
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization of the global metabolic profile of liquiritin in rat plasma, urine, bile and feces based on UHPLC-FT-ICR MS†
Li Ma,
Yangyang Zhao,
Xiaoxue Zhang,
Tianfeng Liu,
Fei Han and
Ran Yin *
*
School of Pharmacy, Shenyang Pharmaceutical University, 103 Wenhua Road, Shenhe District, Shenyang, 110016, China. E-mail: yinran_syphu@163.com
First published on 5th February 2018
Abstract
Liquiritin is a major flavonoid in Radix Glycyrrhizae and it has been reported to possess various pharmacological activities. In the present work, a strategy based on an ultra high performance liquid chromatography combined with Fourier transform ion cyclotron resonance mass spectrometry (UHPLC-FT-ICR MS) method was proposed to systematically characterize the in vivo metabolites of liquiritin for the first time. After oral administration of liquiritin to rats in a single dose of 120 mg kg−1, the rat plasma, urine, feces and bile samples were collected and used to discover metabolites. As a result, besides the parent drug, a total of 76 metabolites (6 phase I and 70 phase II metabolites) of liquiritin were detected and tentatively identified. It was indicated that the metabolic pathways of liquiritin in rats included oxidation, reduction, deglycosylation, isomerization, methylation, glucuronidation and sulfation. In summary, the results could provide valuable information regarding the metabolism of liquiritin in rats, which could contribute to a better understanding of its action mechanism.
1. Introduction
Liquiritin (Fig. 1) is a major flavonoid and marker component of Radix Glycyrrhizae, a widely used herbal medicine called “Gancao” in China.1 It has been reported that liquiritin possess multifold biological activities, such as anti-inflammatory, anti-oxidative, antiviral, antithrombotic, and antidiabetic as well as antidepressant-like effect and neuroprotective activities, and so on.2–4 Also, recent pharmacological research has demonstrated that liquiritin may have a role in the treatment of many diseases, such as neurodegenerative diseases.4 However, to the best of our knowledge, studies on the metabolic profile of liquiritin in vivo still remain unclear. | ||
| Fig. 1 Chemical structure of liquiritin. | ||
As we know, metabolism research on drugs is valuable for better understanding their action mechanism and evaluating their efficacy and safety.5–7 Up to now, although the study involving the metabolism of liquiritin in rats has been reported, however only 22 metabolites of liquiritin have been found in rat plasma, urine and feces at a single dose of 40 mg kg−1 by the LC/qTOF-MS method.8 Moreover, few study on metabolism of liquiritin in rat bile has been reported till now. It suggests that the metabolism of liquiritin in vivo has not yet been fully elucidated. Therefore, it is crucial and valuable to establish a sensitive and effective analytical method for systematic study on the metabolic profile of liquiritin in rat plasma, bile, urine and feces after oral administration.
In recent years, ultra high performance liquid chromatography combined with Fourier transform ion cyclotron resonance mass spectrometry (UHPLC-FT-ICR-MS) has been extensively applied as a robust tool for characterization the prototype components and/or their metabolites in many kinds of complex biological samples profiting from its high sensitivity and selectivity.9–16 Generally, ultra-high mass accuracy (<2.0 ppm in most cases) and excellent resolving power (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000, m/z = 400) of FT-ICR MS can help to identify the metabolite. Consequently, it has been successfully utilized to discover metabolites of compounds and traditional Chinese medicines in rats by us and other groups.17,18
000000, m/z = 400) of FT-ICR MS can help to identify the metabolite. Consequently, it has been successfully utilized to discover metabolites of compounds and traditional Chinese medicines in rats by us and other groups.17,18
In this study, a strategy for systematic study of the metabolic profile of liquiritin in rat plasma, bile, urine and feces after oral administration was illustrated based on UHPLC-FT-ICR-MS for the first time. More important, more metabolites of liquiritin were identified and the potential metabolic pathways in vivo were discovered and proposed by the developed method. And the results of this study may provide meaningful information for further efficacy and safety evaluation of liquiritin.
2. Experimental
2.1 Materials and reagents
The standards of liquiritin (Z02M8X30249), liquiritigenin (W22A7K13544) and isoliquiritigenin (R12A7F19390) were purchased from Shanghai Yuanye Biological Technology Limited Company (Shanghai, China). Methanol (HPLC grade) and acetonitrile (HPLC grade) were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Formic acid (HPLC grade) was purchased from Tianjin Kemiou Chemical Reagent Co., Ltd. (Tianjin, China). Deionized water was purified with a Milli-Q system (Millipore, Milford, MA, USA). All other chemicals and reagents were of analytical grade and commercially available.2.2 Animal experiments
Male Sprague Dawley rats (220–240 g, SPF degree) were provided by the Experimental Animal Center of Shenyang Pharmaceutical University (Shenyang, China). Before administration, the rats were acclimated in a controlled animal room for 7 days with standardized humidity (50 ± 10%), temperature (22 ± 2 °C). Purified water and standard food were provided freely. All rats were fasted for 12 h but with access to water prior to the experiment. All experiments were performed in accordance with the approved animal protocols and guidelines established by Animal Ethics Review Committee for animal experiments of Shenyang Pharmaceutical University (authorization number: SYPU-ICAUC-C2017-0227-203).Twelve rats were randomly divided into groups I and groups II of 6 rats each. Liquiritin was suspended in 0.5% sodium carboxymethyl cellulose and intragastric administrated to rats at a single dose of 120 mg kg−1. Each rat of group I was held in a separate metabolic cage to collect the urine and feces samples from 0 to 24 h after administration of dosing. Blood samples were collected into heparinized polythene tubes at 0.5, 1, 2, 3, 4 and 6 h after dosing from the suborbital vein of rats in group I. And then, it was immediately centrifuged at 8000 rpm for 5 min to obtain plasma samples.
For bile collection, rats in group II were cannulated in the bile duct under general anesthesia, then they were allowed to recover from anesthesia before oral administration and bile samples were collected from 0 to 24 h after dosing. Blank plasma, urine, bile and feces samples of rats were collected prior to administration. All samples were frozen at −80 °C until analysis.
2.3 Sample preparation
To remove the endogenous substances and minimize the loss of trace metabolites, the rat plasma (100 μl) was mixed with 200 μl methanol by vortexing for 5 min. The mixture was centrifuged at 15000 rpm for 5 min to separate precipitated proteins, and then the supernatant solution was dried under nitrogen stream at 30 °C. The residue was redissolved in 100 μl methanol for UHPLC-FT-ICR MS analysis. As for the preparation of urine or bile samples, Waters Sep-Pak® C18 solid-phase extraction (SPE) cartridge (3 cc/500 mg; Waters Co, Milford, MA, USA) was used to concentrate the analytes and minimize the endogenous matrix interference. Before use, SPE cartridges were preprocessed with 1 ml methanol and equilibrated by 1 ml ultrapure water. Briefly, one milliliters of urine or bile samples were subjected to SPE cartridge and eluted with H2O (1 ml) and MeOH (1 ml). The MeOH eluate was collected for UHPLC-FT-ICR MS analysis. The feces samples were dried in the dark on filter paper at ambient temperature. One hundred milligrams of feces samples were weighted and extracted by ultrasonication using 2.5 ml methanol for 0.5 h. And then, it was centrifuged at 15000 rpm for 5 min. Subsequently, the supernatant was injected into the UHPLC-FT-ICR MS system for analysis.
2.4 Instrument and analytical conditions
All analysis were performed on a UHPLC-FT-ICR MS instrument, an Agilent 1260 system coupled with a Bruker Solarix 7.0 T FT-ICR MS system (Bruker, Germany) equipped with an electrospray ionization source (ESI). Chromatographic separation was performed on a Shim-pack XR-ODS C18 column (75 mm × 3.0 mm, 2.2 μm) (SHIMADZU, Japan), the column temperature was maintained at 35 °C, the flow rate was set at 0.50 ml min−1. The mobile phase, consisting of 0.1% formic acid water (A) and acetonitrile (B), was delivered using a liner gradient program as follows: 10–20% (B) in 0–13 min, 20–35% (B) in 13–18 min, 35–50% (B) in 18–20 min, 50–50% (B) in 20–30 min. The injection volume was 5 μl.For MS detection, the instrument was operated in negative ion mode, and full-scan mass rage was 100–1000 Da. The optimal conditions were as follows: a nebulizer gas pressure of 4.0 bar, a dry gas flow rate of 8.0 L min−1, a capillary voltage of −3.5 kV, an end plate off set of −500 V, and a transfer capillary temperature of 250 °C. While in MS/MS experiments, the collision energy was initially set at 20 V of the preferred ions and then adjusted according to the fragments. FT MS control and Bruker Compass-Hystar (Bruker, Germany) were used to control the equipment and for data acquisition and analysis, respectively.
2.5 Data processing
MetabolitePredict software (Version 2.0, Bruker Daltonics) was designed to predict metabolic pathways of molecules based on biotransformation rules applied to the molecule. A metabolite tree representing the relationship of metabolites to each other and visualizing the corresponding structures will be obtained. In this study, it was used to predict metabolism of liquiritin. The default values of prediction parameters and a set of biotransformation rules were set according to the workflow. Based on the results, an in-house metabolite library of liquiritin was established. And then, information on all potential metabolites was verified by manual inspection. Finally, data processing was carried out with DataAnalysis Software (Version 4.1, Bruker Daltonics, Germany).3. Results and discussion
3.1 Metabolic profile analysis based on UHPLC-FT-ICR-MS
All plasma, bile, urine and feces samples were detected and analyzed under the UHPLC-FT-ICR-MS conditions mentioned above. The ultra-high resolution MS informations on the exact mass of the deprotonated molecular ion and fragment ions were obtained. By comparing the blank and medicated biological samples, the prototype compounds and potential metabolites were screening and confirmed. The base peak ion chromatograms (BPCs) of plasma, bile, urine and feces samples were presented in ESI Fig. 1.† In order to exclude the endogenous interferences for metabolites in BPCs, the extract ion chromatograms (EICs) were performed to extract the neat MS data of each metabolite. As a result, a total of 76 metabolites of liquiritin were tentatively identified. The obtained EICs of the prototype (M0) and 76 metabolites (M1–M76) from different matrixes were all displayed in Fig. 2 (plasma) and ESI Fig. 2† (bile, urine and feces). The element composition of each metabolite was calculated by DataAnalysis Software (Bruker, Germany). The maximum mass errors between the observed and calculated values were all below 2.0 ppm in this study, which demonstrated a high level of confidence in the proposed elemental compositions of the metabolites. The chemical structures of metabolites were characterized according to the retention behaviors, element compositions, MS and MS2 data, relevant drug biotransformation knowledge and the fragmentation pattern of the prototype. The data of retention times, calculated and observed masses, mass errors, elemental compositions and characteristic fragment ions of the proposed metabolites were all listed in Table 1 and ESI table.†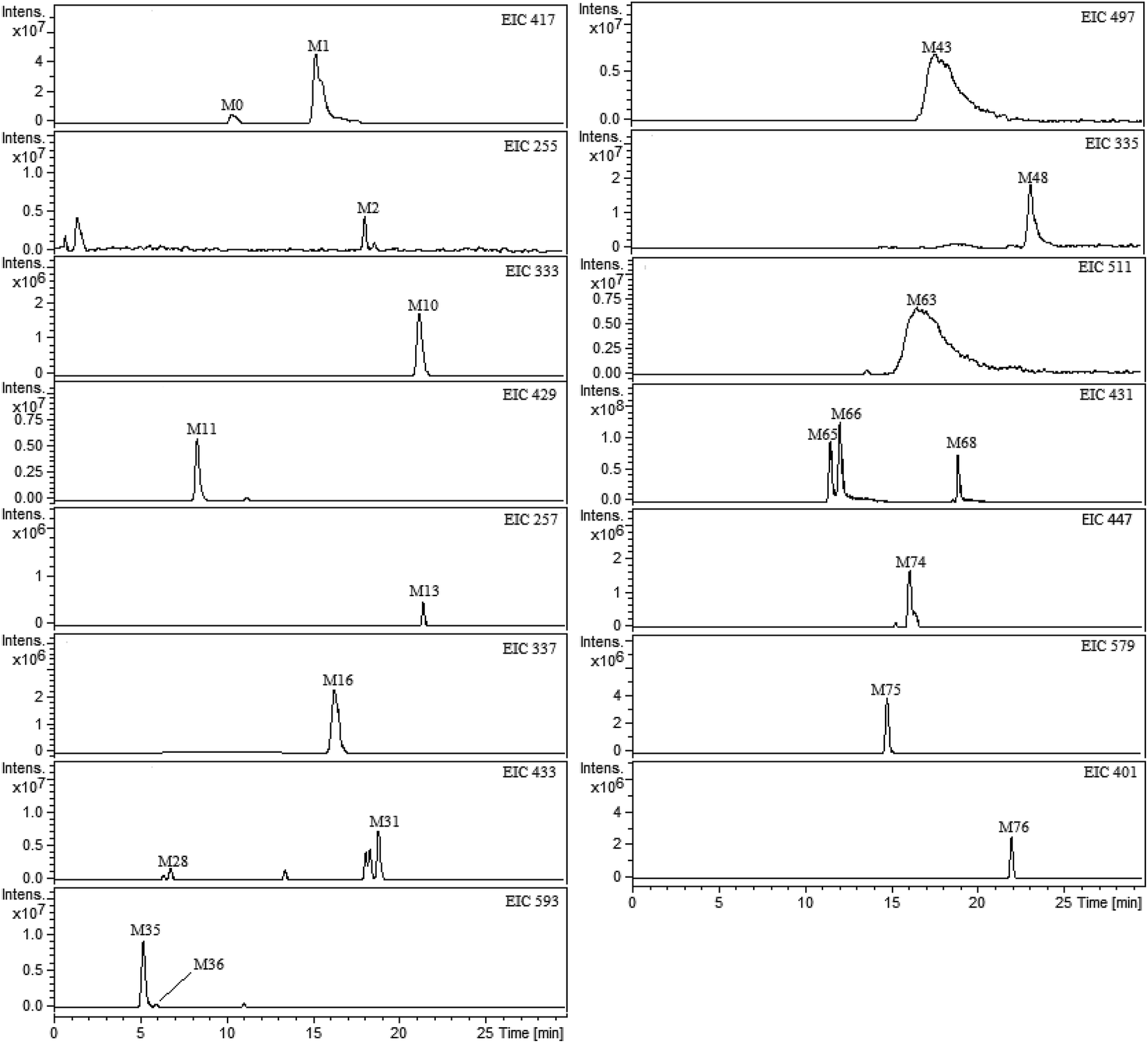 | ||
| Fig. 2 Extracted ion chromatograms (EICs) for metabolites of liquiritin in rat plasma. M0 represents parent drug; M1–M76 represent metabolites of liquiritin. | ||
| No. | tR (min) | [M − H]− | MS/MS | Formula | Metabolite description | Matrix | ||
|---|---|---|---|---|---|---|---|---|
| Calculated mass (Da) | Observed mass (Da) | Error (ppm) | ||||||
| a P, U, B and F represented rat plasma, urine, bile and feces samples respectively. | ||||||||
| M0 | 10.72 | 417.11911 | 417.11920 | 0.22 | 255.06597, 119.05058, 135.00899, 153.01991 | C21H22O9 | Parent | P, U, F |
| M1 | 15.76 | 417.11911 | 417.11855 | 1.32 | 255.06587, 119.05063, 135.00932, 153.01990 | C21H22O9 | Isomerization | P, U, B |
| M2 | 18.50 | 255.06628 | 255.06616 | 0.46 | 135.00843, 119.04989, 153.01898 | C15H12O4 | Deglycosylation | P, U, F |
| M3 | 22.06 | 255.06628 | 255.06605 | 0.93 | 135.00838, 119.04993, 153.01896 | C15H12O4 | Deglycosylation + isomerization | U |
| M13 | 21.74 | 257.08193 | 257.08169 | 0.93 | 153.03969, 119.04991, 137.00838 | C15H14O4 | Deglycosylation + hydrogenation + isomerization | P, U, F |
| M14 | 13.80 | 337.03875 | 337.03838 | 1.08 | 257.08129, 151.03969 | C15H14O7S | Deglycosylation + hydrogenation + isomerization + sulfation | U |
| M15 | 14.81 | 337.03875 | 337.03827 | 1.40 | 257.08127, 151.03970 | C15H14O7S | Deglycosylation + hydrogenation + isomerization + sulfation | U, B |
| M16 | 16.17 | 337.03875 | 337.03840 | 1.03 | 257.08135, 151.03967 | C15H14O7S | Deglycosylation + hydrogenation + isomerization + sulfation | B, P |
| M17 | 24.83 | 337.03875 | 337.03834 | 1.21 | 257.08140, 151.03960 | C15H14O7S | Deglycosylation + hydrogenation + isomerization + sulfation | U |
| M18 | 25.85 | 337.03875 | 337.03862 | 0.37 | 257.08125, 151.03983 | C15H14O7S | Deglycosylation + hydrogenation + isomerization + sulfation | U |
| M19 | 28.12 | 337.03875 | 337.03823 | 1.55 | 257.08121, 151.03976 | C15H14O7S | Deglycosylation + hydrogenation + isomerization + sulfation | U |
| M21 | 5.42 | 513.07084 | 513.07060 | 0.47 | 337.03875, 255.06564, 160.03994 | C21H22O13S | Deglycosylation + hydrogenation + isomerization + sulfation + glucuronidation | U |
| M22 | 9.22 | 513.07084 | 513.07067 | 0.32 | 337.03878, 255.06569, 160.03990 | C21H22O13S | Deglycosylation + hydrogenation + isomerization + sulfation + glucuronidation | B |
| M23 | 16.05 | 513.07084 | 513.07067 | 0.32 | 337.03890, 255.06560, 160.03997 | C21H22O13S | Deglycosylation + hydrogenation + isomerization + sulfation + glucuronidation | U |
| M24 | 19.08 | 513.07084 | 513.07077 | 0.13 | 337.03887, 255.06564, 160.03998 | C21H22O13S | Deglycosylation + hydrogenation + isomerization + sulfation + glucuronidation | U |
| M25 | 20.35 | 513.07084 | 513.07095 | −0.22 | 337.03884, 255.06565, 160.03990 | C21H22O13S | Deglycosylation + hydrogenation + isomerization + sulfation + glucuronidation | U |
| M26 | 3.41 | 433.11402 | 433.11377 | 0.57 | 257.08193, 153.01981 | C21H22O10 | Deglycosylation + hydrogenation + isomerization + glucuronidation | U |
| M27 | 5.56 | 433.11402 | 433.11379 | 0.53 | 257.08190, 153.01989 | C21H22O10 | Deglycosylation + hydrogenation + isomerization + glucuronidation | U |
| M28 | 6.54 | 433.11402 | 433.11396 | 0.14 | 257.08188, 153.01987 | C21H22O10 | Deglycosylation + hydrogenation + isomerization + glucuronidation | P, U, F |
| M29 | 13.04 | 433.11402 | 433.11351 | 1.17 | 257.08198, 153.01980 | C21H22O10 | Deglycosylation + hydrogenation + isomerization + glucuronidation | U |
| M30 | 14.81 | 433.11402 | 433.11367 | 0.80 | 257.08192, 153.01976 | C21H22O10 | Deglycosylation + hydrogenation + isomerization + glucuronidation | U, F |
| M31 | 18.53 | 433.11402 | 433.11359 | 0.99 | 257.08190, 153.01985 | C21H22O10 | Deglycosylation + hydrogenation + isomerization + glucuronidation | U, B, P |
| M34 | 4.85 | 593.15119 | 593.15037 | 1.39 | 417.11842, 255.06594 | C27H30O15 | Isomerization + glucuronidation | U |
| M35 | 5.23 | 593.15119 | 593.15070 | 0.83 | 417.11836, 255.06597 | C27H30O15 | Isomerization + glucuronidation | B, P |
| M36 | 6.17 | 593.15119 | 593.15150 | −0.52 | 417.11835, 255.06598 | C27H30O15 | Isomerization + glucuronidation | P |
| M37 | 8.53 | 593.15119 | 593.15065 | 0.92 | 417.11832, 255.06596 | C27H30O15 | Isomerization + glucuronidation | U |
| M38 | 11.12 | 593.15119 | 593.15098 | 0.37 | 417.11830, 255.06581 | C27H30O15 | Isomerization + glucuronidation | B |
| M39 | 14.11 | 497.07592 | 497.07556 | 0.73 | 255.06598 | C21H22O12S | Isomerization + sulfation | B |
| M40 | 15.92 | 497.07592 | 497.07564 | 0.54 | 255.06599 | C21H22O12S | Isomerization + sulfation | U |
| M41 | 16.93 | 497.07592 | 497.07506 | 1.72 | 255.06590 | C21H22O12S | Isomerization + sulfation | B |
| M42 | 17.73 | 497.07592 | 497.07599 | −0.15 | 255.06597 | C21H22O12S | Isomerization + sulfation | U |
| M43 | 19.09 | 497.07592 | 497.07612 | −0.39 | 255.06594 | C21H22O12S | Isomerization + sulfation | P, U |
| M44 | 13.88 | 335.02310 | 335.02283 | 0.80 | 255.06591 | C15H12O7S | Deglycosylation + isomerization + sulfation | B |
| M45 | 15.31 | 335.02310 | 335.02275 | 1.04 | 255.06588 | C15H12O7S | Deglycosylation + isomerization + sulfation | B |
| M46 | 20.02 | 335.02310 | 335.02284 | 0.77 | 255.06582 | C15H12O7S | Deglycosylation + isomerization + sulfation | U |
| M47 | 21.60 | 335.02310 | 335.02265 | 1.33 | 255.06599 | C15H12O7S | Deglycosylation + isomerization + sulfation | U |
| M48 | 23.13 | 335.02310 | 335.02303 | 0.19 | 255.06596 | C15H12O7S | Deglycosylation + isomerization + sulfation | P |
| M49 | 27.58 | 335.02310 | 335.02266 | 1.29 | 255.06595 | C15H12O7S | Deglycosylation + isomerization + sulfation | U |
| M50 | 28.71 | 335.02310 | 335.02247 | 1.87 | 255.06576 | C15H12O7S | Deglycosylation + isomerization + sulfation | U |
| M59 | 12.18 | 511.05519 | 511.05524 | −0.12 | 335.02310 | C21H20O13S | Deglycosylation + isomerization + sulfation + glucuronidation | U |
| M60 | 12.70 | 511.05519 | 511.05501 | 0.34 | 335.02312 | C21H20O13S | Deglycosylation + isomerization + sulfation + glucuronidation | U |
| M61 | 13.53 | 511.05519 | 511.05485 | 0.66 | 335.02319 | C21H20O13S | Deglycosylation + isomerization + sulfation + glucuronidation | U |
| M62 | 13.90 | 511.05519 | 511.05496 | 0.45 | 335.02334 | C21H20O13S | Deglycosylation + isomerization + sulfation + glucuronidation | B |
| M63 | 14.96 | 511.05519 | 511.05484 | 0.67 | 335.02327 | C21H20O13S | Deglycosylation + isomerization + sulfation + glucuronidation | U, B, P |
| M64 | 10.61 | 431.09837 | 431.09853 | −0.37 | 255.06560, 135.00843, 119.04989, 153.01898 | C21H20O10 | Deglycosylation + isomerization + glucuronidation | U |
| M65 | 11.52 | 431.09837 | 431.09834 | 0.07 | 255.06547, 135.00842, 119.04979, 153.01890 | C21H20O10 | Deglycosylation + isomerization + glucuronidation | U, B, P |
| M66 | 12.85 | 431.09837 | 431.09892 | −1.28 | 255.06523, 135.00813, 119.04940, 153.01897 | C21H20O10 | Deglycosylation + isomerization + glucuronidation | U, B, P |
| M67 | 13.44 | 431.09837 | 431.09875 | −0.88 | 255.06569, 135.00840, 119.04988, 153.01883 | C21H20O10 | Deglycosylation + isomerization + glucuronidation | U |
| M68 | 18.77 | 431.09837 | 431.09875 | −0.88 | 255.06545, 135.00893, 119.04789, 153.01873 | C21H20O10 | Deglycosylation + isomerization + glucuronidation | U, B, P |
| M69 | 6.04 | 607.13046 | 607.13018 | 0.46 | 431.09837, 135.00927, 119.05067, 153.01981 | C27H28O16 | Deglycosylation + isomerization + glucuronidation | U, B |
As shown in Table 1 and ESI table,† the identified 76 metabolites included 6 phase I metabolites (M1, M2, M3, M4, M5, M13) and 70 phase II metabolites. Among them, the parent drug (M0), liquiritigenin (M2) and isoliquiritigenin (M3) were confirmed by comparison with reference standards, and the other 74 metabolites were indentified according to the obtained MS/MS data. Comparing with the previous literatures,8,19 27 potential new metabolites of liquiritin in rats including M4, M5–M12, M20, M32, M33, M51–M58 and M70–M76 were first tentatively identified by the UHPLC-FT-ICR-MS method in this study (ESI table†).
In addition, there were 19, 60, 20 and 6 metabolites were detected in rat plasma, urine, bile and feces, respectively. It was clear that the kidney is the main excretory organ for liquiritin. Furthermore, M1, M31, M63, M65, M66 and M68 were found in plasma, urine and bile samples; M0, M2, M13 and M28 were found in plasma, urine and feces samples, while M10, M36, M48, M75 and M76 were found only in plasma samples, M6, M8, M22, M38, M39, M41, M44, M45 and M62 were found only in bile samples. Other metabolites could be found in different biological sources.
3.2 Mass spectral fragmentation of liquiritin
In this study, negative ion mode was selected to obtain higher responses of liquiritin and other relative compounds in the MS spectra, and authentic liquiritin was characterized by signal of deprotonated molecule ([M − H]−) at m/z 417.11839 (C21H21O9−) through direct injection. The major MS2 product ions of deprotonated molecule were determined at m/z 255.06590 (C15H11O4−), 119.05067 (C8H7O−), 135.00927 (C7H3O3−) and 153.01981 (C8H9O3−), which were consistent with the previous literature.203.3 Identification of metabolites
In this study, a total of 76 metabolites of liquiritin were detected and tentatively identified. Among them, M0 and two metabolites including M2 and M3 were identified unambiguously by comparison with reference standards. In addition, 47 metabolites including M1, M13–M19, M21–M31, M34–M50 and M59–M69 were identified by comparison with previous study,8,19 and the UHPLC-FT-ICR MS data was summarized in Table 1. Furthermore, 27 potential new metabolites including M4–M12, M20, M32, M33, M51–M58 and M70–M76 were speculated by obtained UHPLC-FT-ICR MS/MS data and the proposed fragmentation pattern of liquiritin (M0).M1 with retention time of 15.76 min exhibited the same [M − H]− ion with liquiritin (M0) at m/z 417.11855 (C21H21O9−, 1.32 ppm). In addition, the characteristic ions of M1 at m/z 255.06587 (C15H11O4−), 119.05063 (C8H7O−), 135.00932 (C7H3O3−) and 153.01990 (C8H9O3−) underwent a similar fragmentation pathway as liquiritin in MS/MS spectra. By referring to relevant literature,8 M1 was speculated to be isoliquiritin.20,29
M2 with retention time of 18.50 min displayed a [M − H]− ion at m/z 255.06616 (C15H11O4−, 0.46 ppm) which was 162 Da (hexose) lower than that of M0 (C21H21O9−). M3 with retention time of 22.06 min was detected at the same m/z with M2. In addition, the major MS2 product ions of deprotonated M2 and M3 were also determined at similar m/z to liquiritin. Furthermore, the retention times and the fragmentation pathways of M2 and M3 were in accordance with the standard liquiritigenin and isoliquiritigenin, respectively. Therefore, M2 and M3 were confirmed as liquiritigenin25,28 and isoliquiritigenin,20,26–28,30 respectively. BPCs of standards of liquiritin, liquiritigenin and isoliquiritigenin were displayed in ESI Fig. 3.†
3.3.2.1 M4, M5. Metabolite M4 with retention time of 17.94 min showed a deprotonated molecule ion [M − H]− at m/z 253.05029 (C15H9O4−, 1.34 ppm) which was 2 Da (2H) less than that of liquiritigenin (M2, C15H11O4−). And the MS/MS spectra of M4 exhibited fragment ions at m/z 224.04719 (C14H8O3−) and 135.00929 (C7H3O3−), the loss of 29 Da (CHO) and 118 Da (C8H6O) from M4. So, M4 might be the oxidation metabolite of liquiritigenin. However, the dehydrogenation site is still unclear.
Furthermore, the deprotonated molecule ion of M5 (tR = 20.77 min) was at m/z 269.04530 (C15H9O5−, 0.91 ppm), which was 16 Da (O) higher than M4 (C15H9O4−). In addition, the MS/MS spectra of M5 exhibited fragment ions at m/z 253.05063 (C15H9O4−) and 224.04718 (C14H8O3−). Therefore, M5 was suggested might be the hydroxylated metabolite of M4.
3.3.2.2 M6–M12. The deprotonated molecule ions of M6 (tR = 14.70 min) and M7 (tR = 24.07 min) were observed at m/z 349.00198 (C15H9O8S−, 1.10 ppm) and 349.00210 (C15H9O8S−, 0.75 ppm), respectively. And the molecular weight was 80 Da (SO3) higher than that of M5 (C15H9O5−). Further product ions at m/z 269.04502 (C15H9O5−) and 269.04498 (C15H9O5−) showed the loss of 80 Da (SO3) from M6 and M7. Based on the information above, M6 and M7 were tentatively speculated to be the sulfate conjugates of M5.
Similarly, the deprotonated molecule ions of M8 (tR = 10.27 min), M9 (tR = 19.55 min) and M10 (tR = 21.50 min) were at m/z 333.00707 (C15H9O7S−, 1.13 ppm), 333.00717 (C15H9O7S−, 0.84 ppm) and 333.00740 (C15H9O7S−, 0.15 ppm), respectively. And they showed 80 Da (SO3) higher than M4 (C15H9O4−). In addition, the MS/MS spectra of M8, M9 and M10 exhibited similar fragment ions. So, M8, M9 and M10 were tentatively considered to be the sulfate conjugates of M4.
Metabolites M11 (tR = 8.13 min) and M12 (tR = 9.91 min) showed the same [M − H]− ion at m/z 429.08270 (0.05 ppm) and were deduced as C21H17O10−. In the MS/MS spectra, the fragment ion at m/z 253.05069 (C15H9O4−) and 253.05056 (C15H9O4−) showed a loss of 176 Da (C6H8O6−) from the deprotonated molecule ion, and the further fragmentation behavior was identical to that of M4. Thus, M11 and M12 were tentatively inferred to be the glucuronide conjugates of M4.
3.3.2.3 M20, M32, M33. The [M − H]− ion of metabolite M20 (tR = 23.56 min) was observed at m/z 365.06969 (C17H17O7S−, 0.97 ppm) and the molecular weight was 28 Da (C2H4) higher than that of M14–M19 (C15H13O7S−), which showed M20 might be the dimethylated metabolite of M14–M19.
In addition, the [M − H]− ions of M32 and M33 (tR = 16.33 and 17.63 min) were observed at m/z 447.12953 (C22H23O10−, 0.31 ppm) and m/z 447.12952 (C22H23O10−, 0.33 ppm), respectively. And the molecular weight was 14 Da (CH2) higher than that of M26–M31 (C21H21O10−). Furthermore, their product ions (C16H15O4−) showing the loss of 176 Da (C6H8O6) from the [M − H]− ions were observed. Based on the information above, M32–M33 were tentatively speculated to be the methylated metabolites of M26–M31.
3.3.2.4 M51–M58. The deprotonated molecule ion of M51 (tR = 21.11 min) was at m/z 414.97975 (C15H11O10S2−, 0.39 ppm) which was 80 Da (SO3) higher than M44–M50 (C15H11O7S−), suggesting that M51 might be the sulfate conjugation of M44–M50. Furthermore, the characteristic ion (C15H11O4−) of M2 and M3 was also observed in the MS/MS spectrum of M51. Thus, M51 was tentatively speculated to be the disulfate conjugation of M2 or M3.
Metabolite M52–M55 (tR = 11.18, 20.02, 25.68 and 28.82 min) gave the [M − H]− ion at m/z 351.01794 (0.21 ppm), 351.01784 (0.49 ppm), 351.01798 (0.09 ppm) and 351.01769 (0.90 ppm), respectively. And all of them were deduced as C15H11O8S−, which was 16 Da (O) higher than M44–M50 (C15H11O7S−). In addition, the MS/MS spectra of M52–M55 exhibited fragment ion at m/z 271.060 (C15H11O5−), which was also 16 Da greater than the fragment ion (C15H11O4−) of M44–M50. And the characteristic ion (C15H11O4−) of M52–M55 was also in accordance with that of M44–M50. Therefore, M52–M55 were tentatively inferred to contain a hydroxyl substituent on the M44–M50.
Besides, metabolite M56–M58 (tR = 20.71, 23.04 and 28.57 min) gave the [M − H]− ion at m/z 365.03344 (0.61 ppm), 365.03339 (0.75 ppm) and 365.03310 (1.54 ppm), respectively. And all of them were deduced as C16H13O8S−, and the molecular weight was 14 Da (CH2) higher than that of M52–M55 (C15H11O8S−). In addition, the MS/MS spectra of M56–M58 exhibited fragment ion at m/z 285.076 (C16H13O5−), which was also 14 Da greater than the fragment ion (C15H11O5−) of M52–M55. Based on the information above, M56–M58 were suggested might be the methylated metabolites of M52–M55.
3.3.2.5 M70–M74. Metabolite M70 and M71 (tR = 2.79 and 12.82 min) were speculated as C27H29O16− and displayed the ion at m/z 609.14646 (−0.57 ppm) and 609.14551 (0.99 ppm) respectively, which was 2 Da (2H) higher than that of M69 (C27H27O16−). Therefore, M70 and M71 might be the reduction metabolites of M69.
At the same time, the m/z of metabolite M72 (tR = 19.24 min) was observed at 445.11379 (C22H21O10−, 0.52 ppm) and the molecular weight was 14 Da (CH2) higher than that of M64–M68 (C21H19O10−). Further product ion at m/z 269.04459 (C16H13O4−) showed the loss of 176 Da (C6H8O6) from the [M − H]− ion, which was also 14 Da greater than the fragment ion (C15H11O4−) of M64–M68. So, M72 was tentatively speculated to be the methylated metabolite of M64–M68.
Similarly, metabolite M73 and M74 (tR = 3.29 and 15.82 min) were speculated as C21H19O11− and displayed the ion at m/z 447.09309 (0.43 ppm) and 447.09300 (0.64 ppm), respectively. And all of them were 16 Da (O) higher than that of M64–M68 (C21H19O10−). In addition, the MS/MS spectra of M73–M74 exhibited fragment ion at m/z 271.0602 (C15H11O5−), which was also 16 Da greater than the fragment ion (C15H11O4−) of M64–M68. And the characteristic ion (C15H11O4−) of M73–M74 was also in accordance with that of M64–M68. Based on the information above, M73–M74 were tentatively speculated to be the hydroxylated metabolites of M64–M68.
3.3.2.6 M75, M76. Metabolite M75 (tR = 14.76 min) showed a deprotonated molecule ion at m/z 579.17295 (C27H31O14−, −1.76 ppm) which was 162 Da (C6H10O5) higher than that of liquiritin (M0) and isoliquiritin (M1). Thus, M75 might contain a hexose on the liquiritin (M0) or isoliquiritin (M1).
Metabolite M76 (tR = 21.94 min) was deduced as C20H17O9−, with an accurate [M − H]− ion at m/z 401.08772 (0.22 ppm) which was 148 Da (C5H8O5) higher than that of M2 and M3 (C15H11O4−). Thus, M76 was tentatively considered to contain a pentaose on the M2 or M3.
3.4 Metabolic pathways of liquiritin in rats
The proposed metabolic pathways of liquiritin in rats and deduced structures of metabolites are shown in ESI Fig. 4.† Among the total of 76 metabolites, 6 phase I metabolites (M1, M2, M3, M4, M5, M13) and 70 phase II metabolites were found. The phase I metabolic pathways included oxidation, reduction and deglycosylation. The phase II metabolic pathways contained glucuronidation, sulfation, methylation, pentaose conjugation and hexose conjugation. These results suggested that liquiritin underwent extensive metabolic reactions in rats after oral administration. The phase II metabolism, as we know, it is generally to reduce toxicity or biological activity through conjugation of drugs and/or their phase I metabolites with endogenous substances in vivo, and their water solubility trended to enhance.31 It was verified by the fact that 21 types of phase II reaction involving 70 phase II metabolites were discovered in this study.Moreover, a total of 27 potential new metabolites of liquiritin including M4–M12, M20, M32, M33, M51–M58 and M70–M76 were detected and tentatively identified by UHPLC-FT-ICR-MS this time (ESI Fig. 4†). Among them, M4 and M75 were speculated to be the oxidation metabolite of liquiritigenin (M2) and the hexose conjugation product of M0 or M1, respectively. In addition, all of the remaining 25 metabolites were derived from M2, M3, M4 and M13. For example, M5, M8–M10 and M11–M12 were the metabolites of M4 after hydroxylation, glucuronidation and sulfation, respectively; M20 and M32–M33 were the sulfate conjugates and glucuronide conjugates of M13 after methylation, and so on. Therefore, these metabolic pathways of liquiritin gave a new clue for metabolic profile study of liquiritin in vivo.
Finally, it is noteworthy that liquiritin (M0) could be metabolized to isoliquiritin (M1) via isomerization and liquiritigenin (M2) via deglycosylation in vivo after oral administration, respectively. And at the same time, both of (M1) and (M2) could be gradually metabolized to isoliquiritigenin (M3) via deglycosylation and isomerization, respectively. It indicated that extensive metabolic reactions take place mainly on the forms of these four compounds (M0, M1, M2 and M3) in vivo. These results are well consistent with previous studies.8 Therefore, it could be concluded that M1, M2 and M3 might play a synergistic effect with liquiritin in vivo. That is, it could be concluded that M1, M2 and M3 are the major metabolites of liquiritin. Future studies might be concentrated on the pharmacokinetic study of liquiritin, isoliquiritin, liquiritigenin and isoliquiritigenin in vivo, so as to illustrate the further biotransformation process of liquiritin.
4. Conclusion
This study is designed to investigate the metabolic profile of liquiritin in vivo based on UHPLC-FT-ICR MS for the first time. After intragastric gavage to male rats, 76 metabolites including 6 phase I and 70 phase II metabolites of liquiritin were observed in rat plasma, urine, feces and bile samples. Results showed that metabolic pathways of liquiritin included oxidation, reduction, deglycosylation, isomerization, methylation glucuronidation and sulfation. It could be concluded that liquiritin, isoliquiritin, liquiritigenin and isoliquiritigenin might be interconversion in vivo after oral administration of liquiritin. This study provided important information for clarifying the biotransformation process of liquiritin in vivo and laid an experimental basis for further research and development of liquiritin.Conflicts of interest
The authors declare that they have no conflict of interest.References
- B. Q. Fu, H. Li, X. R. Wang, F. S. C. Lee and S. F. Cui, J. Agric. Food Chem., 2005, 53, 7408–7414 CrossRef CAS PubMed.
- S. L. Jia, X. L. Wu, X. X. Li, X. L. Dai, Z. L. Gao, Z. Lu, Q. S. Zheng and Y. X. Sun, J. Asian Nat. Prod. Res., 2016, 18, 1186–1199 CrossRef CAS PubMed.
- Y. Yang, G. X. Bian and Q. J. Lu, Photochem. Photobiol. Sci., 2008, 33, 931 CAS.
- Z. A. Chen, J. L. Wang, R. T. Liu, J. P. Ren, L. Q. Wen, X. J. Chen and G. X. Bian, Cytotechnology, 2009, 60, 125–132 CrossRef CAS PubMed.
- P. Kamil, Ż. Dorota, J. Marek and P. Elzbieta, Curr. Drug Metab., 2016, 17, 107–117 CrossRef.
- S. Asha and M. Vidyavathi, Biotechnol. Adv., 2009, 27, 16–29 CrossRef CAS PubMed.
- H. Faber, M. Vogel and U. Karst, Anal. Chim. Acta, 2014, 834, 9–21 CrossRef CAS PubMed.
- C. Xiang, X. Qiao, Q. Wang, R. Li, W. Miao, D. Guo and M. Ye, Drug Metab. Dispos., 2011, 39, 1597–1608 CrossRef CAS PubMed.
- K. A. Youdim and K. C. Saunders, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 1326–1336 CrossRef CAS PubMed.
- Y. L. Song, H. X. Yan, J. F. Chen, Y. T. Wang, Y. Jiang and P. F. Tu, J. Pharm. Biomed. Anal., 2014, 89, 183–196 CrossRef CAS PubMed.
- S. Forcisi, F. Moritz, B. Kanawati, D. Tziotis, R. Lehmann and P. Schmitt-Kopplin, J. Chromatogr. A, 2013, 1292, 51–65 CrossRef CAS PubMed.
- Y. Wang, C. Ding, K. Du, Y. Xiao, C. Wu, J. Zhang, H. Qin and G. Du, Rapid Commun. Mass Spectrom., 2009, 23, 2724–2732 CrossRef CAS PubMed.
- M. Harir, M. Frommberger, A. Gaspar, D. Martens, A. Kettrup, M. El Azzouzi and P. Schmitt-Kopplin, Anal. Bioanal. Chem., 2007, 389, 1459–1467 CrossRef CAS PubMed.
- J. F. Xiao, B. Zhou and H. W. Ressom, Trends Anal. Chem., 2012, 32, 1–14 CrossRef CAS PubMed.
- M. Hasegawa, S. Takenaka, M. Kuwamura, J. Yamate and S. Tsuyama, Exp. Toxicol. Pathol., 2007, 59, 115–120 CrossRef CAS PubMed.
- D. F. Smith, A. Kiss, F. E. Leach III, E. W. Robinson, L. Pasa-Tolic and R. M. Heeren, Anal. Bioanal. Chem., 2013, 405, 6069–6076 CrossRef CAS PubMed.
- J. Guan, Q. L. Zhang, R. Rong, F. Han, H. Y. Zhu, Y. L. Zhao, A. H. Song and Z. G. Yu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 993, 60–68 CrossRef PubMed.
- F. Han, Y. T. Li, X. J. Mao, X. S. Zhang, J. Guan, A. H. Song and R. Yin, Anal. Bioanal. Chem., 2016, 408, 1975–1981 CrossRef CAS PubMed.
- G. G. Tan, Z. Y. Lou, X. Dong, W. H. Li, W. T. Liao, Z. Y. Zhu and Y. F. Chai, Chromatographia, 2011, 74, 341–348 CAS.
- W. Zhang, S. Jiang, D. W. Qian, E. X. Shang and J. A. Duan, Biomed. Chromatogr., 2014, 28, 1271–1277 CrossRef CAS PubMed.
- Y. L. Zhou and W. S. Ho, Oncol. Rep., 2013, 31, 298–304 CrossRef PubMed.
- S. Alrushaid, N. M. Davies, S. E. Martinez and C. L. Sayre, Res. Pharm. Sci., 2017, 12, 176–186 CrossRef PubMed.
- Y. Zhang, L. Zhang, Y. Zhang, J. J. Xu, L. L. Sun and S. Z. Li, Biomed. Pharmacother., 2016, 84, 1337–1349 CrossRef CAS PubMed.
- H. Yang, L. Kang, S. H. Kai, L. S. Quan and F. Jun, Anal. Methods, 2014, 6, 1563 RSC.
- S. Q. Dong, H. R. Fan, Q. S. Li, G. L. Wei, Y. Z. Li, C. X. Liu and D. Y. Si, Chin. Herb. Med., 2016, 8, 53–60 CrossRef CAS.
- J. Guo, D. T. Liu, D. J. Nikolic, D. W. Zhn, J. M. Pezzuto and R. B. V. Berrmen, Drug Metab. Dispos., 2008, 36, 461–468 CrossRef CAS PubMed.
- Y. K. Lee, Y. W. Chin, J. K. Bae, J. S. Seo and Y. H. Choi, Planta Med., 2013, 79, 1656–1665 CrossRef CAS PubMed.
- Y. H. Chio, Y. J. Kim, H. S. Chae and Y. W. Chin, Planta Med., 2015, 81, 586–593 CrossRef PubMed.
- Y. Y. Yang, L. Xu, S. Y. Hao, Y. Li and Z. Q. Zhang, J. Anal. Methods Chem., 2012, 6, 1–6 Search PubMed.
- Y. Y. Yang, L. Xu, S. Y. Hao and Z. Q. Zhang, Chem. Eng., 2014, 3, 23–26 Search PubMed.
- G. Zhang, J. Sun, Y. Kano and D. Yuan, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 941, 1–9 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12529a |
| This journal is © The Royal Society of Chemistry 2018 |