
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeted delivery of HES5-siRNA with novel polypeptide-modified nanoparticles for hepatocellular carcinoma therapy†
Yu Xia,
Changbing Wang,
Tiantian Xu,
Yinghua Li,
Min Guo,
Zhengfang Lin,
Mingqi Zhao and
Bing Zhu *
*
Guangzhou Women and children's Medical center, No 318 Renminzhong road, Yuexiu District, Guangzhou, Guangdong 510120, P. R. China. E-mail: zhubing2017@hotmail.com; Tel: +86 020 81322725
First published on 9th January 2018
Abstract
For actively targeted delivery of small interfering RNA (siRNA) to solid tumors, we fabricated functionalized selenium nanoparticles (SeNPs) decorated with the polypeptide RGDfC. Herein, RGDfC was used as tumor-targeted moiety and installed onto the surface of SeNPs to enhance the cellular uptake. RGDfC-SeNPs@siRNA were internalized into the HepG2 cell mainly through clathrin-mediated endocytosis. The active efficacy of the RGDfC-SeNPs@siRNA was confirmed via gene silencing assay, MTT assay and flow cytometry analysis. Owing to the tumor-targeting effect of RGDfC, RGDfC-SeNPs@siRNA achieved an obvious improvement in gene silencing ability, which led to significant growth inhibition of HepG2 cells. Furthermore, treatment with RGDfC-SeNPs@siRNA resulted in greater antitumor efficacy than lipofectamine 2000@siRNA in vitro and in vivo. In addition, the RGDfC-SeNPs@siRNA was almost non-toxic to the key organs of mice. In sum, these findings provide an alternative therapeutic route for targeted cancer treatments.
Introduction
Hepatocellular carcinoma (HCC) is one of the most common primary malignant cancers and the second main cause of cancer-related deaths worldwide.1 Owing to the low percentage of HCC patients that are eligible for surgery and the high tumor recurrence rates, chemotherapy is still a major clinical method for HCC treatment.2 Many traditional chemotherapy drugs have been utilized for HCC therapy, but most of them have been ineffective.3 Recently, gene therapy using small interfering RNA (siRNA) technology has attracted increasing attention in the treatment of cancers because of the enhanced anticancer efficiency and low cytotoxicity.4 Traditionally, viruses have usually been utilized as gene carriers.5 However, viral carriers are prone to immunogenicity and risk of insertional mutagenesis.6 Therefore, non-viral carriers have great potential in the application of gene therapy due to their better safety profile.7 Thus, the design of novel non-viral gene delivery carriers has become a focus in the field of cancer gene therapy.8 The ideal non-viral vector should have the following characteristics: small size below 200 nm, biocompatible, stable enough in blood to protect the nucleic acid, effective delivery of gene into target location, and so on.9Selenium (Se) is a mineral trace element of fundamental importance to humans and animals. The role of Se in potential cancer chemotherapeutic and chemopreventive agents has been supported by many epidemiological, preclinical, and clinical studies.10 The previous studies showed that the Se is degradable in vivo and the degraded Se can be used as a nutrient for many kinds of normal cells or as an antiproliferative agent for many kinds of cancer cells.11 Recently, selenium nanoparticles (SeNPs) have attracted increasing attention in the field of drug delivery vehicles in cancer therapy. Moreover, SeNPs show many merits in gene delivery systems, such as the desired size, high gene-loading capacity, enhanced anticancer effect, controlled release and low cytotoxicity.12 The previous reports indicated that SeNPs were capable of conjugating with siRNA to achieve enhanced anticancer activity in vitro.13 Nevertheless, there is still some deficiency to overcome in this gene delivery vehicle for effectively carrying the genes, such as the lack of tumor-targeting activity.14 To prepare the actively tumor-targeted delivery carrier, the positively charged polypeptide RGDfC was conjugated onto the surfaces of SeNPs to fabricate positively charged nanoparticles, RGDfC-SeNPs, which are not only conducive to binding with siRNA, but also protect the siRNA from rapid degradation by plasma nucleases.15 Moreover, the RGDfC can selectively bind to αvβ3/αvβ5 integrins overexpressed on various cancer cells.16
Hairy and enhancer of split 5 (HES5) is a DNA-binding transcription factor and it can repress the transcription of Hash1 and consequently inhibit cell differentiation.17 Previous studies showed that HES5 plays an important role in the initiation or/and development of cancer, and it has gradually developed into a crucial therapeutic target in tumor treatments.18 Therefore, in this paper, HES5-siRNA was loaded onto the surface of SeNPs modified with tumor-targeted moiety RGDfC to fabricate functionalized SeNPs RGDfC-SeNPs@siRNA, aimed at silencing the HES5 gene and thus inhibiting tumor growth. As expected, the RGDfC-SeNPs@siRNA could selectively accumulate in HepG2 tumors, and presented improved gene silencing effectiveness and antitumor efficacy without obviously observable side effects. Therefore, such RGDfC-conjugated selenium nanoparticles for the delivery of HES5-siRNA to the HepG2 tumor hold great potential for application in hepatocellular carcinoma therapy.
Experimental section
Materials
Sodium selenite (Na2SeO3) and ascorbic acid (vitamin C) were purchased from Guangzhou Chemical Reagent Factory (Guangzhou, PR China). Hoechst 33342 and the Annexin V-FITC Apoptosis Kit were purchased from Sinopharm (Shanghai, PR China). The TUNEL assay kit and cy5.5 were purchased from Beyotime (Shanghai, PR China). The cyclic polypeptide Arg-Gly-Asp-D-Phe-Cys (RGDfC) was obtained from China Polypeptides Co., Ltd. All the antibodies were purchased from CST (Massachusetts, US). The siRNA was obtained from RiboBio (Guangzhou, PR China), and the HES5-siRNA sequence was as follows: 5′-AAGGCTACTCGTGGTGCCT-3'.Synthesis of RGDfC-SeNPs
The RGDfC-conjugated selenium nanoparticles (RGDfC-SeNPs) were synthesized as previously described. Briefly, 5 ml of ascorbic acid (4 mM) was added to 5 ml of sodium selenite (1 mM) and the mixed solution was stirred for 4 h to prepare the selenium nanoparticles (SeNPs); then 1 mg RGDfC dissolved in deionized water was added to the SeNPs solution and the solution was stirred for another 2 h to prepare the selenium nanoparticles conjugated with RGDfC (RGDfC-SeNPs). Then, the mixed solutions were dialyzed (3.5 kDa) to remove the redundant RGDfC, ascorbic acid and sodium selenite. The RGDfC-SeNPs@siRNA complexes were obtained as previously reported.12 Briefly, the RGDfC-SeNPs were dispersed in DNase/RNase-free water, and then vortexed with a siRNA solution for 1 h to form RGDfC-SeNPs@siRNA complexes. The nanoparticles were characterized by transmission electron microscopy (TEM) and Fourier transform infrared (FTIR) spectroscopy. Energy-dispersive X-ray (EDX) spectroscopy was conducted to examine the elemental composition of the nanoparticles. The average sizes and zeta potentials of the nanoparticles were obtained via a Zetasizer Nano analyzer (Malvern, UK).To examine the siRNA loading capability, we established a calibration curve against the FAM-siRNA, and obtained the relative fluorescence intensity of FAM-siRNA in the nanoparticles. The test was performed in a multimode plate reader (EnSpire, PerkinElmer, USA) (for FAM-siRNA: excitation, 465 nm; emission, 520 nm). The loading efficiency and loading content of siRNA were calculated using the following formulas: loading efficiency (%) = (weight of loaded siRNA/weight of siRNA in feed) × 100%; loading content (%) = (weight of loaded siRNA/weight of RGDfC-SeNPs@siRNA) × 100%.
The loading efficiency and loading content of siRNA onto RGDfC-SeNPs were about 82.5% and 7.5 wt%, respectively.
Agarose gel retardation assay
RGDfC-SeNPs@siRNA complexes with various RGDfC-SeNPs/siRNA weight ratios were fabricated and the concentration of siRNA was fixed at 1 μM. Then the RGDfC-SeNPs@siRNA were subjected to gel electrophoresis in 1% agarose gel with 0.5 mg ml−1 ethidium bromide (EB) at 120 mV for 15 min, and the gel was visualized by using a UV gel image system. An electrophoretic mobility assay of RGDfC-SeNPs@siRNA was carried out to explore whether RGDfC-SeNPs had the ability to protect the siRNA in the presence of fetal bovine serum (FBS). RGDfC-SeNPs@siRNA (weight ratio 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was incubated with 50% (v/v) FBS for different amounts of time. Subsequently, an aliquot of each sample was electrophoresed according to a process similar to that described above.
:1) was incubated with 50% (v/v) FBS for different amounts of time. Subsequently, an aliquot of each sample was electrophoresed according to a process similar to that described above.
Cell culture
HepG2 cells and Lo2 cells were purchased from the American Type Culture Collection (ATCC) and incubated in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS and 1% penicillin/streptomycin at 37 °C under a 5% CO2 atmosphere.Cellular uptake of nanoparticles
HepG2 cells were seeded in 24-well plates and incubated overnight. The cells were exposed to the different formulations of FAM-siRNA at an siRNA concentration of 100 nM and incubated for 4 h. Then the cells were washed with phosphate-buffered saline (PBS) and measured by using a flow cytometer (BD FACSCalibur System, San Jose, CA, USA).Transmission electron microscopy (TEM)
To confirm whether nanoparticles were located inside HepG2 cells, the cellular uptake was further investigated by TEM. Briefly, HepG2 cells (2 × 105 cells per ml) were seeded in complete DMEM containing 10% FBS. The cells were gently washed with cold PBS to remove extracellular RGDfC-SeNPs@siRNA after 4 h of incubation with RGDfC-SeNPs@siRNA. Then the cells were harvested and fixed with 2.5% glutaraldehyde solution. Images of samples were captured using a Tecnai transmission electron microscope at 120 kV.The uptake pathways of RGDfC-SeNPs@siRNA
The cellular uptake of nanoparticles in the presence of different uptake inhibitors was investigated using HepG2 cells, which were incubated at 4 °C for 0.5 h without inhibitors or with NaN3 (sodium azide, 3 mg ml−1) + 50 mM 2-deoxy-D-glucose (DOG), nystatin (5 μg ml−1), chlorpromazine (5 μg ml−1) and amiloride (10 μg ml−1) in serum-free medium at 37 °C for 30 min. After that, RGDfC-SeNPs@FAM-siRNA were added to the medium and incubated for another 2 h. Then the cells were washed with cold PBS and measured by flow cytometry.Real-time tracking of endosome/lysosome escape
To follow the internalization and endosome/lysosome release of RGDfC-SeNPs@siRNA, the cells were co-cultured with 80 nM LysoTracker Red for 1.5 h. After washing gently with cold PBS, the cells were stained with Hoechst 33342, and then the cells were co-cultured with RGDfC-SeNPs@FAM-siRNA for 1, 2 and 4 h. Then the excess RGDfC-SeNPs@FAM-siRNA were washed away twice with cold PBS. For real-time tracking imaging, cellular fluorescence was monitored with a Leica SP5 confocal microscope. The LysoTracker Red was excited at 580 nm and the RGDfC-SeNPs@FAM-siRNA were excited at 465 nm.Quantitative real-time PCR (qPCR)
TRIzol reagent (TaKaRa, PR China) was used to measure the total RNA via a standard chloroform-extraction protocol. The StepOne™ PCR System was utilized to analyze the data using the 2−ΔΔCT method. The sequences of the primers employed were: forward 5′-GGAATTCCAATGGCCCCCAGCACTGTG-3′ and reverse 5′-GGGTACCCCACGGCCACAGTGCTGG-3′ for HES5, and forward 5′-ATCCCATCACCATCTTCCAG-3′ and reverse 5′-ATGAGTCCTTCCACGATACC-3′ for GAPDH.MTT assay
Cytotoxicity was determined by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Briefly, 5 × 104 cells were incubated in a 96-well cell culture plate for 24 h. The cells were treated with individual agents in various siRNA equivalent concentrations for another 48 h. Twenty-five microliters of MTT solution with a concentration of 5 mg mL−1 was added into each well for 4 h of incubation at 37 °C. The medium was removed carefully and dimethyl sulfoxide (DMSO) was added to dissolve the formazan dye. The OD value was measured at 570 nm.Flow cytometry analysis
Cell cycle distributions and cell apoptosis in HepG2 cell were analyzed by flow cytometry as follows: briefly, the HepG2 cells were incubated in 12-well plates and allowed to reach 50% confluence, then treated with different formulations of siRNA (at a concentration equivalent to 100 nM of siRNA). The cells were collected after 24 h of treatment for cell cycle distribution analysis or 48 h of treatment for cell apoptosis analysis. Then the collected cells were stained with propidium iodide (PI) or Annexin V-FITC/PI in the dark for 30 min. The stained cells were immediately tested by FACS flow cytometer (BD Biosciences, USA) and the results were analyzed by means of FlowJo software (Tree Star, USA).Western blotting analysis
To determine the protein expression levels, western blotting was carried out as previously reported. The HepG2 cells were cultured in 6-well plates for 12 h to reach 70% confluence and treated with RGDfC-SeNPs@siRNA (at siRNA concentrations equivalent to 25, 50 and 100 nM) for another 24 h, then the cells were collected and lysed with 50 μL Radio-Immunoprecipitation Assay (RIPA) containing 1% phenylmethylsulfonyl fluoride (PMSF). Lysates were collected by centrifugation after 15 minutes. The protein concentration was estimated by means of the BCA protein kit.In vivo imaging of nanoparticles
HepG2 cells (1 × 107) were subcutaneously injected into the abdomens of female BALB/c nude mice (about 5 weeks of age). After tumors reached ∼300 mm3, the tumor-bearing nude mice were subjected to tail-vein injection with the E-mail: RGDfC-SeNPs@cy5.5-siRNA at the siRNA equivalent dose of 0.5 mg kg−1. After 3 h and 6 h post-injection with nanoparticles, the fluorescence images of the mice were recorded at the IVIS imaging system (Xenogen, USA).Xenograft mouse model
BALB/c nude mice at ∼5–6 week ages were used for investigating the in vivo antitumor efficacy. Approximately 1 × 107 HepG2 cells were resuspended in 150 μL saline and subcutaneously injected into the abdomens of mice. When tumors grew to about 100 mm,3 the mice were divided into four groups randomly, then saline (as control group) and different formulations of siRNA (at the siRNA equivalent dose of 0.5 mg kg−1) were injected into mice intravenously once every other day during the experimental period. The volumes of tumors were calculated by the following formula: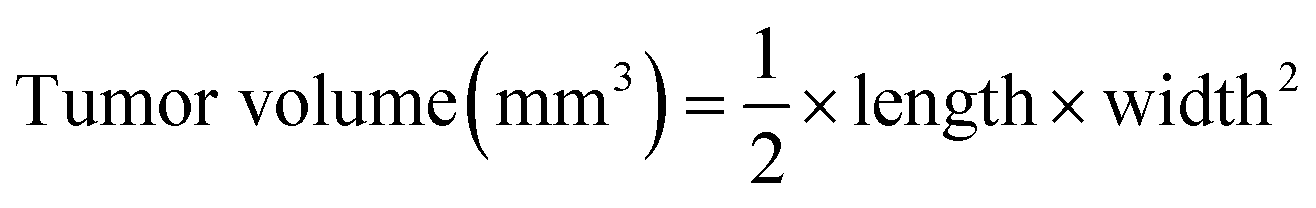
Histology and immunohistochemistry
The tumors stripped from the mice and main organ (liver, heart, kidney, spleen and lung) tissues were fixed with 3.7% paraformaldehyde and sectioned into 6 μm slices. The histological sections of tissues were used for hematoxylin and eosin (H&E) staining. The expression level of Ki67 protein associated with tumor cell growth was examined by immunohistochemical (IHC) methods. The in vivo tumor cell apoptosis was examined by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay using an apoptotic cell detection kit (Beyotime Biotech, Shanghai, PR China) following the manufacturer's protocol. The angiogenesis-related protein CD31 was detected by immunohistochemistry according to manufacturer's protocols. The images of sections were acquired by using a digital microscope (Leica DMi8). All animal experiments were approved by the Ethics Committee of Guangzhou Medical University and performed according to the protocols and guidelines of the Experimental Animal Center of Guangzhou Medical University.Statistical analysis
All the data represent the mean ± standard deviation (S.D.). Student's t-test was performed for comparison between two groups. *p < 0.05 and **p < 0.01 were considered statistically significant and highly significant differences, respectively.Results and discussion
Preparation and characterization of RGDfC-SeNPs
In this study, a novel tumor-targeted delivery carrier, RGDfC-SeNPs, was designed and synthesized. In this gene delivery system, the selenite was reduced to Se atoms by the redox reaction after adding ascorbic acid, and the accumulating Se atoms formed selenium nanoparticles (SeNPs).19 The SeNPs were conjugated with positively charged RGDfC to prepare the tumor-targeting carrier, RGDfC-SeNPs, and then the HES5-siRNA was conjugated with the RGDfC via electrostatic interaction (Scheme 1). Transmission electron microscopy (TEM) images indicated that RGDfC-SeNPs exhibited spherical morphology with a size of about 15 nm (Fig. 1A). Use of a Nano ZS particle analyzer further confirmed the average size of RGDfC-SeNPs (Fig. 1B). As shown in Fig. 1C, the marker signal of the S atom (from the RGDfC molecule) was detected in the energy-dispersive X-ray (EDX) analysis of RGDfC-SeNPs, indicating RGDfC was successfully loaded onto the SeNPs. Then, Fourier transform infrared (FTIR) spectroscopy was utilized to further confirm the conjugation between the SeNPs and RGDfC. As shown in Fig. 1D, peaks characteristic of SeNPs also appeared in the spectrum of RGDfC-SeNPs, and the RGDfC displayed typical amide band peaks at 1680 and 1547 cm−1. After conjugating SeNPs with RGDfC, the obvious characteristic peaks at 1677 cm−1 and 1539 cm−1 from amide bands were also detected, suggesting that the RGDfC was successfully conjugated with SeNPs.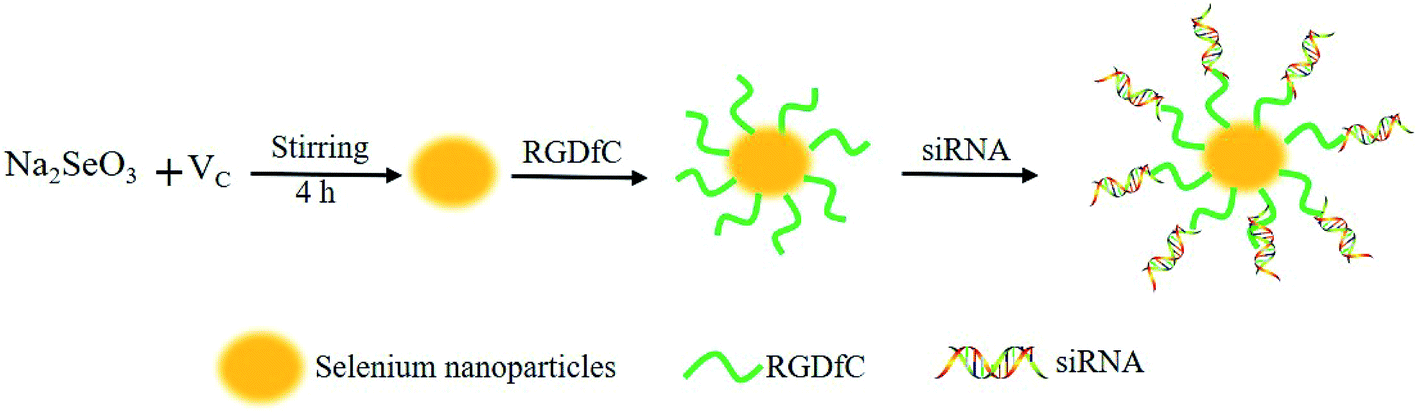 | ||
| Scheme 1 Schematic illustration of the formation of RGDfC-SeNPs@siRNA. | ||
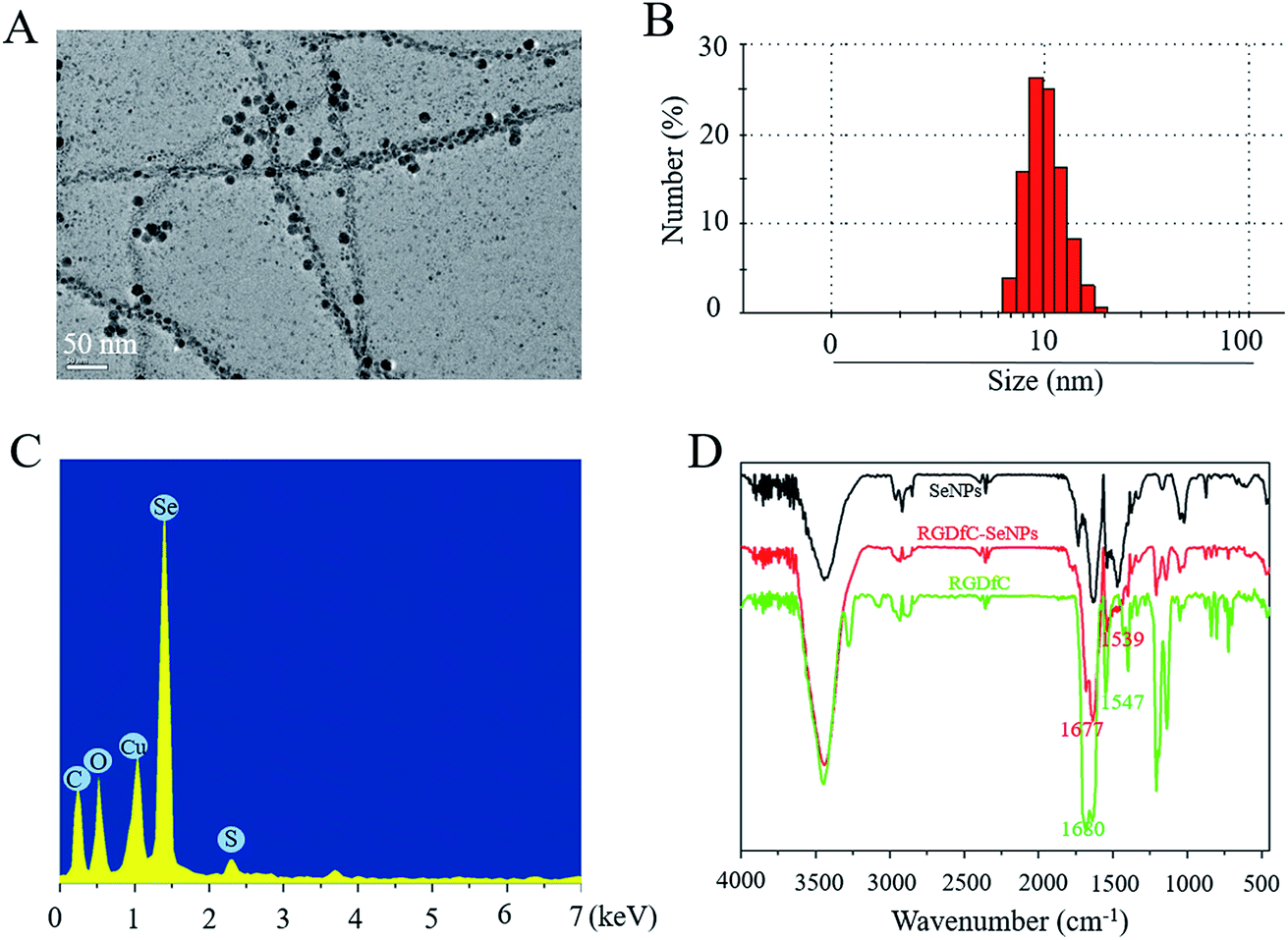 | ||
| Fig. 1 Characterization of RGDfC-SeNPs. (A) Representative TEM image featuring RGDfC-SeNPs. (B) Particle size distributions of RGDfC-SeNPs. (C) EDX analysis of RGDfC-SeNPs. (D) Fourier transform infrared spectra of SeNPs, RGDfC-SeNPs and RGDfC. | ||
The zeta potentials of nanoparticles, RGDfC and siRNA are shown in Fig. 2A; the potential of SeNPs was about −15 mV. After loading the positively charged RGDfC onto the surface of SeNPs, the potential changed from about −15 mV to +10 mV, which was helpful for the nanoparticles to load siRNA. Furthermore, the size distribution of RGDfC-SeNPs kept stable (<20 nm) for 15 days (Fig. 2B). The favorable stability of RGDfC-SeNPs supports their potential application in medical areas.20
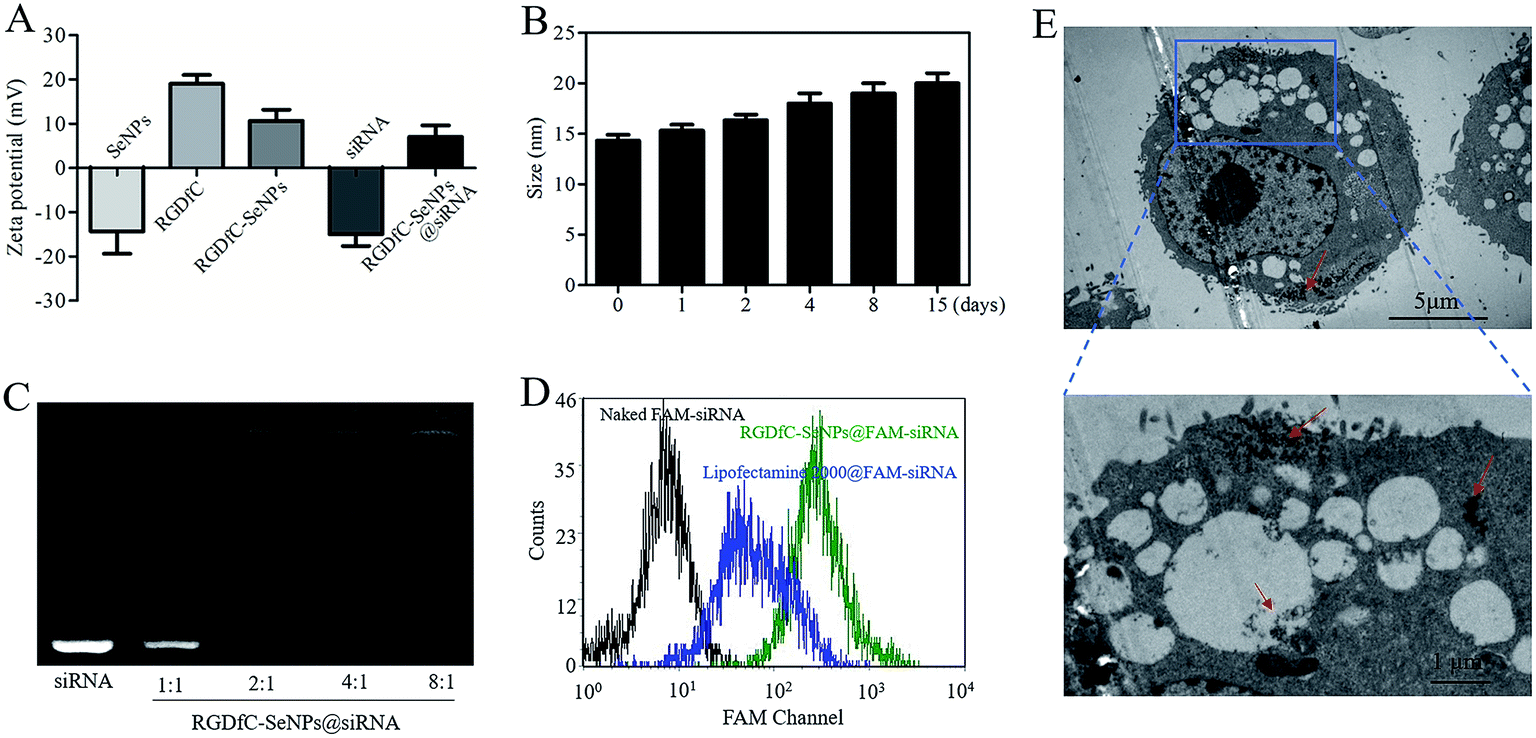 | ||
| Fig. 2 (A) Zeta potentials of SeNPs, RGDfC, RGDfC-SeNPs, siRNA and RGDfC-SeNPs@siRNA. (B) Stability of RGDfC-SeNPs@siRNA in aqueous solutions. (C) Electrophoretic mobility of free siRNA and RGDfC-SeNPs@siRNA complexes with different weight ratios (RGDfC-SeNPs:siRNA). (D) The cellular uptake of naked FAM-siRNA, lipofectamine 2000@FAM-siRNA and RGDfC-SeNPs@FAM-siRNA for 24 h was analyzed by flow cytometry. (E) TEM images of RGDfC-SeNPs@siRNA nanoparticles internalized into HepG2 cells after 4 h of incubation. | ||
siRNA loading capability and serum stability study
Agarose gel electrophoresis was performed to investigate the siRNA loading ability of RGDfC-SeNPs.21 Complete retardation of siRNA migration was achieved at an RGDfC-SeNPs/siRNA weight ratio of 4:1 (Fig. 2C), indicating that RGDfC-SeNPs were capable of binding the siRNA to impede degradation in the process of electrophoresis. The RNAse in serum easily leads to complete degradation of siRNA, thus the increased stability of siRNA in the presence of serum is essential for improving the intracellular transfection efficiency.22,23 For evaluating the capability of the gene carrier RGDfC-SeNPs to protect the siRNA from degradation in serum, RGDfC-SeNPs@siRNA were incubated in DMEM medium with 50% FBS and the stability of RGDfC-SeNPs@siRNA in serum was evaluated via a gel retardation experiment. As shown in Fig. S1,† obvious degradation of naked siRNA occurred after 1 h of incubation in medium containing 50% FBS, and the naked siRNA was almost totally degraded after 2 h of incubation. On the contrary, the siRNA from the RGDfC-SeNPs@siRNA was significantly protected during 2 h of incubation under the same conditions. This finding indicated that the RGDfC-SeNPs could provide valid protection of siRNA from nuclease-mediated degradation.
Cellular uptake of RGDfC-SeNPs@siRNA
To precisely examine the cellular uptake efficiency of siRNA, HepG2 cells were co-cultured with naked FAM-siRNA, lipofectamine 2000@FAM-siRNA and RGDfC-SeNPs@FAM-siRNA for 24 h, and then the cellular uptake efficiency of siRNA was analyzed by FACS. As shown in Fig. 2D, where the fluorescence signal of naked FAM-siRNA was set as control, the fluorescence intensity in the cells cultured with RGDfC-SeNPs@FAM-siRNA was stronger than that in the lipofectamine 2000@FAM-siRNA group, suggesting RGDfC-SeNPs@FAM-siRNA exhibited a high cellular uptake efficiency.TEM is another tool with which to observe the intracellular distribution of nanoparticles.24 As shown in Fig. 2E, TEM images showed that the cells incorporated the RGDfC-SeNPs@siRNA in perinuclear compartments and vesicular structures (red arrows). The enlargement image in Fig. 2E definitely confirmed the efficient cellular uptake and internalization of nanoparticles.
Exploring uptake pathways and intracellular localization of nanoparticles
Previous reports showed that the nanoparticles entered the cells by energy-dependent endocytic routes.25 Thus, we researched whether the cellular uptake of nanoparticles was influenced by the temperature. The cellular uptake of nanoparticles in the HepG2 cells incubated at 4 °C was decreased ∼81.7% (Fig. S2†), suggesting that the RGDfC-SeNPs@siRNA were internalized into the cells in an energy-dependent manner. The pretreatment with NaN3/DOG (cell energy metabolism inhibitor) also significantly decreased the uptake of nanoparticles in HepG2 cells, which indicated that the endocytosis of the RGDfC-SeNPs@siRNA was an energy-dependent active cellular process. Moreover, various endocytic inhibitors were utilized to explore the internalization mechanism of nanoparticles. Amiloride, nystatin and chlorpromazine were reported to suppress micropinocytosis, endocytosis, and caveolae-mediated and clathrin-associated cellular uptake, respectively. After pretreatment with amiloride and nystatin, the cellular uptakes of nanoparticles were decreased approximately 38.6% and 31.7%, respectively. However, the pretreatment with chlorpromazine resulted in a 66.9% decrease in cellular uptake, which indicated that clathrin-mediated endocytosis predominantly played a key role in cellular uptake of the RGDfC-SeNPs@siRNA in HepG2 cells.The co-localization of RGDfC-SeNPs@siRNA and endosomes/lysosomes was investigated by confocal laser scattering microscopy (CLSM) imaging.26 Fig. 3 shows that the RGDfC-SeNPs@siRNA gradually moved toward and became accumulated in the endosomes/lysosomes during 1 h of incubation. After 2 h of incubation, a fraction of the RGDfC-SeNPs@siRNA entered the cytoplasm, indicating that the RGDfC-SeNPs@siRNA might escape from endosomes/lysosomes to the cytoplasm after internalization.
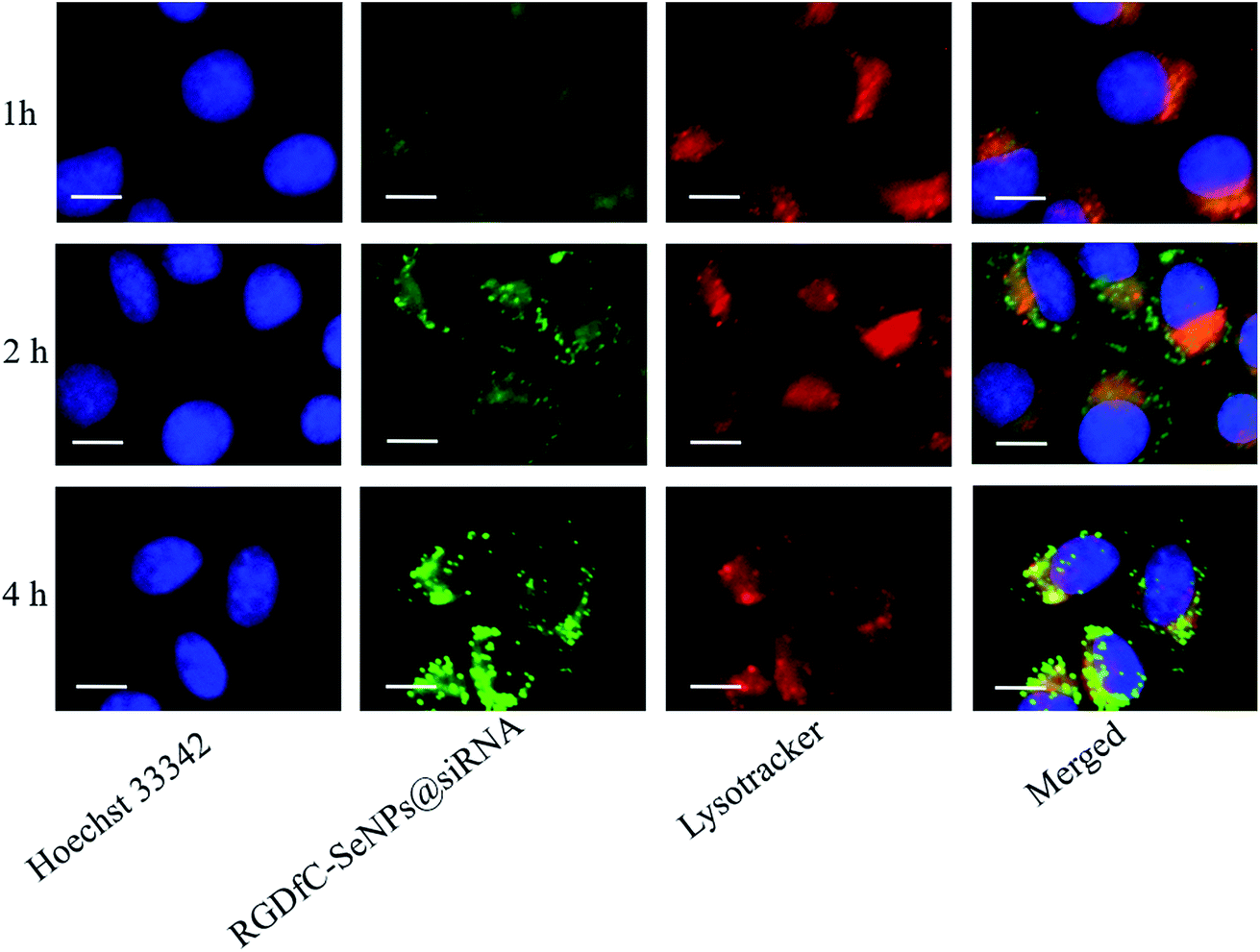 | ||
| Fig. 3 The observation by LSCM of the escape of RGDfC-SeNPs@FAM-siRNA from endosomes/lysosomes after 1, 2 and 4 h of incubation. Scale bar is 10 μm. | ||
Downregulation of gene expression
The tumor-targeted RGDfC-SeNPs were utilized to deliver siRNA into the HepG2 cell and to knockdown the gene expression of HES5 in the HepG2 cell. After the cells were transfected with different formulations of siRNA for 24 h, the mRNA levels of HES5 were assessed by quantitative polymerase chain reaction (qPCR). Fig. 4A shows that RGDfC-SeNPs@siNC failed to silence HES5 expression, while RGDfC-SeNPs@siRNA significantly downregulated the mRNA expression of HES5 in HepG2 cells. It is worth noting that RGDfC-SeNPs@siRNA showed stronger ability to silence the HES5 expression than lipofectamine 2000@siRNA.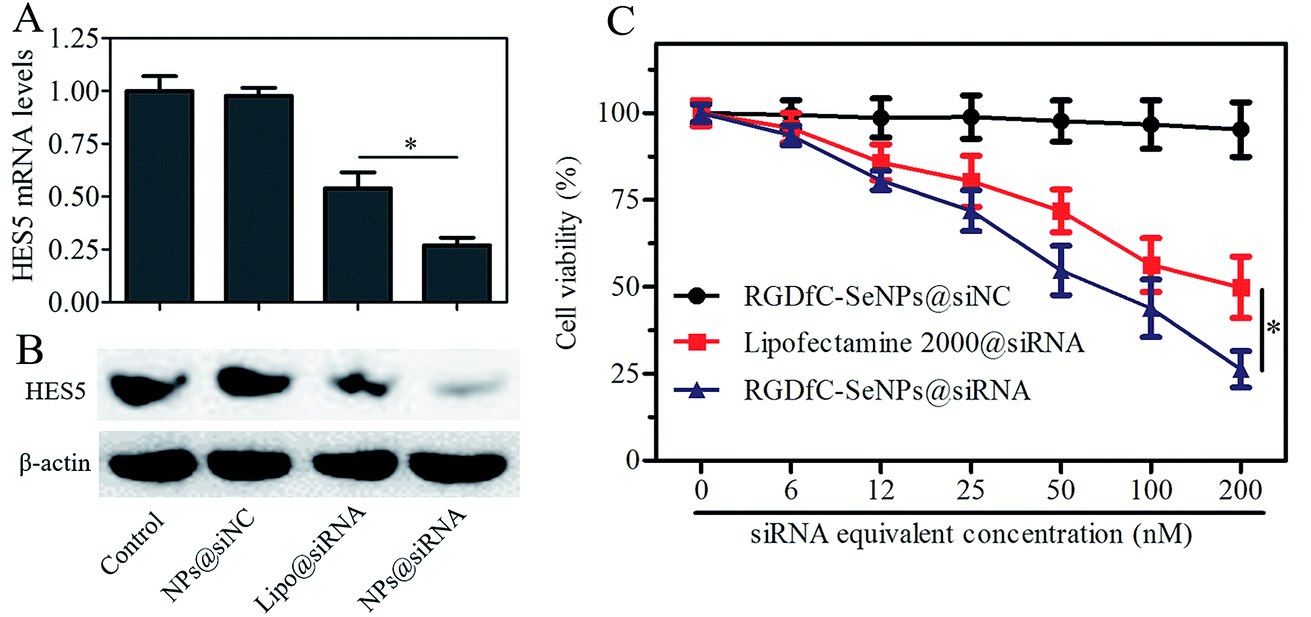 | ||
| Fig. 4 (A) Relative mRNA expression levels of HES5 in cells treated with RGDfC-SeNPs@siNC, lipofectamine 2000@siRNA and RGDfC-SeNPs@siRNA were assessed by quantitative reverse transcription PCR (qRT-PCR). (B) The expression levels of HES5 protein in cells treated with RGDfC-SeNPs@siNC, lipofectamine 2000@siRNA and RGDfC-SeNPs@siRNA were detected using western blotting assay. (C) The viabilities of HepG2 cells treated with different formulations of siRNA (at siRNA equivalent concentrations of ∼0–200 nM) for 48 h. | ||
The protein expression level of HES5 in HepG2 cells was detected via western blotting. Fig. 4B shows that the delivery of HES5-siRNA by actively tumor-targeted nanoparticles RGDfC-SeNPs exhibited stronger activity to inhibit the protein expression of HES5 than lipofectamine 2000@siRNA. However, RGDfC-SeNPs@siNC had no effect on the protein level of HES5, suggesting that no nonspecific gene silencing occurred in this kind of delivery system. The results of qPCR and western blotting both showed that the RGDfC-SeNPs@siRNA could significantly silence the expression of HES5.
In vitro cytotoxicity study
The cytotoxicity of the nanoparticles was studied by MTT assay.27 Fig. 4C shows that the cell viabilities in the RGDfC-SeNPs@siNC treatment group remained at about 94.3% in comparison with the untreated cells, indicating no nonspecific cytotoxicity against HepG2 cells in this gene delivery carrier. Significant inhibitory effects of RGDfC-SeNPs@siRNA on HepG2 cells were observed at the siRNA equivalent concentrations of 6–200 nM, and RGDfC-SeNPs@siRNA showed significantly higher cytotoxicity than lipofectamine 2000@siRNA as expected, suggesting RGDfC-SeNPs had extraordinary advantages for use as gene delivery carrier. The cellular toxicity of RGDfC-SeNPs@siRNA in normal liver cells, Lo2 cells, was also evaluated by MTT assay. As shown in Fig. S3,† compared with untreated cells, the Lo2 cell viability remained at nearly 93% after 48 h incubation with RGDfC-SeNPs@siRNA at the siRNA equivalent concentration of 200 nM, indicating its low toxicity and side effects.Cell cycle arrest and cell apoptosis
The cell cycle distribution of the HepG2 cells after treatment with different formulations of siRNA for 24 h was analyzed by flow cytometry.28 As shown in Fig. 5A, obviously reduced populations at the S and G2/M phases were detected, and meanwhile a prominent cell cycle arrest at G0/G1 phase occurred after the cells were incubated with siRNA in different formulations. Compared with lipofectamine 2000@siRNA, the RGDfC-SeNPs@siRNA displayed slightly greater effects on the cell cycle distribution. Meanwhile, the RGDfC-SeNPs@siNC exhibited no obvious effect on the cell cycle distribution. This finding indicated that the knockdown of HES5 might repress the proliferation of HepG2 cells by inducing cell cycle arrest at the G0/G1 phase.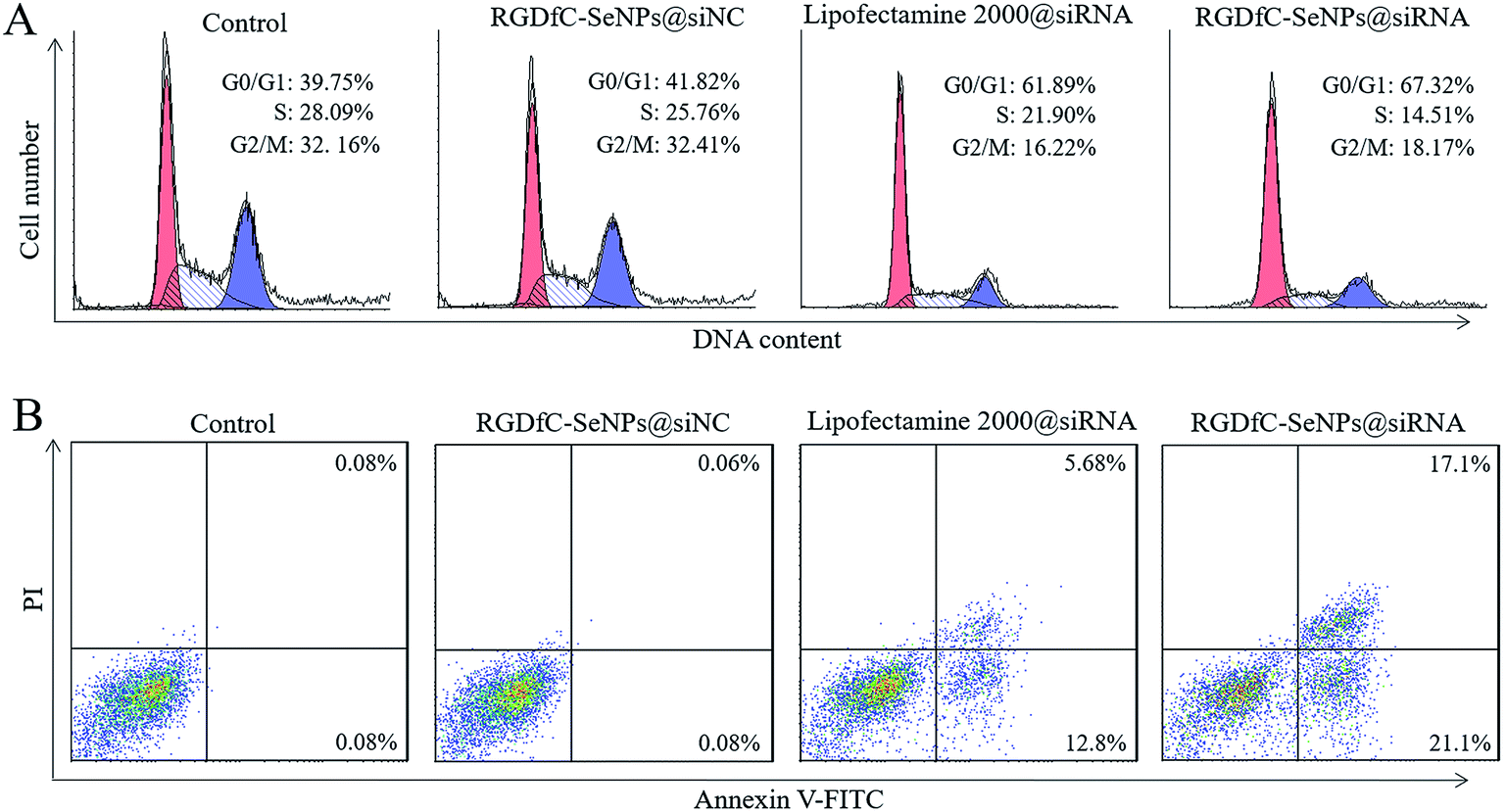 | ||
| Fig. 5 (A) Effects of RGDfC-SeNPs@siNC, lipofectamine 2000@siRNA and RGDfC-SeNPs@siRNA on cell cycle distribution in HepG2 cells. (B) Effects of RGDfC-SeNPs@siNC, lipofectamine 2000@siRNA and RGDfC-SeNPs@siRNA on cell apoptosis in HepG2 cells. | ||
Annexin V-FITC/PI staining was employed to analyze cell apoptosis by flow cytometry.29 As shown in Fig. 5B, treatment with RGDfC-SeNPs@siRNA markedly induced HepG2 cell apoptosis, and gave a higher apoptosis rate (∼38.2%) than lipofectamine 2000@siRNA (∼18.5%). However, no obvious cell apoptosis was observed in the RGDfC-SeNPs@siNC treatment group, suggesting that the silencing of HES5 contributed to the apoptosis of HepG2 cells. FACS analysis showed that RGDfC-SeNPs@siRNA repressed the proliferation of HepG2 cells mainly by arresting the cell cycle at G0/G1, and subsequently resulted in cell apoptosis.
Cell cycle- and apoptosis-related protein expression
Previous studies reported that cyclin D1 was highly expressed in various cancers and regulated the cell cycle progression via the G1/S restriction point.30 The transition from G1 to S phase was primarily regulated through the activation of CDK2/cyclin E complexes.31 p21 protein was a proliferation inhibitor and played a key role in G1 arrest by repressing the activities of CDK-cyclin complex.32 To explore the antiproliferative activity mechanism of RGDfC-SeNPs@siRNA in HepG2 cells, the expression levels of cell cycle-related proteins cyclin D1, CDK2, cyclin E and p21 after 24 h of incubation with RGDfC-SeNPs@siRNA were analyzed via western blotting assay. As shown in Fig. 6A, RGDfC-SeNPs@siRNA at the tested concentrations obviously decreased the expression levels of CDK2, cyclin E and cyclin D1 proteins in the HepG2 cells, whereas the expression level of p21 protein was slightly enhanced. These results revealed that RGDfC-SeNPs@siRNA arrested the cell cycle at G1 phase mainly by suppressing the expression of proteins CDK2, cyclin E and cyclin D1 and upregulating the level of protein p21 in HepG2 cells.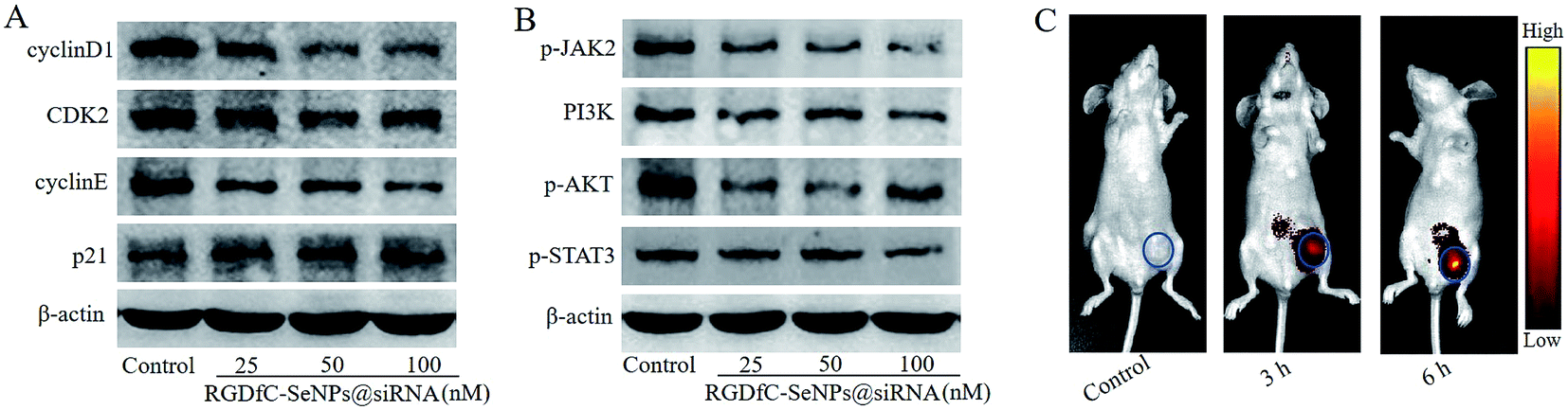 | ||
| Fig. 6 (A) The protein expression levels of cyclin D1, CDK2, cyclin E and p21 in HepG2 cells treated with different concentrations of RGDfC-SeNPs@siRNA were detected via western blotting. (B) The protein expression levels of p-JAK2, PI3K, p-AKT and p-STAT3 in HepG2 cells treated with different concentrations of RGDfC-SeNPs@siRNA were detected via western blotting. (C) In vivo fluorescence images of mice after injecting with RGDfC-SeNPs@cy5.5-siRNA for 3 or 6 h; mice receiving no injection formed the control group. | ||
Disoriented regulation of tumor cell apoptosis was reported to be a key contributor to tumorigenesis. The JAK/STAT pathway plays a significant role in cancer cell proliferation and apoptosis.33 Herein, western blotting experiments were carried out to examine the expression levels of some key proteins in the JAK/STAT pathway, including p-JAK2, PI3K, p-AKT and p-STAT3. Fig. 6B shows that treatment with RGDfC-SeNPs@siRNA resulted in decreased protein expression levels of p-JAK2, PI3K, p-AKT and p-STAT3 in comparison with the control group. The results indicated that RGDfC-SeNPs@siRNA could block JAK2/PI3K/Akt/STAT3 signaling via suppressing the phosphorylation of some key proteins.
In vivo imaging of nanoparticles
Efficacious actively tumor-targeted delivery vehicles should be capable of selectively delivering genes to tumors in order to obtain high-efficiency treatment of cancer. The in vivo distribution of RGDfC-SeNPs@siRNA loaded with cy5.5 was assessed via ex vivo fluorescence imaging after 3 h and 6 h post-injection. Mice without injection with nanoparticles composed the control group. As shown in Fig. 6C, no fluorescence signal was captured from the control group, suggesting that there was no autofluorescence in the tumor-bearing mice. Nevertheless, an obvious fluorescence signal was observed in tumors after 3 h post-injection with RGDfC-SeNPs@cy5.5-siRNA, and significantly higher fluorescence intensity was observed after 6 h post-injection. These results indicated that the RGDfC-SeNPs@siRNA efficiently accumulated in tumors during systemic administration, presumably due to the enhanced avidity of RGDfC ligands for αvβ3/αvβ5 integrin receptors on cancerous cells and also tumor-associated endothelial cells.34,35In vivo anti-cancer activity
The anti-tumor activity of RGDfC-SeNPs@siRNA was evaluated using a HepG2 tumor xenograft model. The mice were intravenously injected with saline, RGDfC-SeNPs, lipofectamine 2000@siRNA and RGDfC-SeNPs@siRNA to test the respective anti-tumor activities. Rapid growth in tumor size over time was observed in the different treatment groups (Fig. 7A). Compared with the group treated with saline, the RGDfC-SeNPs exhibited slight inhibition of tumor growth, which could be explained by the selenium nanoparticles exhibiting slight anti-tumor activity because of the continuous administration of RGDfC-SeNPs. Meanwhile, RGDfC-SeNPs@siRNA showed greater tumor growth inhibition activity than lipofectamine 2000@siRNA. The enhanced antitumor activity in the RGDfC-SeNPs@siRNA treatment group might be attributed to tumor-targeting ability mediated by RGDfC, resulting in higher drug accumulation in the tumor sites. The tumor images (Fig. 7B) also confirmed the remarkable antitumor activity of RGDfC-SeNPs@siRNA. The body weight was measured every other day up to 21 days. No noteworthy body weight loss was recorded after various treatments, indicating the minimal side effects of the drugs at the tested doses (Fig. 7C). To verify whether tumor growth inhibition was related to the downregulation of HES5 induced by RGDfC-SeNPs@siRNA, the expression level of HES5 protein in tumors was determined through western blotting experiment. As shown in Fig. 7D, as expected, RGDfC-SeNPs@siRNA exhibited a greater ability to downregulate the protein expression of HES5 in the tumor than did lipofectamine 2000@siRNA. However, there was no obvious downregulation of HES5 expression level after treatment with RGDfC-SeNPs.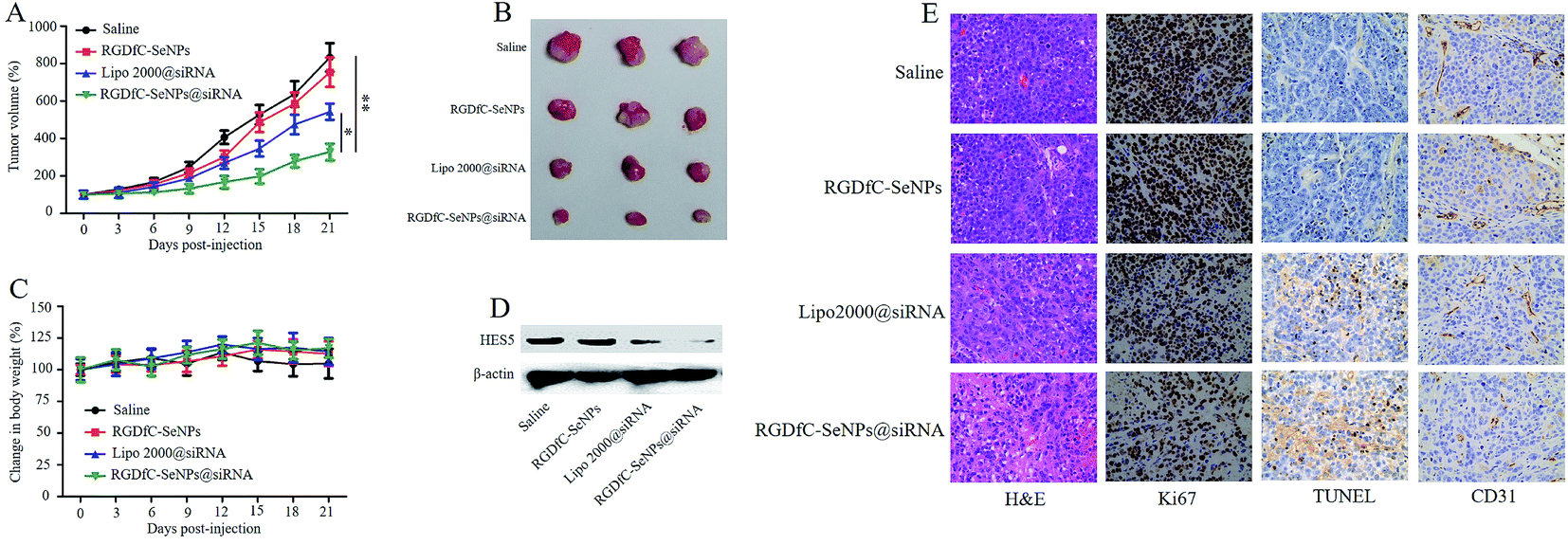 | ||
| Fig. 7 (A) Tumor growth curve of xenograft nude mice bearing HepG2 cells after intravenous administration of saline and various formulations of siRNA. (B) Morphology of all tumors stripped from the mice. (C) The body weight change of mice during the treatments. (D) The expression level of HES5 protein in tumors stripped from the mice after 21 days of administration. (E) H&E, Ki67, TUNEL and CD31 immunohistochemistry analysis of the tumors treated with saline, RGDfC-SeNPs, lipofectamine 2000@siRNA and RGDfC-SeNPs@siRNA. | ||
To investigate the enhanced antitumor and anti-angiogenesis effects in vitro, the tumor tissues were stained with H&E and Ki67, and subjected to TUNEL assay, and the expression of CD31, a marker for neovasculature, was examined. As shown in Fig. 7E, the H&E-stained section of tumor tissue from the saline group appeared to be hypercellular and nuclear polymorphism in the tumor tissue was very evident. In contrast, most of the cancer cells in the RGDfC-SeNPs@siRNA treatment group were lysed and destroyed, indicating that RGDfC-SeNPs@siRNA treatment had induced significant cancer cell apoptosis and necrosis in vivo. Additionally, RGDfC-SeNPs@siRNA treatment resulted in an obvious decrease of Ki67-positive cancer cells, suggesting that the cancer cell proliferation had been significantly suppressed by RGDfC-SeNPs@siRNA. TUNEL assay confirmed that treatment with RGDfC-SeNPs@siRNA led to enhanced apoptosis and cell death, compared with the saline treatment group. Immunostaining with anti-CD31 antibody was used to visualize the formation of microvessels in the tumor mass. As shown in Fig. 7E, the angiogenesis-positive cells with brown color were widespread in the control group. The microvessel density was greatly reduced after treatment with RGDfC-SeNPs@siRNA. As expected, the anti-tumor effect was more pronounced in the RGDfC-SeNPs@siRNA-treated group than the lipofectamine 2000@siRNA-treated group. These results suggested RGDfC-SeNPs@siRNA could inhibit cancer cell proliferation and CD31-induced vessel formation, and induce cancer cell apoptosis in vivo.
Analysis of side effects
Apart from the anti-tumor efficacy, in vivo toxicity assessment is crucial for cancer therapy. Histological analysis of main organs was performed by H&E staining. As shown in Fig. S4,† compared with the saline treatment group, no obvious differences were observed in the RGDfC-SeNPs@siNC, lipofectamine 2000@siRNA or RGDfC-SeNPs@siRNA treatment groups. The integrated results certified that RGDfC-SeNPs@siRNA were well-tolerated in vivo at the tested dose. By analyzing these issues, the RGDfC-SeNPs@siRNA have been shown to have great potential as tumor-targeted delivery system for effective hepatocellular carcinoma therapy with reduced systemic toxicity.Conclusion
In this work we have developed functionalized selenium nanoparticles conjugated with the tumor-targeting moiety polypeptide RGDfC (RGDfC-SeNPs) as a carrier to deliver HES5-siRNA for HCC therapy. RGDfC-SeNPs@siRNA were internalized into cells by clathrin-mediated endocytosis and then escaped from endosomes/lysosomes to the cytoplasm. The tumor-targeted functionalized selenium nanoparticles RGDfC-SeNPs@siRNA exhibited greater enhanced siRNA transfection efficiency than lipofectamine 2000@siRNA and effectively knocked down the expression of HES5 in vitro and in vivo. Meanwhile, RGDfC-SeNPs@siRNA were able to inhibit HepG2 cell proliferation and arrest the cells at the G0/G1 phase. RGDfC-SeNPs@siRNA could induce HepG2 cell apoptosis and inhibit tumor growth via the silencing of the HES5 gene in vivo. The enhanced antitumor activity of RGDfC-SeNPs@siRNA was consistent with its high accumulation in tumors in vivo. Moreover, no obvious in vivo toxicity was observed in the main organs of mice treated with the nanoparticles, suggesting the good compatibility of this gene delivery system. In short, these studies provide a potential strategy for HCC-targeted gene therapy.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the China Postdoctoral Science Foundation (No. 2017M612632), the Technology Planning Project of Guangdong Province (No. 2014A020212697) and the Technology Planning Project of Guangzhou City (No. 201607010120).Notes and references
- R. Zhao, T. Li, G. Zheng, K. Jiang, L. Fan and J. Shao, Biomaterials, 2017, 143, 1–16 CrossRef CAS PubMed.
- R. R. Castillo, A. Baeza and M. Vallet-Regi, Biomater. Sci., 2017, 5, 353–377 RSC.
- X. Du, C. Zhao, M. Zhou, T. Ma, H. Huang, M. Jaroniec, X. Zhang and S. Z. Qiao, Small, 2017, 13, 1602592 CrossRef PubMed.
- Y. Li, H. Wang, K. Wang, Q. Hu, Q. Yao, Y. Shen, G. Yu and G. Tang, Small, 2017, 13, 1602697 CrossRef PubMed.
- A. K. Kozlowska, A. Florczak, M. Smialek, E. Dondajewska, A. Mackiewicz, M. Kortylewski and H. Dams-Kozlowska, Acta Biomater., 2017, 59, 221–233 CrossRef CAS PubMed.
- D. Polyak, A. Krivitsky, A. Scomparin, S. Eliyahu, H. Kalinski, S. Avkin-Nachum and R. Satchi-Fainaro, J. Controlled Release, 2017, 257, 132–143 CrossRef CAS PubMed.
- C. Ma, L. Shi, Y. Huang, L. Shen, H. Peng, X. Zhu and G. Zhou, Biomater. Sci., 2017, 5, 494–501 RSC.
- L. Zhou, Y. Xi, M. Yu, M. Wang, Y. Guo, P. Li, P. X. Ma and B. Lei, Acta Biomater., 2017, 58, 90–101 CrossRef CAS PubMed.
- A. D. Tagalakis, R. Maeshima, C. Yu-Wai-Man, J. Meng, F. Syed, L. P. Wu, A. M. Aldossary, D. McCarthy, S. M. Moghimi and S. L. Hart, Acta Biomater., 2017, 51, 351–362 CrossRef CAS PubMed.
- Y. Xia, P. You, F. Xu, J. Liu and F. Xing, Nanoscale Res. Lett., 2015, 10, 349–363 CrossRef PubMed.
- W. Zheng, C. Cao, Y. Liu, Q. Yu, C. Zheng, D. Sun, X. Ren and J. Liu, Acta Biomater., 2015, 11, 368–380 CrossRef CAS PubMed.
- Y. Xia, Z. Lin, Y. Li, M. Zhao, C. Wang, M. Guo, B. Zhang and B. Zhu, J. Mater. Chem. B, 2017, 5, 6941–6952 RSC.
- Y. Li, Z. Lin, M. Zhao, T. Xu, C. Wang, H. Xia, H. Wang and B. Zhu, Int. J. Nanomed., 2016, 11, 3065–3076 CrossRef CAS PubMed.
- Z. Ruan, L. Liu, W. Jiang, S. Li, Y. Wang and L. Yan, Biomater. Sci., 2017, 5, 313–321 RSC.
- J. Y. Lee, C. Crake, B. Teo, D. Carugo, M. de Saint Victor, A. Seth and E. Stride, Adv. Healthcare Mater., 2017, 6, 1601246 CrossRef PubMed.
- M. Gujrati, A. M. Vaidya, M. Mack, D. Snyder, A. Malamas and Z. R. Lu, Adv. Healthcare Mater., 2016, 5, 2882 CrossRef CAS PubMed.
- G. Zhu, W. Shi, H. Fan, X. Zhang, J. Xu, Y. Chen, Z. Xu, T. Tao and C. Cheng, Exp. Mol. Pathol., 2015, 99, 474–484 CrossRef CAS PubMed.
- G. Xu, M. Zhang, H. Zhu and J. Xu, Gene, 2017, 604, 33–40 CrossRef CAS PubMed.
- S. J. Lee, M. J. Kim, I. C. Kwon and T. M. Roberts, Adv. Drug Delivery Rev., 2016, 104, 2–15 CrossRef CAS PubMed.
- V. B. Kumar, H. Medhi, Z. Yong and P. Paik, Nanomedicine, 2016, 12, 579–588 CrossRef CAS PubMed.
- Z. Wang, G. Liu, H. Zheng and X. Chen, Biotechnol. Adv., 2014, 32, 831–843 CrossRef CAS PubMed.
- R. Muralidharan, A. Babu, N. Amreddy, K. Basalingappa, M. Mehta, A. Chen, Y. D. Zhao, U. B. Kompella, A. Munshi and R. Ramesh, J. Nanobiotechnol., 2016, 14, 47–64 CrossRef PubMed.
- H. Zhu, S. Zhang, Y. Ling, G. Meng, Y. Yang and W. Zhang, J. Controlled Release, 2015, 220, 529–544 CrossRef CAS PubMed.
- W. Liao, W. Li, T. Zhang, M. Kirberger, J. Liu, P. Wang, W. Chen and Y. Wang, Biomater. Sci., 2016, 4, 1051–1061 RSC.
- M. W. Amjad, P. Kesharwani, M. C. I. Mohd Amin and A. K. Iyer, Prog. Polym. Sci., 2017, 64, 154–181 CrossRef CAS.
- W. Ngamcherdtrakul, J. Morry, S. Gu, D. J. Castro, S. M. Goodyear, T. Sangvanich, M. M. Reda, R. Lee, S. A. Mihelic, B. L. Beckman, Z. Hu, J. W. Gray and W. Yantasee, Adv. Funct. Mater., 2015, 25, 2646–2659 CrossRef CAS PubMed.
- Y. Xia, Q. Chen, X. Qin, D. Sun, J. Zhang and J. Liu, New J. Chem., 2013, 37, 3706–3715 RSC.
- X. Ding, Y. Su, C. Wang, F. Zhang, K. Chen, Y. Wang, M. Li and W. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 23353–23369 CAS.
- Z. Zhou, H. Li, K. Wang, Q. Guo, C. Li, H. Jiang, Y. Hu, D. Oupicky and M. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 14576–14589 CAS.
- L. Wu, J. Zheng, P. Chen, Q. Liu and Y. Yuan, Biomed. Pharmacother., 2017, 90, 705–712 CrossRef CAS PubMed.
- Q. Xia, Y. Zhao, J. Wang, W. Qiao, D. Zhang, H. Yin, D. Xu and F. Chen, Proteomics: Clin. Appl., 2017, 11, 1600099 CrossRef PubMed.
- G. He, J. Kuang, J. Koomen, R. Kobayashi, A. R. Khokhar and Z. H. Siddik, Br. J. Cancer, 2013, 109, 2378–2388 CrossRef CAS PubMed.
- L. Zhang, X. Huo, Y. Liao, F. Yang, L. Gao and L. Cao, Sci. Rep., 2017, 7, 1669 CrossRef PubMed.
- H. Y. Ren, B. Chen, G. L. Huang, Y. Liu and D. Y. Shen, Mol. Med. Rep., 2016, 14, 3602–3608 CrossRef CAS PubMed.
- J. Zhang, T. Wang, S. Mu, L. D. Olerile, X. Yu and N. Zhang, Nanomedicine, 2017, 12, 911–925 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12461a |
| This journal is © The Royal Society of Chemistry 2018 |