
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient mesoporous polymer supported phosphine-gold(I) complex catalysts for amination of allylic alcohols and intramolecular cyclization reactions†
Huoliang Gu‡
,
Xiong Sun‡,
Yong Wang,
Haihong Wu * and
Peng Wu*
* and
Peng Wu*
Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, North Zhongshan Rd. 3663, Shanghai, 200062, China. E-mail: hhwu@chem.ecnu.edu.cn; pwu@chem.ecnu.edu.cn; Fax: +86-21-6223-8510
First published on 8th January 2018
Abstract
A series of novel heterogeneous gold(I) catalysts were synthesized by immobilizing gold(I) complexes on ordered mesoporous polymer FDU-15 and characterized by XRD, N2 adsorption–desorption, FT-IR, TEM, EDS, etc. The catalytic activities of these catalysts were evaluated by the amination reactions of allylic alcohols. Among the catalysts investigated, FDU-(p-CF3Ph)2PAuCl (3d) was identified as the most efficient catalyst. Compared to the homogeneous catalyst, the enhanced catalytic activity of the heterogeneous gold(I) catalyst is closely related to the mesoporous structure of FDU-15. The catalytic system was suitable for a broad range of substrates and can be easily recovered and recycled at least twelve times without significant loss of catalytic activity. In addition, the catalytic performance of 3d was further examined for intramolecular cyclization for the synthesis of heterocyclic compounds.
1. Introduction
The great potential of homogeneous gold(I) complex catalysts for chemical transformations has attracted increasing interest in numerous fields of research.1–6 In this context, numerous homogeneous gold(I) complex-catalyzed reactions have been explored for the construction of carbon–carbon or carbon–heteroatom bonds, which are prevalent in many important pharmaceutical and bioactive natural products.7–13 However, homogeneous catalysts often suffer from several drawbacks related to the handling of sensitive metal–ligand complexes, deactivation during reaction and difficulty in recovery and reuse of expensive reagents, which hinders their practical applications in industrial processes. One of the most promising approaches to overcome these drawbacks is to generate heterogeneous gold(I) catalysts.14–20The use of heterogeneous catalysts has multiple advantages in both industrial and environmental concerns. Various types of materials, such as mesoporous silica and amorphous polymer, have been employed to support the gold(I) catalysts. Corma et al.21 have immobilized a gold–carbene complex on mesostructured silicates and applied them for the hydrogenation of alkenes and the Suzuki reaction. Yu et al.22 have described a polystyrene-immobilized gold(I) complex, which shows remarkable catalytic activities in three model transformations. Toste et al.23 have developed silica-supported cationic gold(I) complexes, which exhibit superior reactivity in intramolecular additions of allenes with alkynes. Cai et al.24 have developed an efficient and easily recoverable magnetic nanoparticle supported gold(I) catalyst for the direct reductive amination reaction, which proceeded at room temperature. Very recently, Shi et al.25 reported a PS-TA-Au(I) catalyst with good activity and chemoselectivity. Asensio et al.26 developed silica-immobilized NHC-gold(I) complexes, which are efficient in hydration and cyclization. In our previous study, we have successfully immobilized a chiral gold catalyst and applied it for the asymmetric cycloaddition of 2-alkynyl-2-alken-1-ones with nitrones.27 Despite these significant advances, the design and development of a new heterogeneous gold(I) catalyst system is still challenging.
The nature of support plays an important role in governing the catalytic activities of supported gold(I) species on it. The mesoporous material has a suitable pore size for easy functionalization, particularly with large molecules, such as phosphine linkers and long-chain alkyl alkoxysiloxanes. Moreover, the ordered mesopores with a larger specific surface area can enable uniform distribution of anchored functional groups and benefit to mass transfer. The discovery of a class of mesoporous resol resin, which is purely an organic framework, opens up new opportunities for the synthesis of novel heterogeneous gold(I) catalysts. In comparison to conventional silica-based MCM-41, SBA-15 and organic–inorganic hybrid mesoporous materials, this ordered mesopolymer shares the advantages of both mesostructures and conventional organic polymers.28,29 Its uniform mesopores with strong hydrophobicity allow for the easy access of organic reactants to the active sites within the channels. Based on this concept, we have succeeded in creating several heterogeneous catalysts, which show good catalytic activities in some chemical transformations.30–35 Herein, we further explored the performance of FDU-type mesopolymer as the support for homogeneous gold(I) catalysts. We synthesized a series of new supported gold(I) catalysts and studied their catalytic activities in amination of allylic alcohols36 and intramolecular cyclization.37,38 These reactions were chosen because of their high atomic-efficiency and the heterocyclic products are structural fragments, which are widely found in many pharmaceutical active compounds.
2. Experimental
2.1 General
All reactions were carried out under an atmosphere of Ar in flame-dried glassware with magnetic stirring. 1H NMR spectra and 13C NMR spectra were recorded on a Bruker 500 MHz spectrometer in DMSO-d6 or CDCl3. All signals are reported in ppm with the internal TMS signal at 0 ppm as a standard. Data for 1H NMR spectra are reported as follows: chemical shift (ppm, referenced to TMS; s = singlet, d = doublet, t = triplet, dd = doublet of doublets, m = multiplet), coupling constant (Hz), and integration. Data for 13C NMR are reported in terms of chemical shift (ppm) relative to residual solvent peak (CDCl3: 77.0 ppm). Reactions were monitored by thin layer chromatography (TLC) using silica gel plates. Flash column chromatography was performed over silica gel (200–300 mesh). Dichloromethane and toluene were freshly distilled from CaH2; THF and dioxane were freshly distilled from sodium metal prior to use. The X-ray diffraction (XRD) patterns were collected on a Bruker D8 ADVANCE instrument using Cu-Kα radiation (λ = 1.5418 Å) at 35 kV and 30 mA. Nitrogen adsorption–desorption isotherms were measured on a Quantachrome Autosorb-3B instrument after evacuating the samples at 423 K for 6 h. The specific surface areas were evaluated using the Brunauer–Emmett–Teller (BET) method and the pore distribution was calculated by the BJH method from adsorption branches of isotherms. The TEM images were recorded using a JEOL-JEM-2010 microscope after the specimens were dispersed in ethanol and placed on holed-copper grids. The Au loading was determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES).2.2 Preparation of FDU-type mesopolymer and FDU-CH2Cl
The FDU-type mesopolymer was prepared according to previously reported procedures.28 Then, FDU-type material was chloromethylated with chloromethyl methyl ether to obtain FDU-CH2Cl using AlCl3 as a catalyst.342.3 Typical methods for preparation of catalysts 3a–3c
Ar2PCl (7 mmol) was mixed with Li metal (14 mmol) in anhydrous THF (20 mL) under argon atmosphere at −5 °C; the mixture was stirred at r.t. for 8 h. The resultant solution was mixed with FDU-CH2Cl (1 g) and stirred overnight at r.t. to obtain FDU-PPh2. Following this, FDU-PPh2 (1.5 g) was mixed with Me2SAuCl (0.14 mmol) in THF (10 mL) under argon atmosphere, and stirred at r.t. overnight to obtain the catalysts, which were denoted as FDU-Ph2PAuCl, FDU-(o-OMePh)2PAuCl and FDU-(p-OMePh)2PAuCl (3a–3c). The Au loadings were determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES).2.4 The preparation of catalyst 3d
(p-CF3Ph)2PCl (7 mmol) was mixed with metal Li (14 mmol) in anhydrous THF (20 mL) under argon atmosphere at −5 °C; the mixture was stirred at r.t. for 1 h. The resultant solution was mixed with FDU-CH2Cl (1 g) and stirred 10 h at 40 °C to obtain a ligand-functionalized material, FDU-(p-CF3Ph)2P. Following this, FDU-(p-CF3Ph)2P (0.5 g) was mixed with Me2SAuCl (0.15 g) in anhydrous THF (15 mL) under argon atmosphere, and stirred at r.t. overnight to obtain the catalyst, which was denoted as FDU-(p-CF3Ph)2PAuCl (3d).2.5 General procedure for amination of allylic alcohols and reused of the catalyst
Under argon atmosphere, a mixture of catalyst 3d (6.9 mg, 0.05 mol%) and AgOTf (0.13 mg, 0.05 mol%) in dry solvent (1 mL) was magnetically stirred for 10 min at room temperature. A solution of allylic alcohols 4 (1 mmol) and amine 5 (2 mmol) in dry solvent (1 mL) was added into the aforementioned suspension and stirred under argon atmosphere. The reaction was monitored by TLC. When the reaction was over, the catalyst was recovered by simple filtration. The filtrate was diluted with water and brine; the product was extracted with ethyl acetate (3 × 10 mL). The combined organic phase was dried over anhydrous MgSO4 and concentrated under vacuum. The residue was purified by column chromatography on silica gel affording the product 6. The catalyst was washed with ethyl acetate and water and then suspended in acetone for 30 min under ultrasonication. Then, the catalyst was filtered, dried at 50 °C under vacuum overnight and reused for the next cycle.2.6 General procedure for the preparation of 2,3-dihydro-4H-pyran-4-ones
Under an argon atmosphere, AgOTf (5.8 mg, 5 mol%) was added into a suspension of 3d (316.5 mg, 5 mol%) in 4 mL toluene at room temperature. The mixture was stirred for 10 min to generate the cationic gold catalyst. After the substrate 7 (0.45 mmol) was added, the reaction mixture was stirred at room temperature. The reaction was monitored by TLC. When the reaction was over, the catalyst was recovered by simple filtration. The filtrate was diluted with brine, and the product was extracted with ethyl acetate. The combined organic phase was dried over anhydrous MgSO4 and concentrated under vacuum. The residue was purified by column chromatography on silica gel affording the product 8a–k.2.7 Synthesis of substituted furan catalyzed by 3d
AgOTf (0.1 mol%) was added to a solution of 3d (0.1 mol%) in dry toluene at room temperature under an argon atmosphere. The mixture was stirred for 10 min to generate the gold(I) catalyst. After the substrate 9 was added, the reaction mixture was further stirred at room temperature. The reaction was monitored by TLC. When the reaction was completed, the catalyst was recovered by simple filtration. The filtrate was diluted with water and brine, and the product was extracted with ethyl acetate. The combined organic phase was dried over anhydrous MgSO4 and concentrated under vacuum. The residue was purified by column chromatography on silica gel to afford the product 10.3. Results and discussion
3.1 Synthesis and characterization of catalysts
The strategy for the synthesis of FDU-type mesopolymer supported gold(I) catalysts 3a–3d are shown in Scheme 1. The chloromethylated materials, FDU-CH2Cl (1), were first prepared by anchoring the –CH2Cl groups to the benzene rings of the mesopolymers using AlCl3 and then functionalizing with phosphine ligands to afford ligand-modified materials 2a–2d. When 2a–2d were subsequently stirred in anhydrous THF solution containing gold precursor under argon atmosphere, the gold(I) ions were coordinated to phosphine ligands to afford the catalysts 3a–3d. | ||
| Scheme 1 The strategy for the synthesis FDU-type mesopolymer supported gold(I) catalysts 3a–3d. | ||
The powder X-ray diffraction (XRD) patterns of FDU-15 mesopolymer and the modified mesoporous materials are shown in Fig. 1 and S1 (ESI†). Although the diffraction peaks gradually decreased in intensity after stepwise modifications, all samples still showed well defined (10), (11), (20) planes of a 2D hexagonal structure (P6mm) for the FDU-15 mesopolymer, which proved that the mesostructures were well preserved in the entire process of catalyst preparation.
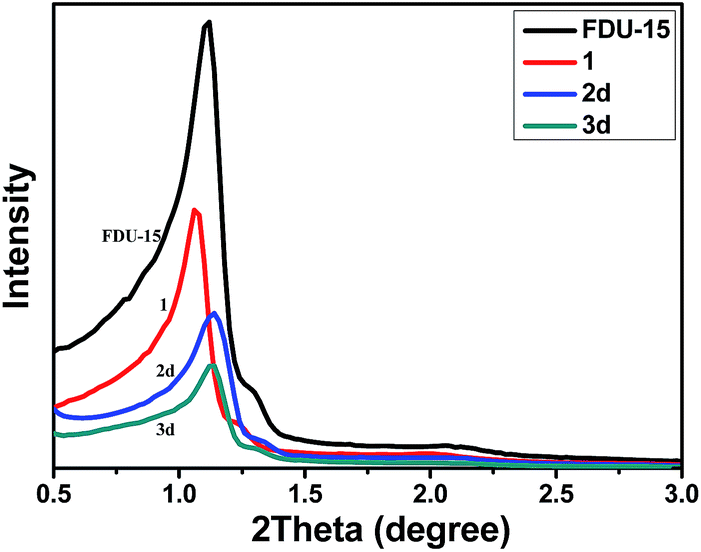 | ||
| Fig. 1 Powder XRD patterns of (a) FDU-15, (b) 1, (c) 2d and (d) 3d. | ||
Fig. 2 and S2† show the N2 adsorption–desorption isotherms and the corresponding pore size distributions of the parent and grafted FDU-15 samples. All samples displayed a typical type IV isotherm with apparent H2 hysteresis loops, implying that the samples possessed a mesoporous structure. The specific surface area (SBET), pore diameter (Dp) and pore volume (Vp) were calculated using the BJH method from the desorption branches of the N2 isotherm (Table 1). After post-modification, a gradual decrease in the BET surface areas, pore volume and average pore diameter was noted, presumably attributed to the incorporation of the organic groups (phosphine ligand) into the channels of FDU-type mesopolymer.
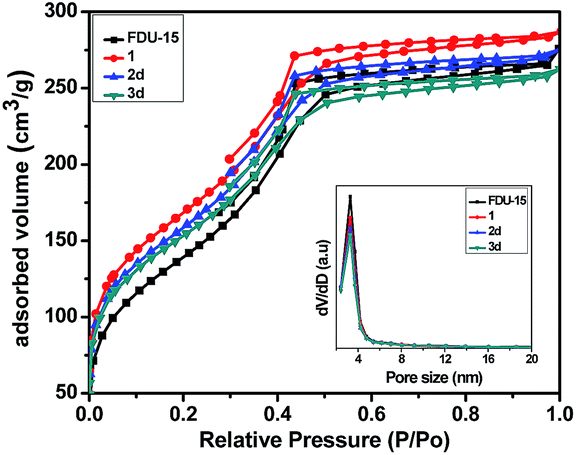 | ||
| Fig. 2 The nitrogen adsorption–desorption isotherms and BJH pore size distribution curves of FDU-15, 1, 2d and 3d. | ||
| Sample | SBET (m2 g−1) | Vp (cm3 g−1) | Dpb (nm) | Auc (wt%) |
|---|---|---|---|---|
| a Given by N2 sorption at 77 K.b By BJH analysis.c ICP analysis. | ||||
| FDU-15 | 466 | 0.47 | 3.3 | — |
| 1 | 443 | 0.43 | 3.3 | — |
| 2a | 421 | 0.41 | 3.2 | — |
| 2b | 405 | 0.40 | 3.3 | — |
| 2c | 417 | 0.40 | 3.3 | — |
| 2d | 432 | 0.30 | 3.3 | — |
| 3a | 417 | 0.40 | 3.2 | 2.6 |
| 3b | 383 | 0.30 | 3.3 | 0.98 |
| 3c | 397 | 0.40 | 3.3 | 1.0 |
| 3d | 400 | 0.30 | 3.3 | 1.4 |
Transmission electron microscopy (TEM) images (Fig. 3) displayed highly ordered mesoporous arrays and long-range mesoporous channels for the parent and functionalized mesopolymers. The mesopore array of 3d was a typical 2D hexagonal mesostructure, which verified that well-ordered mesoporous structures were maintained precisely after the gradual chemical grafting.
 | ||
| Fig. 3 TEM images of (a) FDU-15, (b) 1 (FDU-CH2Cl), (c) 2d and (d) 3d. | ||
The samples before and after grafting were further characterized by FT-IR (Fig. S3†). The band at 3437 cm−1 is assigned to the phenolic hydroxyl stretching vibration. Comparing 1 with the parent FDU-type material, a new band appeared at 698 cm−1 after chloromethylation treatment, which is attributed to the stretching vibration of the C–Cl bond.35 For sample 2d, the new bands developed at 1065 cm−1 and 1167 cm−1 are assigned to the C–F bond stretching vibration. The band at 1323 cm−1 is assigned to the characteristic P-C vibration. These results demonstrated that the phosphine ligands were successfully introduced in the mesoporous polymers. No visible change was observed in IR spectra after further modification with Me2SAuCl.
In addition, the samples before and after functionalization were characterized by EDS spectroscopy (Fig. S4†) and solid state 13C NMR spectroscopy (Fig. S5†). The characteristic peak of elemental Cl was observed after chloromethylation (Fig. S4b†). After the functionalization with chemical ligand and homogeneous gold catalyst, new characteristic peaks of elemental F and Au were noticed (Fig. S4c–d†). According to the 13C NMR spectra, all samples show distinct signals at 35, 129 and 151 ppm, which are assigned to the methylene bridges, phenolic hydroxyl substitutions and other carbons in the phenol ring, respectively. Compared to FDU-15, the ligand-functionalized sample displayed a new band at 65 ppm, which corresponds to the methylene bridges between the phenol rings and ligands. The signals of the carbons in –CF3 groups are not visible, which may overlap with the other carbon in the phenol ring. These spectra further confirmed the successful functionalization of FDU-CH2Cl with the ligand and the homogeneous gold catalyst. Moreover, the binding energy at 85.1 eV of Au XPS spectrum (Fig. S6†) for fresh catalyst-3d clearly confirms the presence of Au(I) species. The Au loadings were determined to be 2.6, 0.98, 1.0, 1.4 wt% for catalysts 3a, 3b, 3c, 3d, respectively (Table 1).
3.2 Catalytic activity
To evaluate the catalytic activities of FDU-supported gold(I) catalysts, the amination of trans-1,3-diphenyl-2-propen-1-ol with p-toluene sulphonamide was studied as a model reaction. Meeting atom economy demand, this reaction is an environment-friendly process since it generates water as the only by-product. The results summarized in Table 2 show the influence of the reaction parameters.| Entry | Catalyst | Au amount (mol%) | Solvent | Time (h) | Yieldb (%) |
|---|---|---|---|---|---|
a Reaction conditions: 1 mmol trans-1,3-diphenyl-2-propen-1-ol, 2 mmol p-toluene sulfonamide, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cat. (mol% Au)/AgOTf, 2 mL solvent, argon atmosphere.b Isolated yield.c Data in parentheses were yields avoiding light. :1 cat. (mol% Au)/AgOTf, 2 mL solvent, argon atmosphere.b Isolated yield.c Data in parentheses were yields avoiding light. |
|||||
| 1 | Ph3PAuCl | 0.5 | Dioxane | 1.5 | 67 (85)c |
| 2 | 3a | 0.1 | Dioxane | 1.5 | 89 |
| 3 | 3b | 0.1 | Dioxane | 4 | 90 |
| 4 | 3c | 0.1 | Dioxane | 4 | 90 |
| 5 | 3d | 0.1 | Dioxane | 1.5 | 95 |
| 6 | 3a | 0.05 | Dioxane | 7 | 43 |
| 7 | 3b | 0.05 | Dioxane | 20 | 38 |
| 8 | 3c | 0.05 | Dioxane | 20 | 41 |
| 9 | 3d | 0.05 | Dioxane | 1.5 | 91 |
| 10 | 3d | 0.01 | Dioxane | 8 | 50 |
| 11 | 3d | 0.05 | MeCN | 1.5 | 50 |
| 12 | 3d | 0.05 | MeNO2 | 3 | 75 |
| 13 | 3d | 0.05 | THF | 3 | Trace |
| 14 | 3d | 0.05 | DMF | 3 | n.r. |
| 15 | 3d | 0.05 | DMSO | 3 | n.r. |
| 16 | SBA-PPh2AuCl | 0.5 | Dioxane | 1.5 | 73 |
| 17 | PS-PPh2AuCl | 0.5 | Dioxane | 1.5 | 78 |
| 18 | AgOTf | 0.1 | Dioxane | 1.5 | n.r (29)c |
| 19 | 2d | 0.1 | Dioxane | 24 | n.r |
The homogeneous Ph3PAuCl catalyst (0.5 mol%) afforded 67% product yield (Table 2, entry 1) in dioxane. In contrast, the immobilized catalyst 3a afforded a 89% yield with 0.1 mol% Au loading (Table 2, entry 2). Other immobilized catalysts, 3b–d, also showed excellent yield (Table 2, entries 3–5) though a much lower catalyst amount was used. In particular, sample 3d could afford a 95% product yield in 1.5 h. The catalyst bearing electron-donating group – OMe in phosphine ligand required a longer reaction time to give a good product yield (Table 2, entries 3 and 4). When the catalyst amount was decreased to 0.05 mol%, all catalysts showed detrimental activity except for 3d (Table 2, entries 6–9). The high activity of 3d might be due to the strong electron-withdrawing effect of the –CF3 group in the phosphine ligand.39–42 Further, decreasing the amount of 3d by tenfold to 0.01 mol% could still lead to a 50% yield after reaction for 8 h (Table 2, entry 10). Thus, 3d was chosen for the subsequent study due to its high catalytic activity. The solvent-effect on the reaction was also studied (Table 2, entries 11–15). Among several commonly used solvents, the reaction proceeding in dioxane afforded the best yield. When homogeneous gold was supported on polystyrene43 and SBA-15,44,45 both catalysts showed lower activity than FDU-15 supported catalysts (Table 2, entries 16 and 17). No reaction was observed on using AgOTf as the catalyst. Interestingly, AgOTf could afford 29% product yield under dark condition (Table 2, entry 18), which is probably due to the light-sensitive nature of AgOTf.46 This light-dependent activity was also observed for homogeneous Ph3PAuCl, which also provises better reactivity under dark conditions (Table 2, entry 2). This behavior effectively explains the enhanced activity of immobilized catalysts compared with the homogenous counterpart since the mesoporous wall may protect both AgOTf and Au-catalyst from light-driven decomposition. This detrimental effect could be prevented by the mesoporous wall of FDU-15, allowing for the formation of active species Ph3PAuOTf.47 It should be noted that no reaction was observed for sample 2d, which did not contain any Au (Table 2, entry 19), further emphasizing the catalytic role of Au(I) centers.
Under the optimized reaction conditions, we then investigated the reactivity of different allylic alcohols and amine. The results are summarized in Table 3. Both the electron-withdrawing and electron-donating substituted aryl sulfonamides afforded the desired products in moderate to high yields (Table 3, entries 1–4). However, the use of electron-withdrawing groups required longer reaction times and higher temperatures. Aliphatic sulfonamide and benzyl carbamate also gave excellent yields (Table 3, entries 5 and 6). Even reactions starting with p-NO2 and p-Cl substituted-aniline could proceed smoothly, affording the corresponding products with a high yield after a prolonged reaction time (Table 3, entries 7 and 8). Next, we investigated the scope of different allylic alcohols (4b–4d) using 5a or 5b as the coupling partner in most cases (entries 9–14). All these reactions proceeded at a lower rate than that of 4a and resulted in moderate product yields at higher temperatures. The results show that 4b afforded a 2:1 mixture of the α- and γ-substituted allylic sulfonamides at 51% overall yield (Table 3, entry 9). The reaction of 5b also gave a mixture of the α- and γ-substituted products (Table 3, entry 10). The γ-products may be obtained via hydroamination reaction of the C–C double bond pathways.48
|
||||||
|---|---|---|---|---|---|---|
| Entry | Allylic alcohols | Nucleophine | T (°C) | Time (h) | Product | Yieldb (%) |
| a Reaction conditions: 1 mmol allylic alcohols, 2 mmol nucleophile, 3d (0.05 mol% Au), 0.05 mol% AgOTf, 2 mL dioxane, argon atmosphere.b Isolated yield.c 0.2 mol% 3d, 0.2 mol% AgOTf.d The ratio of α-product to γ-product. | ||||||
| 1 | 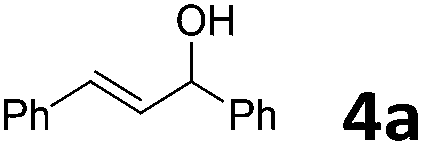 |
 |
30 | 1.5 | 6a | 91 |
| 2 | 4a | 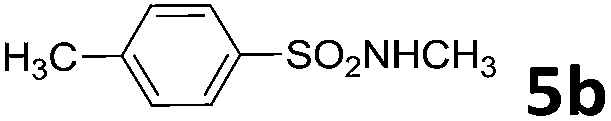 |
30 | 4 | 6b | 91 |
| 3 | 4a | 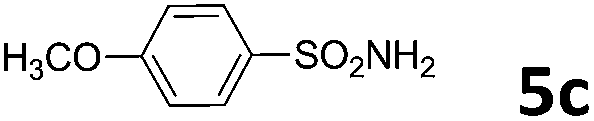 |
30 | 1.5 | 6c | 81 |
| 4c | 4a |  |
50 | 26 | 6d | 77 |
| 5 | 4a | CH3SO2NH2 5e | 50 | 7.5 | 6e | 86 |
| 6c | 4a | CbzNH2 5f | 30 | 2 | 6f | 98 |
| 7c | 4a | 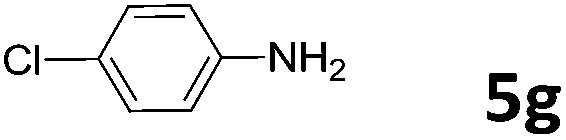 |
50 | 26 | 6g | 93 |
| 8 | 4a |  |
30 | 12 | 6h | 98 |
| 9 | 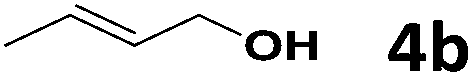 |
5a | 85 | 53 | 6i, 6i′ | 51 (2:1)d |
| 10 | 4b | 5b | 85 | 53 | 6j, 6j′ | 48 (3:1)d |
| 11 | 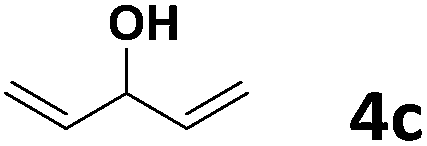 |
5a | 50 | 25 | 6k | 48 |
| 12 | 4c | 5b | 50 | 25 | 6l | 42 |
| 13 | 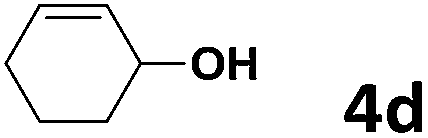 |
5a | 50 | 8 | 6m | 56 |
| 14 | 4d | 5b | 50 | 9 | 6n | 70 |
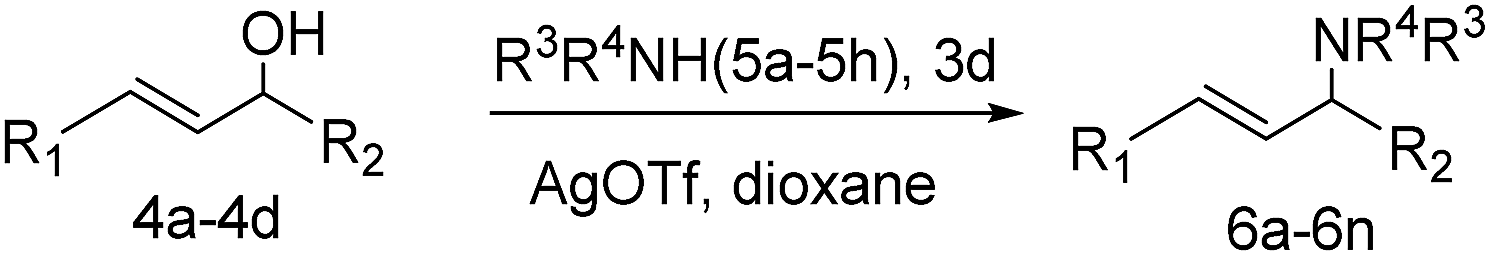
The recyclability of catalyst 3d was also examined in the amination reaction of trans-1,3-diphenyl-2-propen-1-ol with p-toluenesulfonamide. The catalyst was recovered by simple filtration after reaction, washed with ethyl acetate and acetone and then dried in vacuum. The dried catalysts were directly used for the next catalytic cycle. As shown in Fig. 4, catalyst 3d could be reused for at least twelve times without significant decrease in the isolated yield. We compared catalyst 3d before and after 12 runs with different characterization in XPS, XRD and BET (Fig. S6–S8†). It can be clearly observed that the oxidation state of Au(I) was well retained for the spent catalyst (Fig. S6†).49 The XRD pattern indicated that the ordered structure of organic frameworks collapsed to some extent after these runs (Fig. S7†). The BET result also indicated that the pore size and specific surface area decreased, in line with the collapse of the framework, which is probably responsible for the sharp decrease of product yield after twelve runs.
 | ||
| Fig. 4 The reusability of sample 3d in the amination reaction of trans-1,3-diphenyl-2-propen-1-ol with p-toluenesulfonamide. | ||
We further examined the utility of 3d in the intramolecular cyclization for the synthesis of substituted 2,3-dihydro-4H-pyran-4-ones. The dihydropyranones moiety as one of the most important components has been observed in many naturally occurring products and displays a variety of biological activities.50–60 After preliminary screening of the reaction conditions, catalyst 3d could efficiently catalyze the intramolecular cyclization of 7a, which provided results comparable to those obtained using homogeneous catalysts (Table 4, entry1). Benzene ring substituted 2,3-dihydro-4H-pyran-4-ones compounds 8a–k were also synthesized in good yields using this heterogeneous catalyst(Table 4).
|
||||
|---|---|---|---|---|
| Entry | R | Product | Time (h) | Yieldb (%) |
| a Reaction conditions: 0.45 mmol 7; 3d (5 mol% Au), and 5 mol% AgOTf; 4 mL toluene; argon atmosphere.b Isolated yield. | ||||
| 1 | H 7a | 8a | 8 | 81 |
| 2 | 4-OCH3 7b | 8b | 12 | 80 |
| 3 | 3-OCH3 7c | 8c | 12 | 72 |
| 4 | 2-OCH3 7d | 8d | 12 | 73 |
| 5 | 4-CF3 7e | 8e | 48 | 84 |
| 6 | 4-Cl 7f | 8f | 12 | 56 |
| 7 | 3-Cl 7g | 8g | 24 | 80 |
| 8 | 2-Cl 7h | 8h | 12 | 58 |
| 9 | 4-CN 7i | 8i | 24 | 90 |
| 10 | 4-NO2 7j | 8j | 24 | 83 |
| 11 | 4-Br 7k | 8k | 12 | 78 |
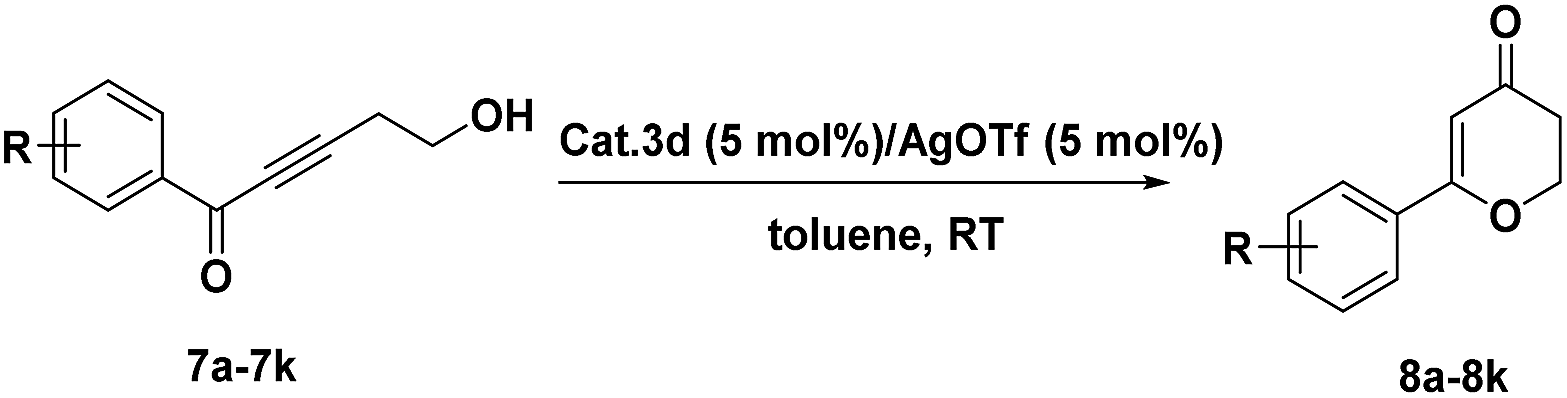
The reaction of substituted groups at the para position of the aryl ring proceeded efficiently (Table 4, entries 2, 5, 6, 9, 10 and 11). The reactions of substrates comprising strong electron-withdrawing groups such as –CF3, –NO2 and –CN all gave the corresponding products in high yield (Table 4, entries 5, 9 and 10). The reactions were also carried out using the substrates with substituted groups on the other positions of the aryl ring (Table 4, entries 3, 4, 7 and 8). The aryl ring containing a chloro group at the meta position afforded a higher yield than those at the para and meta positions (Table 4, entries 6–8), while the methoxyl group led to a slightly lower yield (Table 4, entries 2–4). The catalyst can also be easily recycled.
Finally, we used 3d as the catalyst for the synthesis of five-membered heterocyclic compounds. Furans are important intermediates, which widely serve as key structural subunits in numerous natural products, industrial organic syntheses and material science.61–63 The reaction of propargyl alcohol 9 afforded the corresponding substituted furan 10 at 92% yield after 6 h, which shows similar activity to the homogeneous catalyst (Scheme 2).
 | ||
| Scheme 2 The intramolecular cyclization of 9 to 10. | ||
4. Conclusions
In summary, we have developed a series of ordered mesopolymers FDU supported gold(I) catalysts (3a–3d). Catalyst 3d showed the best catalytic activity in amination of allylic alcohols with different nucleophiles. Compared with homogeneous gold(I) catalyst, the heterogeneous gold(I) catalysts exhibited enhanced catalytic activity. The present study suggests the potential structural cooperation of the support material could enhance the stability. Moreover, the reaction also performed efficiently at a lower catalyst loading (0.05 mol%), which may be explained by the high dispersion of active sites in the mesopolymers and the catalysts can be easily recovered and recycled without significant decrease in activity. Catalyst 3d was also applicable to other organic transformations, affording good yields. Further research for application of 3d and other heterogeneous gold catalysts are now in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support by NSFC (21373088, 21533002), China Ministry of Science and Technology (2016YFA0202804).References
- A. Hoffmann-Roder and N. Krause, Org. Biomol. Chem., 2005, 3, 387–391 Search PubMed.
- G. J. Hutchings, Catal. Today, 2005, 100, 55–61 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- A. Stephen and K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef PubMed.
- Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239–3265 CrossRef CAS PubMed.
- N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776–1782 RSC.
- T. Yao, X. Zhang and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 11164–11165 CrossRef CAS PubMed.
- Y. Liu, F. Song, Z. Song, M. Liu and B. Yan, Org. Lett., 2005, 7, 5409–5412 CrossRef CAS PubMed.
- M. H. Suhre, M. Reif and S. F. Kirsch, Org. Lett., 2005, 7, 3925–3927 CrossRef CAS PubMed.
- B. D. Sherry, L. Maus, B. N. Laforteza and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 8132–8133 CrossRef CAS PubMed.
- G. Zhang, L. Cui, Y. Wang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 1474–1475 CrossRef CAS PubMed.
- H. F. Jonsson, S. Evjen and A. Fiksdahl, J. Am. Chem. Soc., 2017, 19, 2202–2205 CAS.
- M. J. Harper, E. J. Emmett, J. F. Bower and C. A. Russell, J. Am. Chem. Soc., 2017, 13, 12386–12389 CrossRef PubMed.
- A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS PubMed.
- C. M. Crudden, D. Allen, M. D. Mikoluk and J. Sun, Chem. Commun., 2001, 1154–1155 RSC.
- A. M. Liu, K. Hidajat and S. Kawi, J. Mol. Catal. A: Chem., 2001, 168, 303–306 CrossRef CAS.
- D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615–3640 CrossRef CAS PubMed.
- A. Taguchi and F. Schüth, Microporous Mesoporous Mater., 2005, 77, 1–45 CrossRef CAS.
- M. Guino and K. K. Hii, Chem. Soc. Rev., 2007, 36, 608–617 RSC.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- A. Corma, E. Gutiérrez-Puebla, M. Iglesias, A. Monge, S. Pérez-Ferreras and F. Sánchez, Adv. Synth. Catal., 2006, 348, 1899–1907 CrossRef CAS.
- W. Cao and B. Yu, Adv. Synth. Catal., 2011, 353, 1903–1907 CrossRef CAS.
- X.-Z. Shu, S. C. Nguyen, Y. He, F. Oba, Q. Zhang, C. Canlas, G. A. Somorjai, A. P. Alivisatos and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 7083–7086 CrossRef CAS PubMed.
- W. Yang, L. Wei, F. Yi and M. Cai, Catal. Sci. Technol., 2016, 6, 4554–4564 CAS.
- R. Cai, X. Ye, Q. Sun, Q. He, Y. He, S. Ma and X. Shi, ACS Catal., 2017, 7, 1087–1092 CrossRef CAS.
- J. T. Sarmiento, S. Suárez-Pantiga, A. Olmos, T. Varea and G. Asensio, ACS Catal., 2017, 7, 7146–7155 CrossRef CAS.
- M. Chen, Z.-M. Zhang, Z. Yu, H. Qiu, B. Ma, H.-H. Wu and J. Zhang, ACS Catal., 2105, 5, 7488–7492 CrossRef.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., 2005, 117, 7215–7221 CrossRef.
- F. Zhang, Y. Meng, D. Gu, Y. Yan, C. Yu, B. Tu and D. Zhao, J. Am. Chem. Soc., 2005, 127, 13508–13509 CrossRef CAS PubMed.
- R. Xing, N. Liu, Y. Liu, H. Wu, Y. Jiang, L. Chen, M. He and P. Wu, Adv. Funct. Mater., 2007, 17, 2455–2461 CrossRef CAS.
- R. Xing, Y. Liu, H. Wu, X. Li, M. He and P. Wu, Chem. Commun., 2008, 6297–6299 RSC.
- S. Yan, Y. Gao, R. Xing, Y. Shen, Y. Liu, P. Wu and H. Wu, Tetrahedron, 2008, 64, 6294–6299 CrossRef CAS.
- R. Xing, H. Wu, X. Li, Z. Zhao, Y. Liu, L. Chen and P. Wu, J. Mater. Chem., 2009, 19, 4004–4011 RSC.
- C. Yao, H. Li, H. Wu, Y. Liu and P. Wu, Catal. Commun., 2009, 10, 1099–1102 CrossRef CAS.
- W. Zhang, Q. Wang, H. Wu, P. Wu and M. He, Green Chem., 2014, 16, 4767–4774 RSC.
- X. Giner, P. Trillo and C. Nájera, J. Organomet. Chem., 2011, 696, 357–361 CrossRef CAS.
- M. Egi, K. Azechi, M. Saneto, K. Shimizu and S. Akai, J. Org. Chem., 2010, 75, 2123–2126 CrossRef CAS PubMed.
- M. Egi, K. Azechi and S. Akai, Org. Lett., 2009, 11, 5002–5005 CrossRef CAS PubMed.
- J. P. Markham, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 9708–9709 CrossRef CAS PubMed.
- P. Dubé and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 12062–12063 CrossRef PubMed.
- D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS PubMed.
- W. Wang, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2012, 134, 5697–5705 CrossRef CAS PubMed.
- M. Egi, K. Azechi and S. Akai, Adv. Synth. Catal., 2011, 353, 287–290 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- X. Feng, M. Yan, T. Zhang, Y. Liu, M. Bao and X. Feng, Green Chem., 2010, 12, 1758–1766 RSC.
- Q. Dong, N. Li, R. Qiu, J. Wang, C. Guo and X. Xu, J. Organomet. Chem., 2015, 799–800, 122–127 CrossRef CAS.
- J. Zhang, C.-G. Yang and C. He, J. Am. Chem. Soc., 2006, 128, 1798–1799 CrossRef CAS PubMed.
- P. Mukherjee and R. A. Widenhoefer, Org. Lett., 2010, 12, 1184–1187 CrossRef CAS PubMed.
- E. Aguiló, R. Gavara, J. Carlos Lima, J. Llorca and L. Rodríguez, J. Mater. Chem. C, 2013, 1, 5538–5547 RSC.
- M. M. Faul and B. E. Huff, Chem. Rev., 2000, 100, 2407–2474 CrossRef CAS PubMed.
- C. Yao, Z. Xiao, R. Liu, T. Li, W. Jiao and C. Yu, Chem.–Eur. J., 2013, 19, 456–459 CrossRef CAS PubMed.
- A. Ilangovan and P. Sakthivel, RSC Adv., 2014, 4, 55150–55161 RSC.
- R. Aloise Pilli, L. Fernando Toneto Novaes, V. Maria Teixeira Carneiro, R. Lopes Drekener and C. Martins Avila, Curr. Org. Chem., 2015, 12, 523–529 Search PubMed.
- R. S. Coleman and E. B. Grant, Tetrahedron Lett., 1990, 31, 3677–3680 CrossRef CAS.
- J. D. Winkler and K. Oh, Org. Lett., 2005, 7, 2421–2423 CrossRef CAS PubMed.
- L. Candish and D. W. Lupton, Chem. Sci., 2012, 3, 380–383 RSC.
- M. Reiter, S. Ropp and V. Gouverneur, Org. Lett., 2004, 6, 91–94 CrossRef CAS PubMed.
- M. Reiter, H. Turner, R. Mills-Webb and V. Gouverneur, J. Org. Chem., 2005, 70, 8478–8485 CrossRef CAS PubMed.
- X.-Z. Shu, X.-Y. Liu, K.-G. Ji, H.-Q. Xiao and Y.-M. Liang, Chem.–Eur. J., 2008, 14, 5282–5289 CrossRef CAS PubMed.
- F. Yang, K. G. Ji, S.-C. Zhao, S. Ali, Y. Y. Ye, X. Y. Liu and Y. M. Liang, Chem.–Eur. J., 2012, 18, 6470–6474 CrossRef CAS PubMed.
- L. Zhu, J. Luo and R. Hong, Org. Lett., 2014, 16, 2162–2165 CrossRef CAS PubMed.
- R. C. Brown, Angew. Chem., Int. Ed., 2005, 44, 850–852 CrossRef CAS PubMed.
- M. Zhang, H. F. Jiang, H. Neumann, M. Beller and P. H. Dixneuf, Angew. Chem., Int. Ed., 2009, 48, 1681–1684 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12498h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |