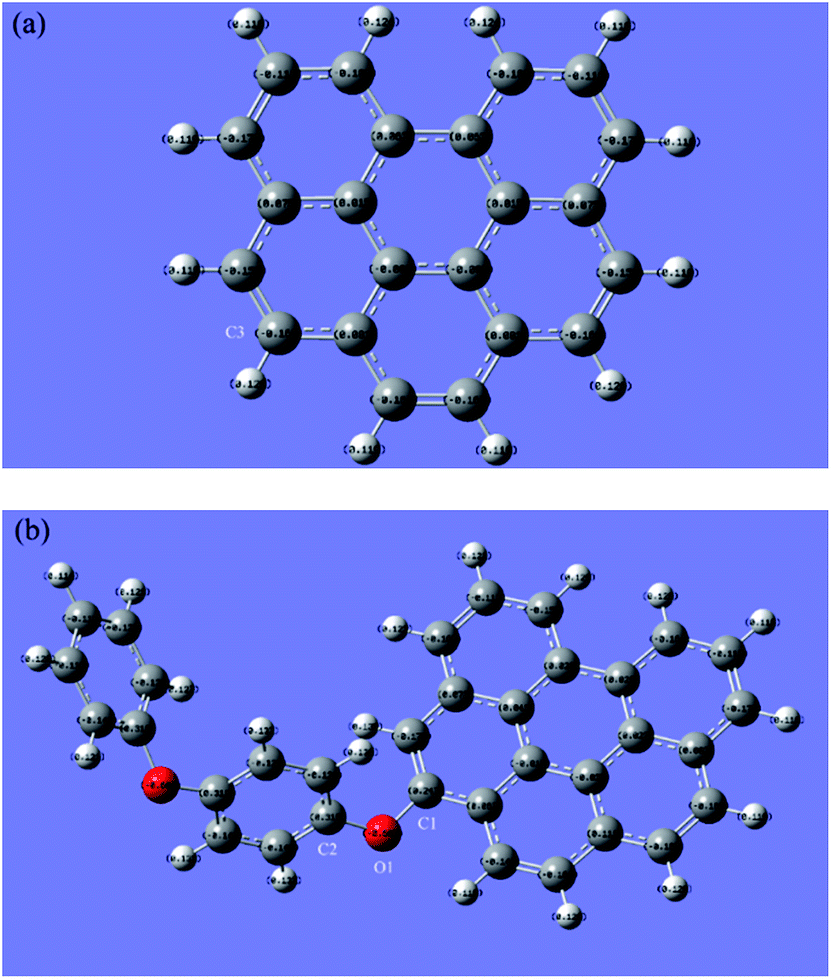 | ||
| Fig. 2 Charge distribution of (a) graphene and (b) rGO-4-PPNc. The numbers on the circles are Mulliken charge values for the atoms. Grey, red and white circles represent C, O and H atoms, respectively. | ||
Fig. 3 shows the two-dimensional valence-electron density color-filled maps of rGO-4-PP Nc in the C1–O1–C2 plane. Obviously, the largest electron distribution area appears around the O atom, and the narrow electron distribution area appears around the C atoms. In addition, the maximum valence-electron density around the O atom is as high as ∼1.15 a.u. compared with the C atoms (C1 and C2) in the C–O–C linker, which suggests that there is an electron-rich area around the O atom and there are electron-deficient areas around the adjacent C atoms.
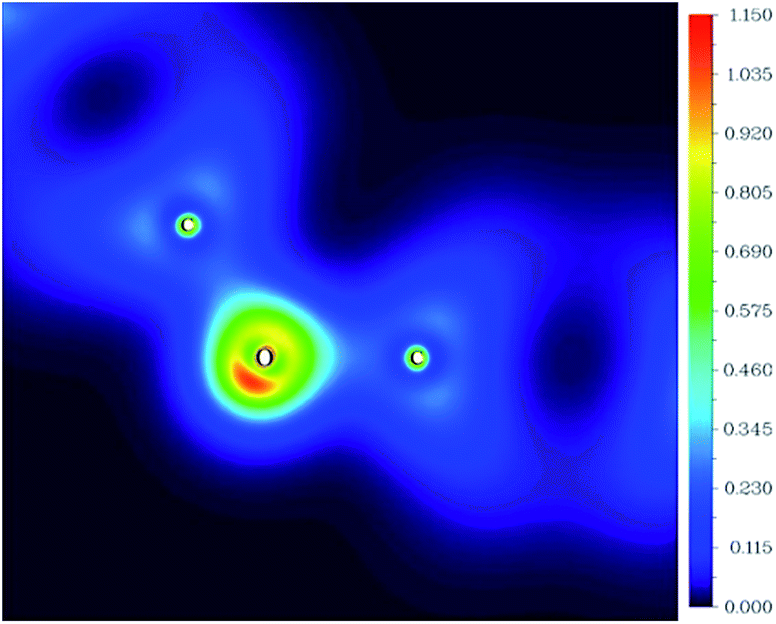 | ||
| Fig. 3 The two-dimensional valence-electron density color-filled map of rGO-4-PP Nc in the C1–O1–C2 plane. The length unit is Angstrom. | ||
3.2. HOMO/LUMO analysis for reactivity
Since we have known that there are significant differences of electronic properties between the rGO-4-PP Nc and graphene, it is very necessary to compare their chemical reactivity, which could be used to predict the catalytic performance to a certain extent. The HOMO (highest occupied molecular orbital)–LUMO (lowest unoccupied molecular orbital) gap has been widely used as an indicator of kinetic stability, and it is generally recognized that the smaller the gap is, the lower the kinetic stability and the higher the chemical reactivity are within a certain range, because it indicates energetically favorable for the LUMO to obtain electrons or for the HOMO to lose electrons. The results of graphene, rGO-4-PP Nc and 4-PP that we have calculated for comparison are listed in Table 1. From Table 1, it is clear that the α,β-HOMO–LUMO gap of 2.23 and 1.88 eV of rGO-4-PP Nc are considerably smaller than the values 3.55 and 5.60 eV of the graphene and 4-PP, respectively. Hence it could be considered that the chemical reactivity of the rGO-4-PP Nc has been significantly improved. We also obtain the HOMO and LUMO spatial distributions of α and β electron for the graphene and rGO-4-PP Nc structures, as shown in Fig. 4. For the graphene, both HOMO and LUMO have the same spatial distribution for α electron and β electron, and the spatial distribution of them are delocalized. For the rGO-4-PP Nc, however, it is obvious that part of the HOMO and LUMO of α electron as well as the LUMO of β electron are localized in the oxygen atom next to the graphene.| Spin | G | rGO-4-PP Nc | 4-PP | |||
|---|---|---|---|---|---|---|
| α | β | α | β | α | β | |
| a HOMO–LUMO gap is the difference between LUMO and HOMO energy levels. | ||||||
| HOMO | −5.26 | −5.26 | −3.24 | −5.67 | −5.92 | −5.92 |
| LUMO | −1.71 | −1.71 | −1.01 | −3.79 | −0.32 | −0.32 |
| HOMO–LUMO gap | 3.55 | 3.55 | 2.23 | 1.88 | 5.60 | 5.60 |
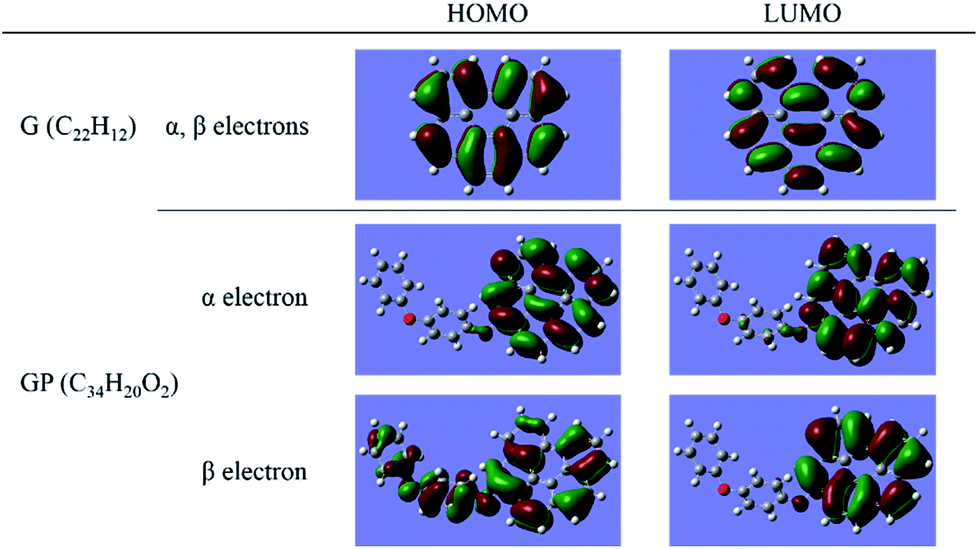 | ||
| Fig. 4 HOMO and LUMO spatial distributions of α electron and β electron for graphene and rGO-4-PP Nc. | ||
3.3. DFT calculations for interaction process of active centers and H2O2
As we have known that the O1 atom in the catalyst has a considerably low atomic charge (Fig. 2b) and there is a significant electron-rich area around it besides part of the HOMO and LUMO localized there, it is reasonable to regards that area as the potential active site. Electrostatic potential (ESP) distribution on the van der Waals surface has long been used to predict the reaction sites.25,26 The negative and positive regions of ESP are usually expected to be the active sites for the reduction and oxidation reaction, respectively. A common method of investigating the distribution of ESP is to project that onto the molecular surface with different colors according to its values. This type of graphical analysis is very intuitive, as shown in Fig. 5.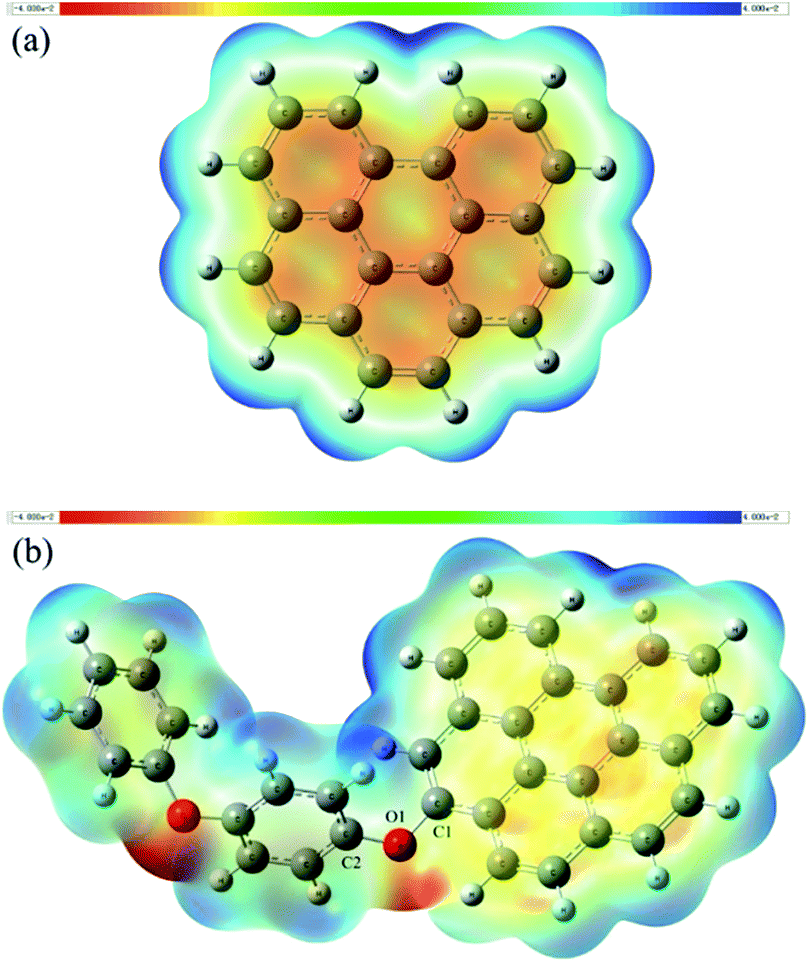 | ||
| Fig. 5 Electrostatic potential (ESP) distribution of (a) graphene and (b) rGO-4-PP Nc. | ||
The red and blue colours represent the most negative potential (electron-rich) and positive potential (electron-deficient) parts of the graphene and rGO-4-PP Nc. Analysis of this result clearly indicate that the ESP distribution of graphene is very uniform for the identical elements, which is significantly different from the situations of rGO-4-PP Nc. In rGO-4-PP Nc, the most electronegative parts are the regions around the two oxygen atoms that are the electron rich areas (red) as well, hence we name these oxygen atoms, especially the O1, as the electron-rich oxygen centers. In addition, it is noted that the negative potential among the graphene carbocyclic rings in Fig. 5b is obviously lower than that in Fig. 5a, which indicates that the π-electron has transferred from the neighboring carbocyclic rings to the surrounding of the O1 atom. The most electropositive and electron deficient regions (blue) of rGO-4-PP Nc are mainly located around the hydrogen atoms that are surrounding the carbocyclic rings near the side of O1. However, it is not appropriate to regard these H atoms as active sites because they are added deliberately for the stability of the fragment whereas the real graphene has an infinite plane-extended structure. Besides, the carbon atoms near O1 also have the relatively positive potential.
According to all the results we have discussed above, the O1 atom as the electron-rich oxygen center is exactly the active site we expected, and the carbon atoms near it (especially C1 and C2) could be regarded as the electron-deficient carbon center of active site for their relatively significant positive charge and low electron density (Fig. 2b and 3).
To clarify the interaction process of H2O2 and the catalyst, the H2O2 adsorption on the dual reaction centers of rGO-4-PP Nc was theoretically modeled through DFT calculations. Generally, the H2O2 can both obtain and lose electrons to realize the reduction and oxidation reaction, respectively. The reduction process results in the formation of ˙OH that is the traditional reactive oxygen species (ROS) in Fenton reaction while the oxidation process results in the formation of HO2˙/O2˙− which would easily transform to O2 and causes the invalid decomposition of H2O2. As for heterogeneous Fenton process, the subsequent reactions can be hardly carried out unless the H2O2 absorbs to the active sites of catalyst. Fig. 6a and b shows the results of our optimum calculations, which we placed the H2O2 around the active sites initially.
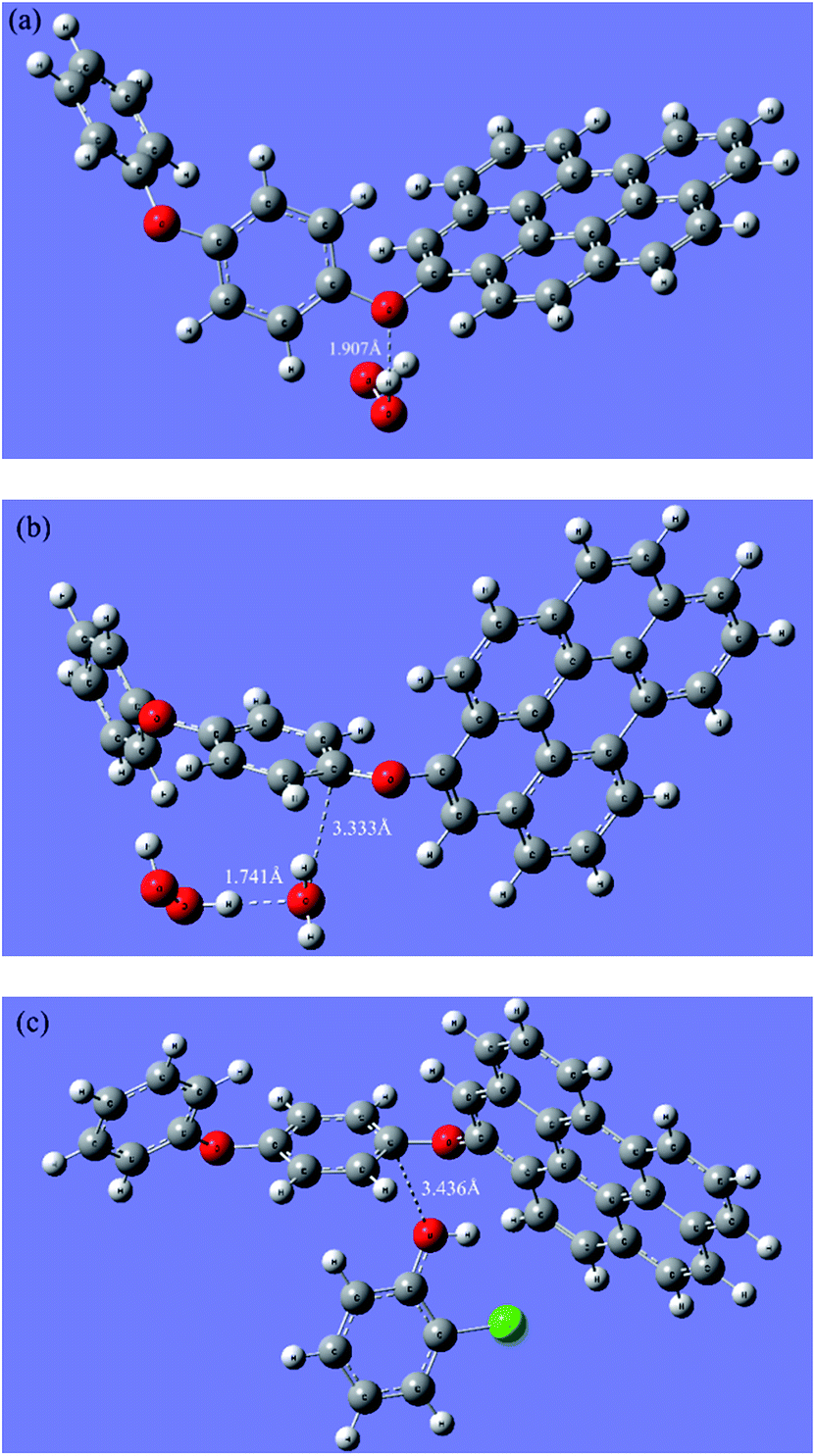 | ||
| Fig. 6 Optimized structures of H2O2 absorbing to (a) the electron-rich oxygen center and (b) the electron-deficient carbon center on rGO-4-PP Nc. (c) Optimum structures of 2-CP absorbing to the electron-deficient carbon center on rGO-4-PP Nc. | ||
In the electron-rich oxygen center (Fig. 6a), H2O2 is connected to the O atom directly by a hydrogen bond (H–O–O–H⋯O) whose bond length is 1.907 Å. However, on the electron-deficient center (Fig. 6b), H2O2 does not directly adsorb on the C atom, but connects the O of H2O, which adsorb to C atom by a C⋯O bond (3.333 Å), with the H atom of H2O2 through a hydrogen bond (1.741 Å). These optimum structures which are relatively energetically stable confirm that the H2O2 can indeed absorb to the active sites as we expected and the dual reaction centers mechanism was reasonable.
In the presence of pollutants, the results are completely different. As shown in Fig. 6c, the 2-CP molecule can directly absorb onto the C atom by its O atom of phenolic hydroxyl, and the C⋯O bond length is 3.436 Å. The connection length between 2-CP and the electron-deficient center is much closer than that of the H2O2 adsorption mode on the electron-deficient centers (3.333 + 1.741 Å), which suggests that the electron transfer from the electron-rich pollutants to the electron-deficient centers of rGO-4-PP Nc are more quickly and easily than that from H2O2 to the electron-deficient centers of rGO-4-PP Nc. This results indicate that the electron-rich pollutants can be used as electron-donors to prevent H2O2 from being ineffective oxidized and decomposed in the formed dual-reaction-center Fenton-like system.
3.4. Experimental evidence for creation of dual reaction centers and its Fenton-like reaction mechanisms
The physical rGO-4-PP Nc sample was prepared by combining molecular doping and copolymerization process. As a reference, GO was synthesized by a modified Hummers method.X-ray diffraction (XRD) was employed to investigate the structures of the GO and rGO-4-PP Nc, as shown in Fig. 7a. The sharp diffraction peak at 10.4° for GO is a typical characteristic (001) XRD peak, which corresponds to a slice gap of 9.0 Å between the individual GO sheets.27 In addition, no diffraction peak corresponding to graphite was observed, indicating that the graphite was entirely oxidized to GO sheets.28 Very differently, this peak disappeared on rGO-4-PP Nc, and two obvious diffraction peaks at 26.0° and 42.9° were observed, implying that GO substrate was fully reduced to rGO state. However, the positions of these peaks were inconsistent with the standard XRD peaks of rGO,29 indicating that the obtained rGO-4-PP Nc possessed a completely new structure compared to GO and rGO. Fig. 7b shows the FTIR spectra of the catalyst rGO-4-PP Nc. Three marked peaks were observed at about 1211.2 cm−1, 1571.9 cm−1 and 1732.0 cm−1 that are attributed to the C–O–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO groups respectively. This result suggested that the three kinds of chemical bonds exist in the rGO-4-PP Nc structure, implying the successful synthesis of the Fenton-like catalyst by forming the C–O–C bridges in the specific configuration.
C and CO groups respectively. This result suggested that the three kinds of chemical bonds exist in the rGO-4-PP Nc structure, implying the successful synthesis of the Fenton-like catalyst by forming the C–O–C bridges in the specific configuration.
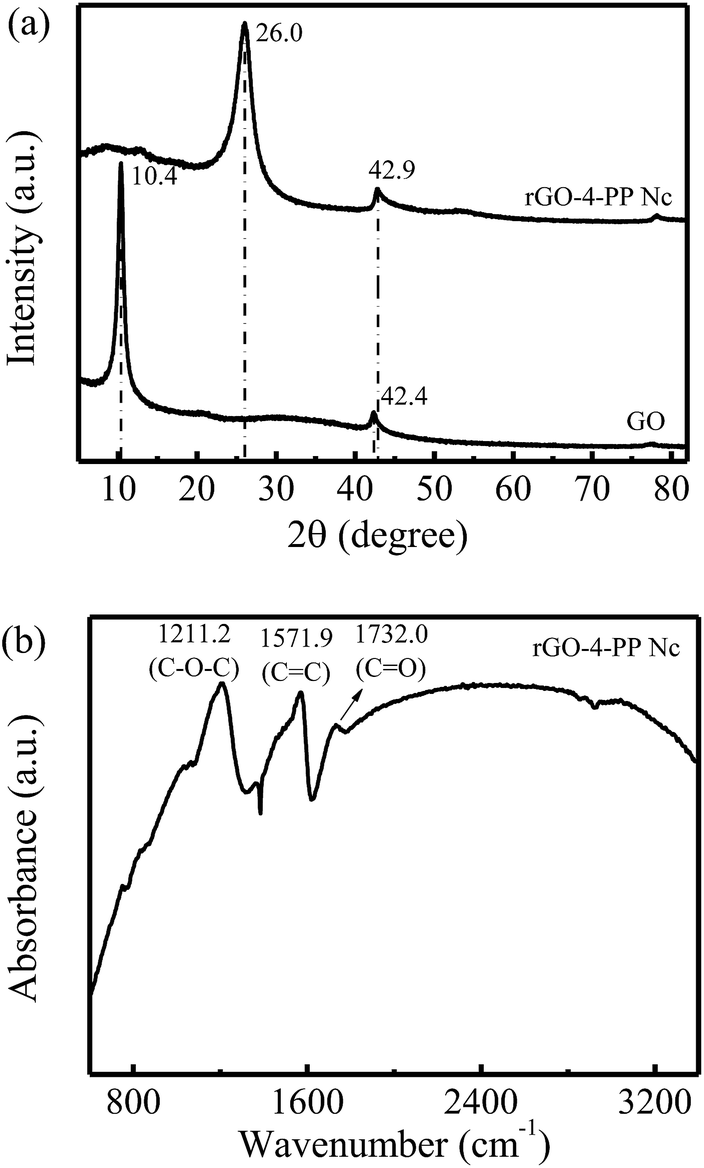 | ||
| Fig. 7 Characterization of catalysts. (a) XRD patterns of the GO and rGO-4-PP Nc. (b) FTIR spectra of rGO-4-PP Nc. | ||
The presence of single electrons in the rGO-4-PP Nc sample was investigated by EPR spectroscopy. As shown in Fig. 8, a Lorentzian line centering at g = 2.0037 was observed for rGO-4-PP Nc, indicating the generation of unpaired electrons on the catalyst,30 which was mainly due to the bonding of the negatively-charged O and edge C atoms carrying π-electronic spins.31 This result revealed that the formed C–O–C bonding bridges in rGO-4-PP Nc gathered many free electrons around the doping O, forming electron-rich centers in rGO-4-PP Nc. This is the evidence of the successful construction of the dual reaction centers.
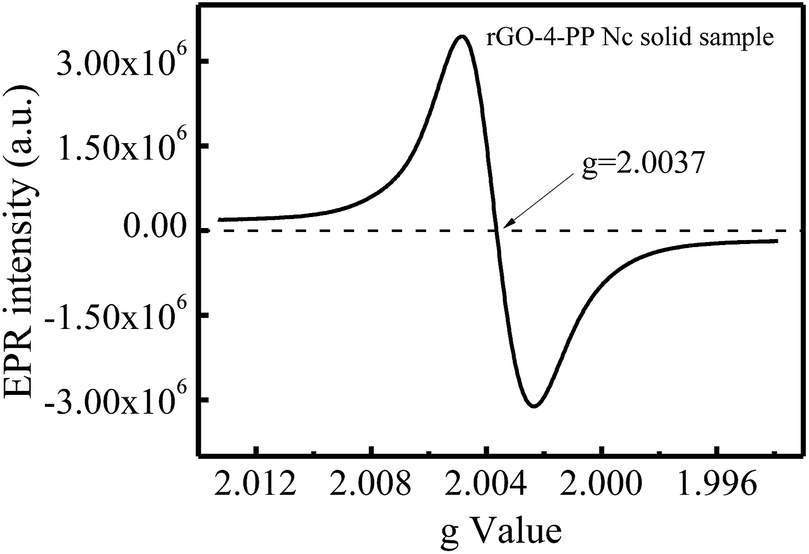 | ||
| Fig. 8 EPR spectra of the rGO-4-PP Nc solid sample. | ||
Fig. 9 showed the results of the BMPO-trapped EPR spectra that could further verify the dual-reaction-center mechanism. In the absence of H2O2, four distinct signals of BMPO-HO2˙/O2˙− were observed in the methanol dispersions of rGO-4-PP Nc (Fig. 9a), indicating that the large amounts of free electrons around the doping O in C–O–C of rGO-4-PP Nc could efficiently reduce the dissolved O2 to HO2˙/O2˙−. However, no signals of BMPO-˙OH appeared in the rGO-4-PP Nc aqueous suspensions without H2O2 (Fig. 9b), indicating that the H2O could not be oxidized to generate ˙OH at the electron-deficient centers. With the addition of H2O2, four evident BMPO-˙OH signals appeared in the rGO-4-PP Nc aqueous suspension (Fig. 9c), which confirmed that H2O2 could be rapidly and efficiently reduced to ˙OH by the single electrons on the electron-rich sites of rGO-4-PP Nc. In addition, four distinct characteristic signals of BMPO-HO2˙/O2˙−were also observed in the system of rGO-4-PP Nc/H2O2 (Fig. 9d), indicating that H2O2 could also be activated at the electron-deficient centers. These results verified the successful creation of the dual reaction centers and revealed the Fenton-like reaction mechanisms.
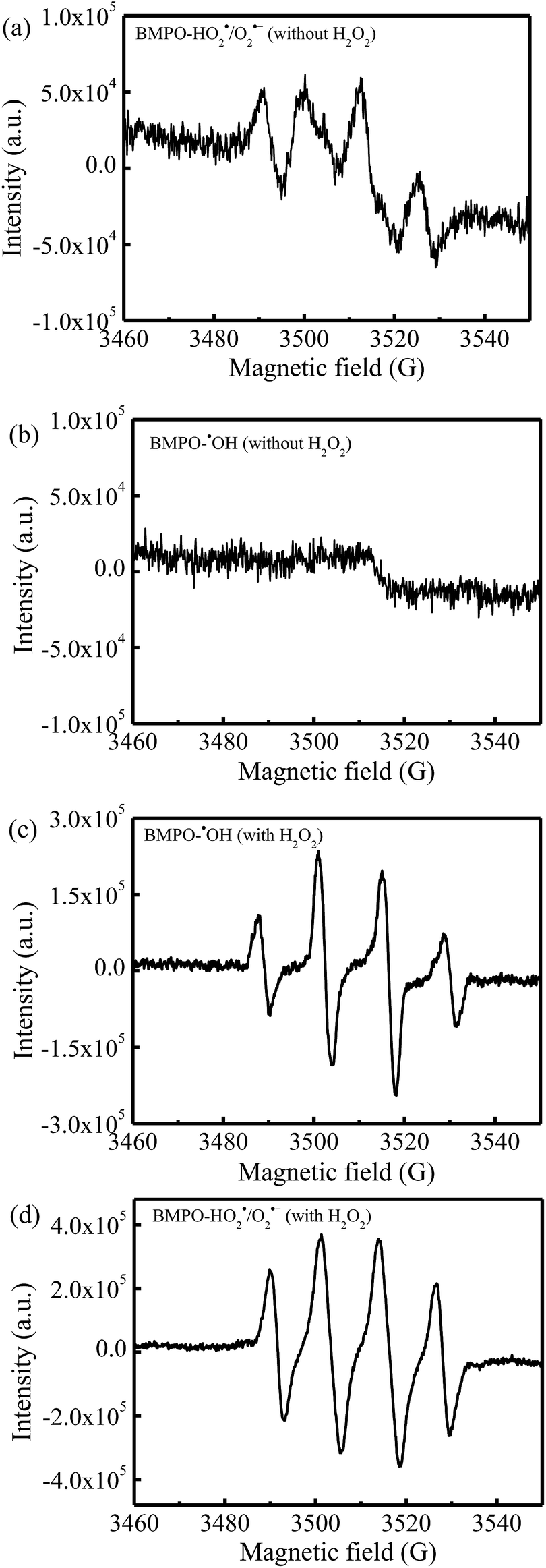 | ||
| Fig. 9 BMPO spin-trapping EPR spectra for (a) HO2˙/O2˙− and (b) ˙OH in the rGO-4-PP Nc suspensions without H2O2, and (c) ˙OH and (d) HO2˙/O2˙− in the rGO-4-PP Nc suspensions with H2O2. | ||
To further investigate the conversion paths of H2O2 on the dual reaction centers in the presence of pollutants, BPA (with phenolic –OH groups) and PHT (without phenolic –OH groups) were selected as the target compounds. The pure water suspension with catalyst was adopted as control. As shown in Fig. 10, in the pure water suspension, ∼8.3% of H2O2 was decomposed within 210 min accompanied by a high reaction rate. However, in PHT suspension, the H2O2 decomposition was very slow and only ∼1.0% of H2O2 was decomposed in the same time interval. This decomposition degree increased to 4.0% in the BPA suspension, and the reaction rate was significantly faster than that in the PHT suspension. For all the three kinds of suspensions, H2O2 could be absorbed on the electron-rich oxygen centers and be reduced to ˙OH rapidly. However, the reaction processes were apparently different in the electron-deficient carbon centers. As shown in Fig. 6b, H2O could be absorbed on the electron-deficient carbon center when there is no pollutant in the system, which is prone to be converted to –OH on the catalyst; hence the H2O2 can either absorb to the carbon center directly or be connected by a –OH/H2O. Whichever process is carried out, H2O2 is the only electron donor in the systems without the pollutants. Thus, the decomposition reactions of H2O2 were carried out simultaneously at both the electron-rich and -deficient centers, which caused its reaction rate to be the highest of the three. After adding pollutants (BPA or PHT) in the systems, the electron-rich organic compounds occupied the electron-deficient reaction centers and might act as electron donors, which prevented H2O2 from contacting with the electron-deficient centers and avoided the useless decomposition of H2O2. Thus, the BPA and PHT systems exhibited a lower rate for H2O2 decomposition. However, the apparent difference for H2O2 decomposition in the BPA and PTH systems was caused by the structural properties of the two pollutants. During the reactions, BPA (with phenolic –OH groups) could absorb on the electron-deficient centers and complex with the C sites through its phenolic –OH groups which was able to transfer the electron more quickly, while the PHT without the phenolic –OH groups could only absorb on the center but not form the complexation with the C sites, which could not rapidly transfer the electrons to the electron-deficient centers. Thus, the activity for H2O2 decomposition in the presence of PHT was lower than that in the presence of BPA, resulting in the higher decomposition rate in the BPA suspension being observed.
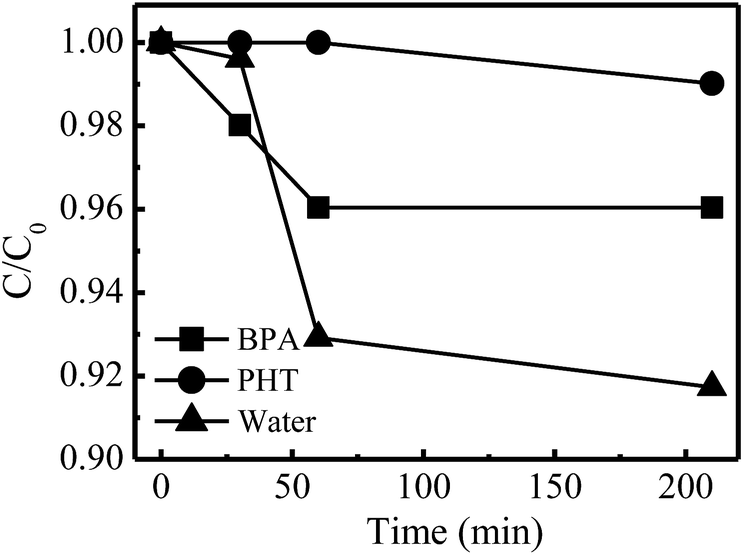 | ||
| Fig. 10 H2O2 decomposition curve of pure water, BPA and PHT suspensions with catalyst (initial BPA/PHT concentration 10 mg L−1, initial H2O2 concentration 10 mM, catalyst concentration 1.0 g L−1, temperature 35 °C, natural pH). | ||
Above results confirmed the formation of the dual reaction centers in the rGO-4-PP Nc and clarified the interaction mechanism of the catalyst with H2O2 and pollutants, which indicated that we can rationally predict the property, performance and the Fenton-like reaction process of the metal-free catalyst through theoretical approaches.
3.5. Fenton-catalytic performance of rGO-4-PP Nc
To investigate the performance of the physical rGO-4-PP Nc catalyst, the typical aromatic compound 2-CP (10 ppm) with phenolic –OH group was chosen as the degradation target under the natural pH conditions (pH = ∼6.8). At the adsorption equilibrium point, ∼29.3% of 2-CP was adsorbed on the surface of rGO-4-PP Nc. After adding H2O2 (Fig. 11a), the degradation rate of 2-CP in the rGO-4-PP Nc suspension could reach 86.4% within 120 min, which was considerably higher than that in the suspensions of graphite, GO and rGO.32 We also carried out the activity evaluation experiments using BPA (10 ppm) as target pollutant. In the neutral mild conditions, the degradation rate of BPA within 120 min could reach ∼75%. The effect of the initial pH value on 2-CP degradation in rGO-4-PP Nc suspension was also determined as presented in Fig. 11a. At pH 3.7, the catalyst shows the highest activity that the degradation rate of 2-CP could reach 91.8% within 120 min. The degradation rate slightly decreased with the increase of the initial pH, but it still could reach ∼78% within 120 min at pH 9.2, indicating that the rGO-4-PP Nc had a wide pH application range. The reusability of rGO-4-PP Nc was examined after getting filtered, washed and dried. As shown in Fig. 11b, the activity of rGO-4-PP Nc for 2-CP degradation did not evidently decrease after 8 continuous cycles of the degradation experiments, which suggested that rGO-4-PP Nc was a stable and efficient catalyst.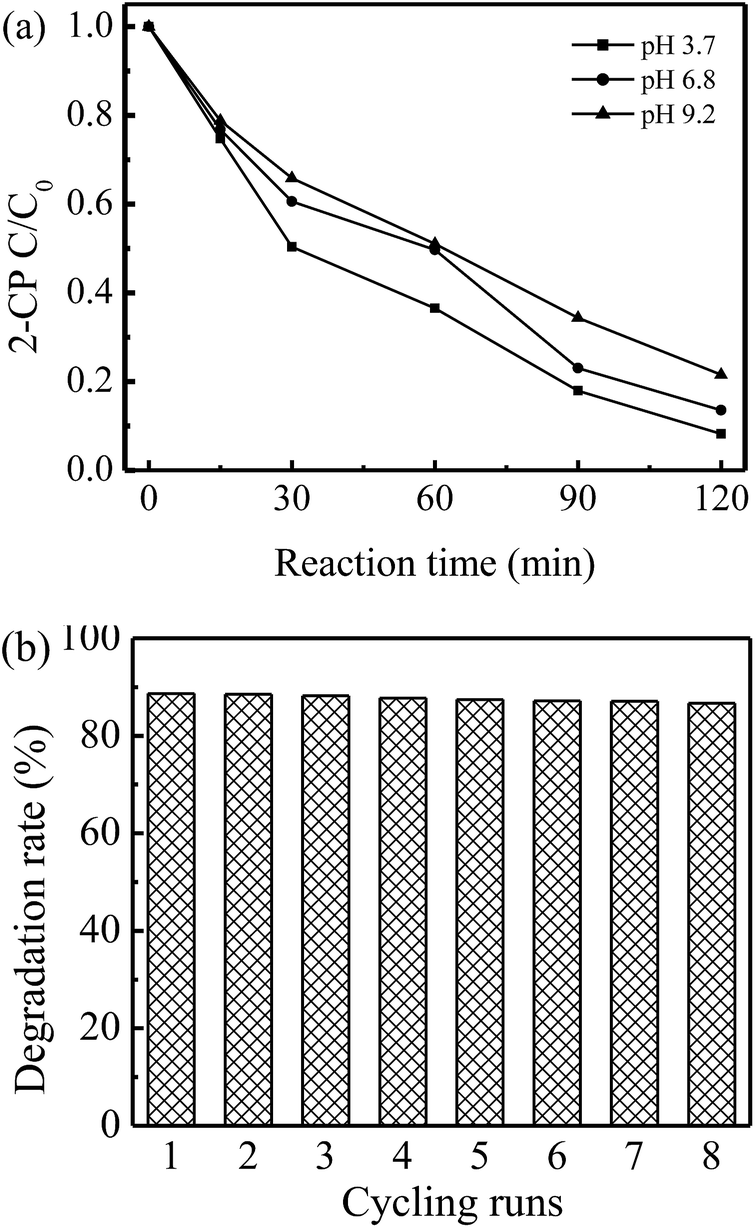 | ||
| Fig. 11 (a) Degradation of 2-CP in rGO-4-PPNc Fenton-like systems under different pH conditions. (b) Reusability of rGO-4-PP Nc for 2-CP degradation at neutral conditions. | ||
In addition, we also carried out the XRD and FTIR measurement of rGO-4-PP Nc after Fenton-like reaction. As shown in Fig. 12a, two obvious XRD diffraction peaks at 26.0° and 43.2° are observed for rGO-4-PP Nc after Fenton-like reaction, which are almost the same with the fresh rGO-4-PP Nc sample (Fig. 7a). Fig. 12b shows the results of FTIR spectra of rGO-4-PP Nc before and after the Fenton-like reaction. Three marked peaks of C–O–C, CC and CO groups, which are the typical characteristics of the fresh sample, are also observed after the Fenton-like reaction. These results suggest that the structure of rGO-4-PP Nc is not changed after the Fenton-like reaction, further revealing the excellent stability and reusability of rGO-4-PP Nc.
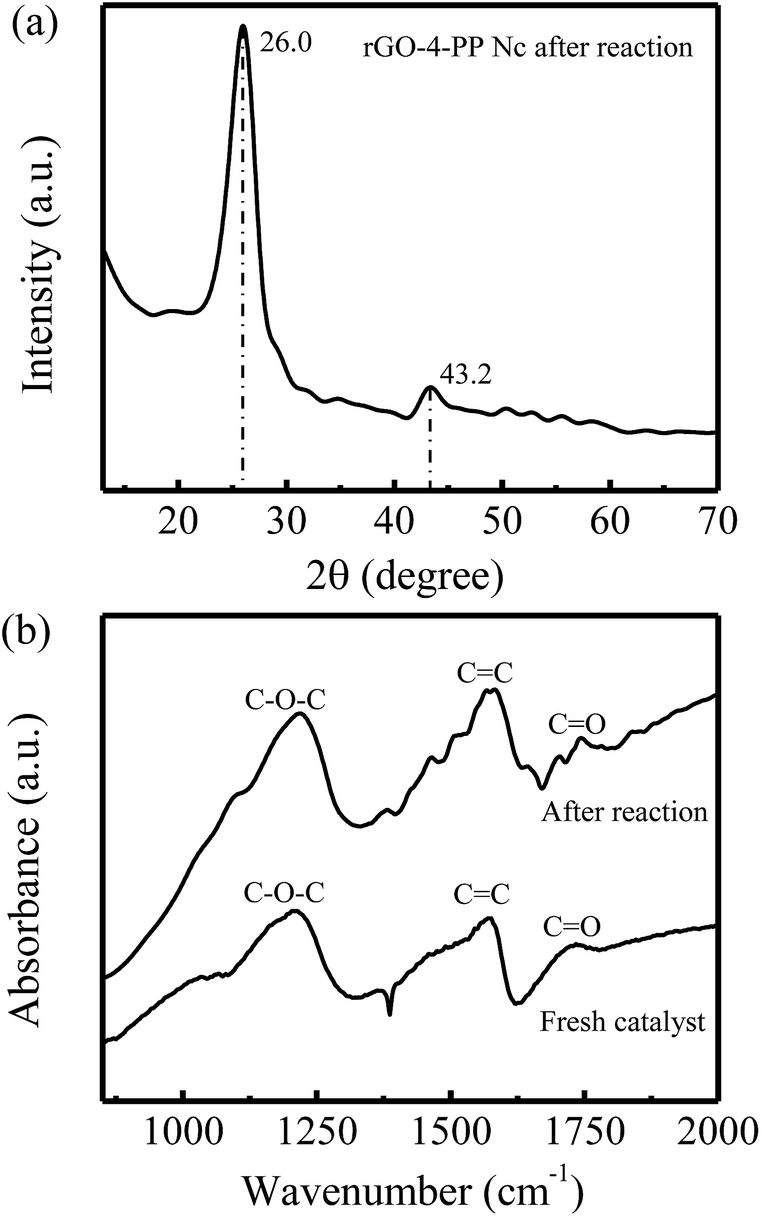 | ||
| Fig. 12 (a) XRD patterns and (b) FTIR spectra of rGO–4-PP Nc after the Fenton-catalytic reaction. | ||
4. Conclusions
The electronic properties and active sites of the 4-PP-modified graphene material were studied using the DFT method by different approaches. The results of the atom charge, electron density, HOMO/LUMO analysis and ESP distribution confirmed the nonuniform distribution of the electrons and the formation of the dual reaction centers for our catalyst models. The actual catalyst, named rGO-4-PP Nc, was also prepared via a surface complexation process and characterized by XRD, FTIR and EPR. The adsorption models and the H2O2 decomposition experiments verified the dual-reaction-center mechanism that the H2O2 could absorb to the electron-rich oxygen center easily while the pollutants compete with the H2O2 to complex with the electron-deficient carbon center, avoiding the useless decomposition of H2O2. Thus, rGO-4-PP Nc shows an excellent performance for pollutant removal. This study offers a new perspective to investigate the potential activity of catalyst theoretically, which is meaningful for finding new metal-free catalyst not only for the Fenton reaction, but also for the other AOPs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Plan (2016YFA0203200), the National Natural Science Foundation of China (51538013) and the Science Starting Foundation of Guangzhou University (2700050302).References
- S. Navalon, M. de Miguel, R. Martin, M. Alvaro and H. Garcia, J. Am. Chem. Soc., 2011, 133, 2218–2226 CrossRef PubMed
.
- R. Martín, M. Álvaro, J. R. Herance and H. García, ACS Nano, 2010, 4, 65–74 CrossRef PubMed
- W. P. Li, C. H. Su, Y. C. Chang, Y. J. Lin and C. S. Yeh, ACS Nano, 2016, 10, 2017–2027 CrossRef CAS PubMed
- A. M. Nowicka, U. Hasse, M. Hermes and F. Scholz, Angew. Chem., Int. Ed. Engl., 2010, 49, 1061–1063 CrossRef PubMed
- P. V. Nidheesh, RSC Adv., 2015, 5, 40552–40577 RSC
- L. Lyu, L. L. Zhang and C. Hu, Chem. Eng. J., 2015, 274, 298–306 CrossRef
- W. J. Song, M. M. Cheng, J. H. Ma, W. H. Ma, C. C. Chen and J. C. Zhao, Environ. Sci. Technol., 2006, 40, 4782–4787 CrossRef PubMed
- S. Navalon, R. Martin, M. Alvaro and H. Garcia, Angew. Chem., Int. Ed., 2010, 49, 8403–8407 CrossRef PubMed
- L. Lyu, L. L. Zhang, Q. Y. Wang, Y. L. Nie and C. Hu, Environ. Sci. Technol., 2015, 49, 8639–8647 CrossRef PubMed
- L. Lyu, L. L. Zhang, C. Hu and M. Yang, J. Mater. Chem. A, 2016, 4, 8610–8619 CAS
- L. Lyu, L. L. Zhang and C. Hu, Environ. Sci.: Nano, 2016, 3, 1483–1492 RSC
- L. Lyu, L. Zhang, G. He, H. He and C. Hu, J. Mater. Chem. A, 2017, 5, 7153–7164 CAS
- M. D. Bhatt, G. Lee and J. S. Lee, J. Phys. Chem. C, 2016, 120, 26435–26441 CAS
- E. Vayner and A. B. Anderson, J. Phys. Chem. C, 2007, 111, 9330–9336 CAS
- L. Yu, X. Pan, X. Cao, P. Hu and X. Bao, J. Catal., 2011, 282, 183–190 CrossRef CAS
- M. D. Esrafili, R. Mohammad-Valipour, S. M. Mousavi-Khoshdel and P. Nematollahi, ChemPhysChem, 2015, 16, 3719–3727 CrossRef CAS PubMed
- M. D. Esrafili and L. Dinparast, Chem. Phys. Lett., 2017, 682, 49–54 CrossRef CAS
- D. Düzenli, J. Phys. Chem. C, 2016, 120, 20149–20157 Search PubMed
- L. H. Thomas, E. Cheung, A. O. F. Jones, A. A. Kallay, M. H. Lemee-Cailleau, G. J. McIntyre and C. C. Wilson, Cryst. Growth Des., 2012, 12, 1746–1751 CAS
- Gaussian, Inc., Wallingford CT, 2016
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS PubMed
- H. Bader, V. Sturzenegger and J. Hoigné, Water Res., 1988, 22, 1109–1115 CrossRef CAS
- P. Politzer, P. R. Laurence and K. Jayasuriya, Environ. Health Perspect., 1985, 61, 191–202 CrossRef CAS PubMed
- P. Sjoberg and P. Politzer, J. Phys. Chem., 1990, 94, 3959–3961 CrossRef
- J. M. Wan, M. Wei, Z. W. Hu, Z. Q. Peng, B. Wang, D. Y. Feng and Y. W. Shen, Int. J. Hydrogen Energy, 2016, 41, 14692–14703 CrossRef
- U. Alam, M. Fleisch, I. Kretschmer, D. Bahnemann and M. Muneer, Appl. Catal., B, 2017, 218, 758–769 CrossRef CAS
- Z. Guo, Z. Y. Wang, H. H. Wang, G. Q. Huang and M. M. Li, Mater. Sci. Eng., C, 2015, 57, 197–204 CrossRef CAS PubMed
- X. Pan and Y.-J. Xu, J. Phys. Chem. C, 2013, 117, 17996–18005 CAS
- D. W. Boukhvalov, V. Y. Osipov, A. I. Shames, K. Takai, T. Hayashi and T. Enoki, Carbon, 2016, 107, 800–810 CrossRef CAS
- L. Lyu, G. F. Yu, L. L. Zhang, C. Hu and Y. Sun, Environ. Sci. Technol. DOI:10.1021/acs.est.7b04865
| This journal is © The Royal Society of Chemistry 2018 |