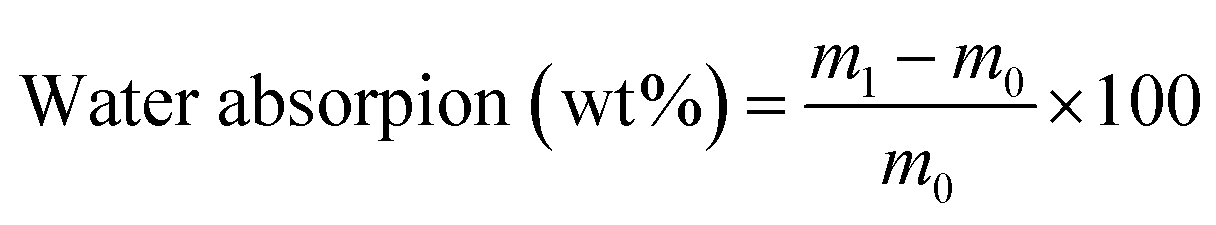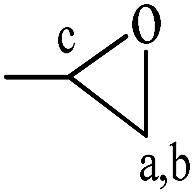DOI:
10.1039/C7RA12180F
(Paper)
RSC Adv., 2018,
8, 8615-8623
Synthesis of a novel CO2-based alcohol amine compound and its usage in obtaining a water- and solvent-resistant coating
Received
7th November 2017
, Accepted 29th January 2018
First published on 23rd February 2018
Abstract
A five-membered cyclo-carbonate, prepared by cycloaddition reaction from CO2 and 1,4-butanediol diglycidyl ether, was reacted with excessive diamine and formed a urethane group-containing new product. Structural characterization was performed for the new alcohol amine, which can be applied to the manufacture of polyurethane coatings as a chain extender. The new chain extender-based polyurethane coatings exhibited excellent water, salt, and solvent resistance and promising mechanical strength. Importantly, the unique performance of the prepared polyurethane coatings should be ascribed to the introduction of a hydroxyl group in the polyurethane molecule. The strengthened hydrogen bonding enlarged the cohesion of the polyurethane coatings and prohibited the solvents from permeating.
1. Introduction
Large amounts of greenhouse gases have caused a series of environmental problems worldwide and have been a topic of widespread concern. However, the main gas produced, CO2, has been regarded as an environmentally friendly C1 building block due to its abundance and nontoxicity. In this context, enormous efforts have been devoted to the development of CO2 control over the past decades. One strategy for controlling the enormous CO2 resource is chemical fixation, such as its usage in the synthesis of polymers.
Polyurethanes (PUs) with interesting tailored properties and excellent flexibility, elasticity, and mechanical properties1–3 have been used in many fields.4–12 Moreover, polyurethane coatings have become one of the main varieties in the coating industry because of their excellent properties.13–17 Although polyurethane coatings have excellent abrasion resistance, toughness, and weather resistance with wide applications,18–23 further improvement of PU coatings is still of great significance, and especially the creation of a coating that is water, salt, and solvent resistant.
Various techniques have been developed to improve the water, salt, and solvent resistance of PU coatings, such as adding hydrophobic particles, introducing hydrophobic fluorine- or siloxane-containing compounds into the polyurethane backbone, and chemical modification of raw materials. In addition to these, a new method consisting of strengthening the hydrogen bonds in the crosslinking system might be helpful to improve the environmental resistance of PU coatings. It has been reported that the intra-molecular hydrogen bonding between the hydroxyl group on the β-carbon atom and the adjacent carbonyl could endow materials with outstanding mechanical properties accompanied with excellent resistance to solvents, water, and chemicals. The resistance was ascribed to the unique structure of the seven-membered ring formed, which can bring about a “blockage” of the carbonyl oxygen that considerably lowers the susceptibility of the entire urethane group to hydrolysis. From another perspective, the stabilizing effect was thought to occur in the intramolecular hydrogen bond system, and it strengthened the interaction of polymer molecules due to the redistribution of charges that arise from the formation of tautomeric resonance structures.23,24
From the above reports, it can be speculated that the direct reaction between di- or polycarbonates and excessive diamines, such as ethyl diamine, would produce an amine-terminated compound. Additionally, a hydroxyl group is formed on the β-carbon atom with the ring opening reaction. Thus, the compound can be regarded as one type of amine alcohol. Because of the much faster reaction rate of isocyanate with an amine group other than a hydroxyl group, the hydroxyl can be reserved when the amine alcohol is used as a chain extender for polyurethane manufacture. Enhancement of the hydrogen bonding in PU enables PU to attain superior performance.
The aim of this work is to provide a new chemical fixation method for CO2 and explore the effect of introduced hydroxyl groups on the performance of polyurethane coatings, especially on the water, salt, and organic solvent resistance. Moreover, a novel alcohol amine chain extender with a secondary or primary hydroxyl group was designed and synthesized for the first time.23 The obtained alcohol amine was further analyzed and tested for its ability to function in the preparation of polyurethane coatings. The related water, salt, and solvent resistance of the final PU coatings was investigated in detail, as well as its microstructures and other properties.
2. Experimental
2.1 Materials
1,2-Ethylenediamine (EDA) and N,N-dimethylformamide (DMF) were obtained from Sinopharm Chemical Reagent Co., Ltd.; 1,4-butanediol diglycidyl ether (BDDE) was supplied by Wuhan Yuanchen Technology Co., Ltd.; and 4,4-diphenylmethane diisocyanate (MDI) was prepared by Yantai Wan Hua Co., Ltd. Polytetramethylene ether glycol (PTMG, Mn = 1000) was supplied by Yantai Hua Da Chemical Industry Co., Ltd. The catalyst basic ion-exchange resin was purchased from Tianjin Resin Technology Co., Ltd. All the raw materials were used as received without further purification.
2.2 Preparation of the aliphatic primary amine chain extender (BDCE)
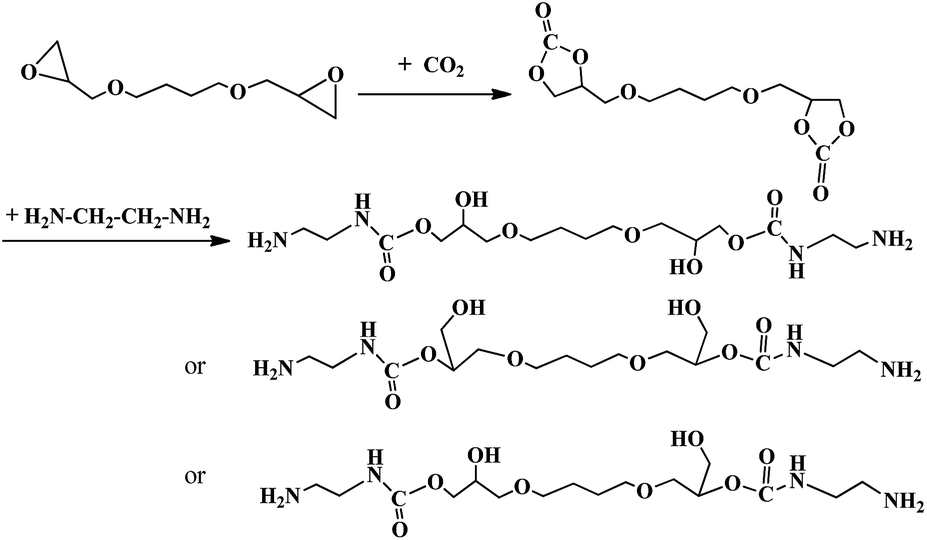
Briefly, the NH2-terminated chain extender was fabricated by aminolysis of the five-member bis(cyclic carbonate)(BDDE-5CC) and diamine (EDA) as shown in Scheme 1. Firstly, BDDE-5CC was prepared via a cycloaddition reaction from CO2 and BDDE. A portion of BDDE and the basic ion-exchange resin catalyst (10 wt% of BDDE) was charged in a high-pressure autoclave. After purging by N2 for 10 min and heating the mixture to 120 °C, CO2 under pressure was pumped into the autoclave. Then, the pressure was maintained at 1 MPa, and the contents of the autoclave were reacted for 30 h under stirring. Secondly, BDDE-5CC and EDA in twice an amount as necessary to ensure complete transformation of BDDE-5CC were added to a four-necked round-bottom flask equipped with a thermometer, mechanical stirrer, reflux condenser, and feeding inlets. Then, the solution was heated to 90 °C and reacted for 6 h under stirring. After that, the mixture was subject to a vacuum at 70 °C for 8 h to remove the residual EDA, and finally, the NH2-terminated chain-extender was obtained. Structures of the products were characterized by FTIR spectroscopy and 1H NMR spectroscopy. Additionally, conversion and the selectivity of each reaction were determined by chemical titration and further confirmed and calculated using the 1H NMR spectra.
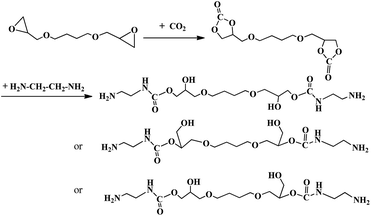 |
| | Scheme 1 Scheme of preparation of BDCE through polyaddition of BDDE-5CC and EDA. | |
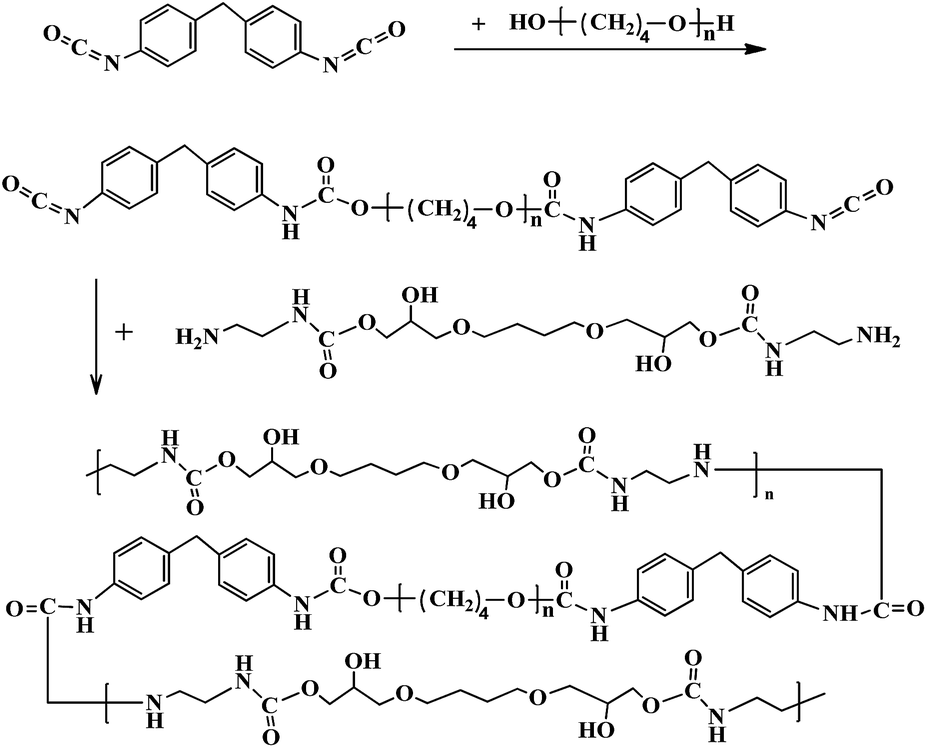
2.3 Preparation of the PU coatings
The PU coatings were prepared by polymerization of the prepolymer and chain extender in DMF solution as shown in Scheme 2. Prepolymers with an –NCO content of 7.0 wt% were synthesized between dehydrated PTMG and MDI at 90 °C for 3 h and defoamed for 0.5 h under vacuum. Note that DMF was used as a solvent to endow the coating solution with the appropriate viscosity to control the reaction rate. Afterward, the solution was immediately coated onto the substrates by a flowing method or blading the coating in the air and allowing it to cross-link in an oven at 140 °C for 2 min.
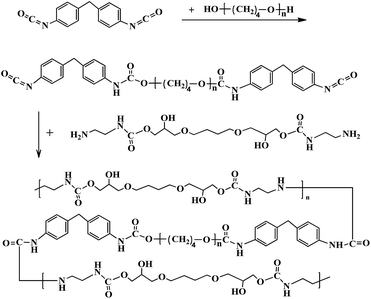 |
| | Scheme 2 Extension of the PU prepolymers with chain extenders. | |
2.4 Surface pretreatment of mild steel panels
The mild steel substrates with dimensions 120 mm × 50 mm × 0.3 mm were first deoiled in acetone solvent for 15 min and then immersed in degreasing solution with weakly alkaline sodium salts and surfactants for 10 min. Finally, these substrates were further washed under water and dried in an oven at 90 °C for a certain amount of time.
2.5 Characterization
2.5.1 Fourier transform infrared spectroscopy (FTIR). Raw material BDDE, the products BDDE-5CC, NH2-terminated chain-extender, prepolymer, and the final coating were directly smeared onto a KBr crystal disk for the FTIR test and recorded on a NICOLET-380 Fourier transform infrared instrument (Thermo Electron Co., Ltd. Massachusetts, USA) with a spectral resolution of 4 cm−1 and 32 scans taken for each recording.
2.5.2 1H NMR spectroscopy. 1H NMR of the samples was performed using a Bruker Avance 400 MHz spectrometer, and DMSO was used as the solvent with toluene as the internal standard substance to determine the amounts of the components. The chemical shifts were reported in parts per million relative to tetramethylsilane.
2.5.3 Water contact angles. Water contact angles (CAs) were measured at 25.0 ± 0.1 °C by a DSA25 drop shape analyzer (KRÜSS GmbH, Hamburg, Germany). The sample was placed on an adjustable lift table and quickly moved to the required measuring height. Then, the dispensed drop was positioned with the correct shape under uniform lighting, and contact angles were reliably measured. Each reported contact angle represents the average value of five measurements.
2.5.4 Basic properties. The basic properties of the coatings such as impact strength, adhesion to the substrate, flexibility, and pencil hardness were measured according to the corresponding standards with the instruments, a QCJ-120 paint film impact tester, QFZ-II adhesion tester, QTX-1731 flexible tester, and VF2378 pencil hardness tester, respectively.25–28
2.5.5 Thermal properties. The thermal stability of coatings was determined with a TA-60WS thermal analysis workstation (Shimadzu Co., Ltd. Kyoto, Japan) instrument. A 3–4 mg sample was heated from 30 °C to 500 °C at a heating rate of 10 °C min−1 under nitrogen with a flow of 30 mL min−1. Thermal properties were evaluated by the differential scanning calorimetry (DSC) test using a TA Instruments DSC Q2000 under nitrogen atmosphere with a flow rate of 50 mL min−1. The sample was scanned from −80 to 260 °C at a heating rate of 10 °C min−1.
2.5.6 Water and solvent resistance. Resistance to acid, alkali, and salt was measured by placing coated tin plates in a 5 wt% HCl solution, 5 wt% NaOH solution, and 10 wt% NaCl solution, respectively, for 24. For salt resistance, a partially coated sample was immersed in the NaCl solution for comparison. Water absorption was used to evaluate the water resistance of the coatings and was calculated by the following equation:| |
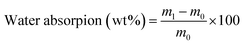 | (1) |
where m0 is the original weight of the dry coating film and m1 is the weight of the sample immersed in water for 24 h at room temperature.
2.5.7 Content of free amino and hydroxyl groups. To determine the content of free –NH2 groups and –OH groups in the chain extender, the titration method with hydrochloric acid and back titration with acetic anhydride and sodium hydroxide was used. In detail, firstly, approximately 0.1 g chain extenders was weighed in an Erlenmeyer flask and then 40 mL ethanol was added. After the sample dissolved, 2–3 droplets of a 1 wt% bromophenol blue-ethanol indicator solution were added. A calibrated HCl solution with a concentration of approximately 0.1 mol L−1 was used to titrate the amine compound, and the process was completed when the color of the system changed to yellow. The –NH2 content was calculated as follows:| | |
x = (c × V × 0.0561 × 1000)/m
| (2) |
where V is the volume of HCl solution used for titration, and c and m represent the concentration of HCl solution and the weight of the chain extenders, respectively.
3. Results and discussion
3.1 Preparation and characterization of the CO2-based NH2-terminated chain extender
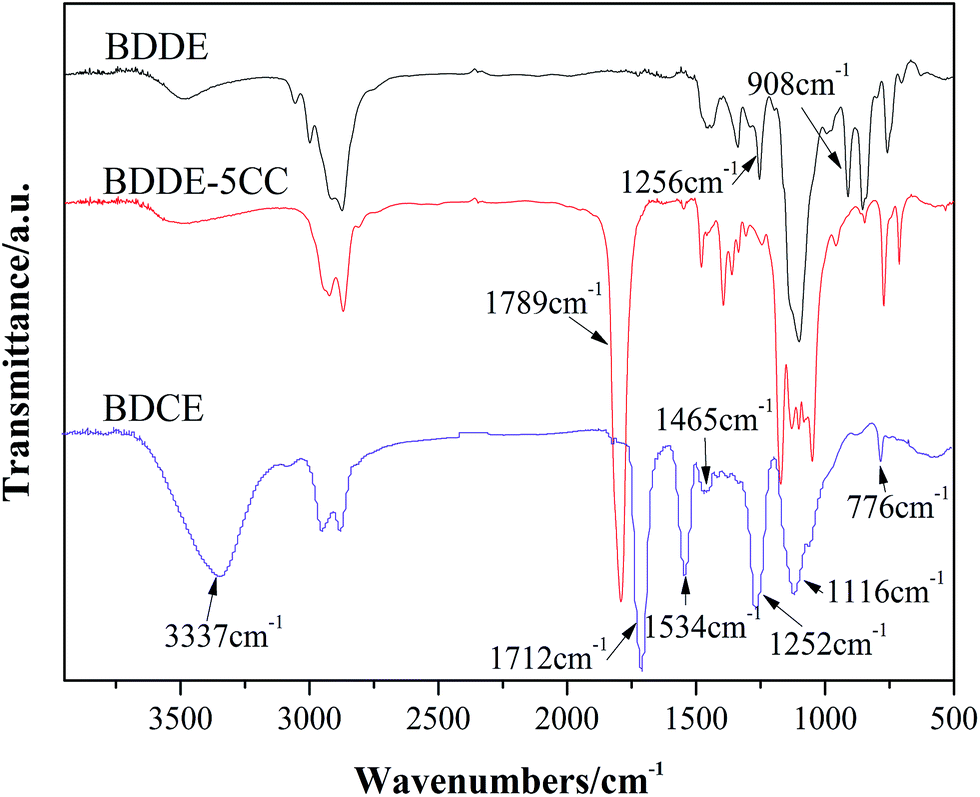
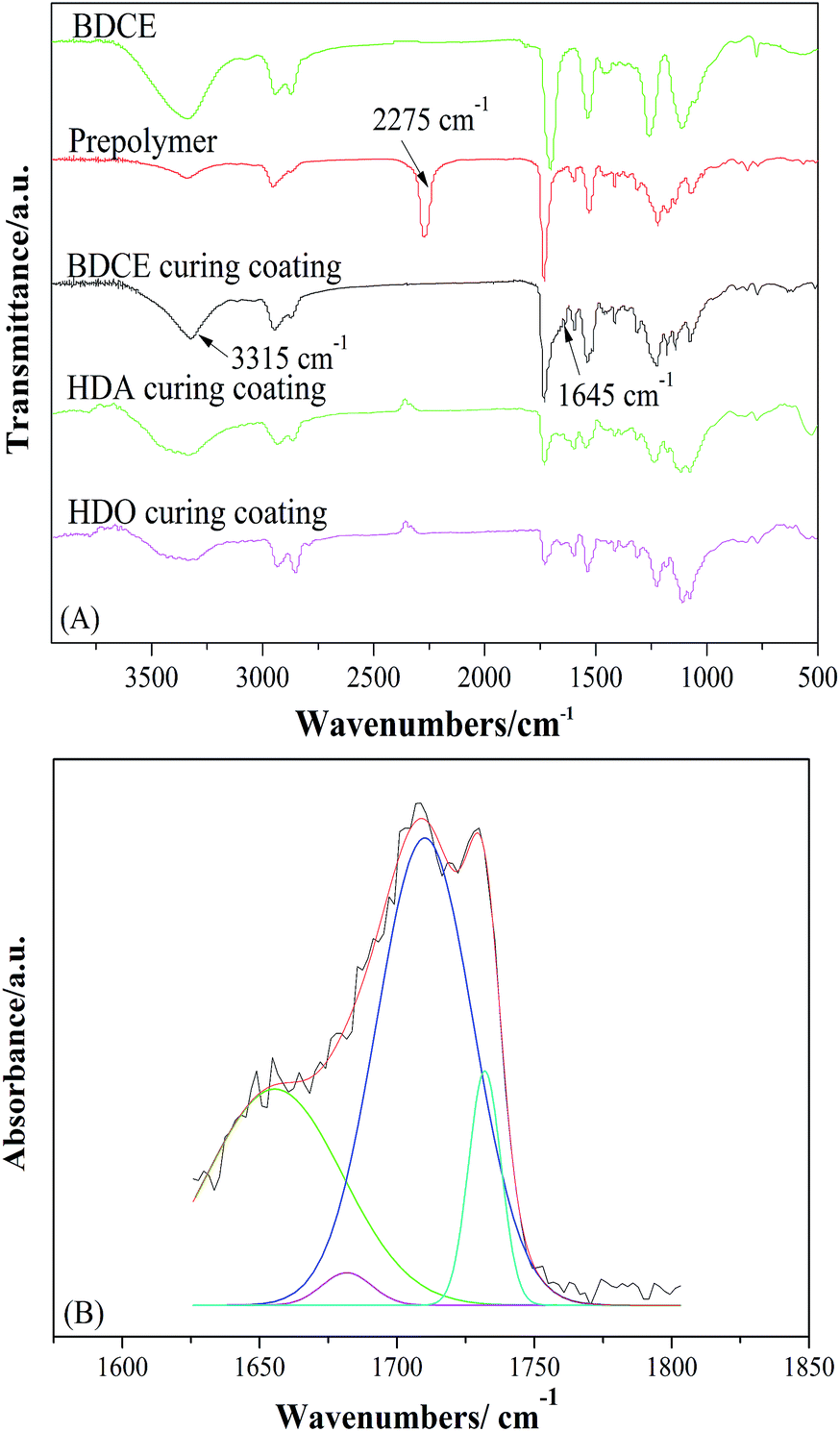
The synthesis of BDDE-5CC and BDCE was carried out according to Scheme 1, which could be further used as a chain extender for polyurethane manufacture due to the high reactivity between amine and isocyanate. Their structure was characterized by FTIR spectroscopy, and the result is shown in Fig. 1.
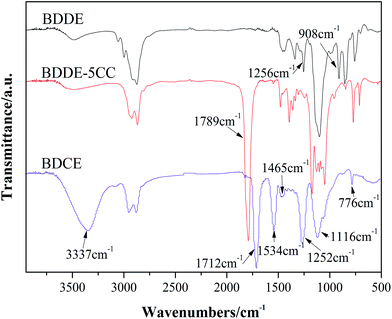 |
| | Fig. 1 FTIR spectra of prepared cyclo-carbonate and NH2-terminated chain extender. | |
Comparing BDDE-5CC with BDDE, a new peak appeared at 1789 cm−1 corresponding to the carbonyl of the cyclic carbonate moieties. Additionally, the characteristic absorption peaks at 908 cm−1 and 1256 cm−1 belonging to asymmetric and symmetric stretching vibrations of the epoxy group disappeared, indicating that the epoxy groups in BDDE reacted with CO2 and were successfully converted to cyclic carbonates. Furthermore, for BDCE, an obvious band at approximately 1712 cm−1 representing the stretching vibration peak of the carbonyl in the urethane groups, typical absorption peaks at 1534 cm−1 of the N–H in-plane bending vibration peak of primary amine, and at 1252 cm−1 of the C–N stretching vibration peak of fatty amine appeared. In addition, the broad peak at 3337 cm−1 was assigned to the aggregation of stretching vibration of inter- and intramolecular hydrogen bonds caused by amino and hydroxyl groups in the BDCE molecule, and the peaks at 1465 cm−1, 1116 cm−1, and 776 cm−1 corresponded to the in-plane bending vibration peak of the hydroxyl group, C–O stretching vibration peak, and outside bending vibration peak of hydroxyl groups, respectively. The titration results and theoretical values of the content of free amino and hydroxyl groups in the chain extender could be performed as additional evidence for the formation of BDCE. The small differences in amine and hydroxyl values from the theoretical values might be caused by the residual trace ethylene diamine in the chain extender and analysis error. The above results confirmed the presence of terminated amino and hydroxyl groups and further verified that BDCE with the desired structure was synthesized.
3.2 Preparation of the PU coatings
Variation of the absorption peak of isocyanate groups in prepolymers, amino groups in chain extender BDCE, and the urethane and urea group in the cured coating were observed (Fig. 2(A)). The primary amine group had particularly high reactivity with the isocyanate group. The absorption band at 2275 cm−1, associated with the isocyanate groups in prepolymers, disappeared with the addition of chain extender BDCE, which indicated the completion of the chain extension reaction between the –NH2 of BDCE and the –NCO of prepolymers. The appearance of a band at 1645 cm−1 representing the stretching vibration of the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in –NH–CO–NH– proved that the chain extension between the prepolymer and BDCE performed as expected. Furthermore, the sharp absorption peak at 3315 cm−1 in the BDCE curing coating was assigned to the stretching vibration peak of NH in the urea and urethane group and hydroxyl groups, meaning that hydroxyl groups were preserved after the crosslinking–solidifying reaction, which was also proved by 1H NMR characterization as described below. Note that in the (1,6-hexanediamine) HDA and (1,6-hexanediol) HDO curing coatings, the peaks curved in the same position compared to BDCE, further proving the existence of hydroxyl groups in the coatings.
O in –NH–CO–NH– proved that the chain extension between the prepolymer and BDCE performed as expected. Furthermore, the sharp absorption peak at 3315 cm−1 in the BDCE curing coating was assigned to the stretching vibration peak of NH in the urea and urethane group and hydroxyl groups, meaning that hydroxyl groups were preserved after the crosslinking–solidifying reaction, which was also proved by 1H NMR characterization as described below. Note that in the (1,6-hexanediamine) HDA and (1,6-hexanediol) HDO curing coatings, the peaks curved in the same position compared to BDCE, further proving the existence of hydroxyl groups in the coatings.
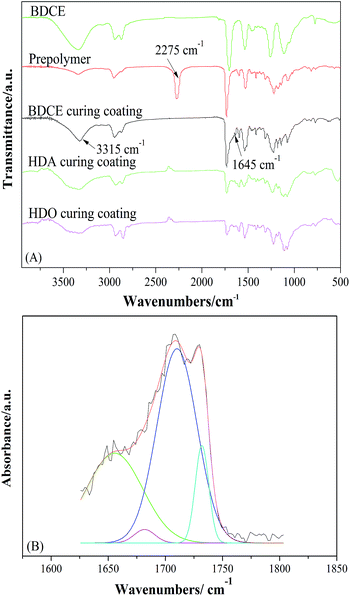 |
| | Fig. 2 FTIR spectra of prepared chain extender BDCE, prepolymer, and the curried coating. | |
Curve fitting of the carbonyl region was used to study the urea, urethane group, and the hydrogen bonding in the PU coatings (Fig. 2(B)). After fitting, the carbonyl stretching region was divided into four bands at 1662 cm−1, 1680 cm−1, 1710 cm−1, and 1731 cm−1, which represent the stretching of hydrogen-bonded carbonyl groups in urea, free urea carbonyl groups, hydrogen-bonded carbonyl groups in urethane, and free urethane carbonyl groups, respectively. As exhibited, the carbonyl groups of both the urea and urethane groups exist in the BDCE coating system. The obvious absorption peaks of hydrogen-bonded carbonyl groups proved the formation of intramolecular and intermolecular hydrogen bonds. The strengthened hydrogen bonding might facilitate segmental motion reduction, micro-phase separation, and some other behaviors. In addition, stretching vibration peaks of NH in the coating curing by different chain extenders were at 3315 cm−1, 3340 cm−1, and 3358 cm−1, respectively. They were lower than the free NH stretching vibration, which was considered to be higher than 3400 cm−1, and that peak occurred because of the formation of hydrogen bonds between NH and the carbonyl oxygen.29 Moreover, the formation of hydrogen bonds led to decreases in the chemical bond force constant of atoms participating in the formation of hydrogen bonds, and therefore, the absorption frequency shifted to the low wavenumbers.
3.3 1H NMR spectra of BDDE, BDDE-5CC, and synthesized BDCE
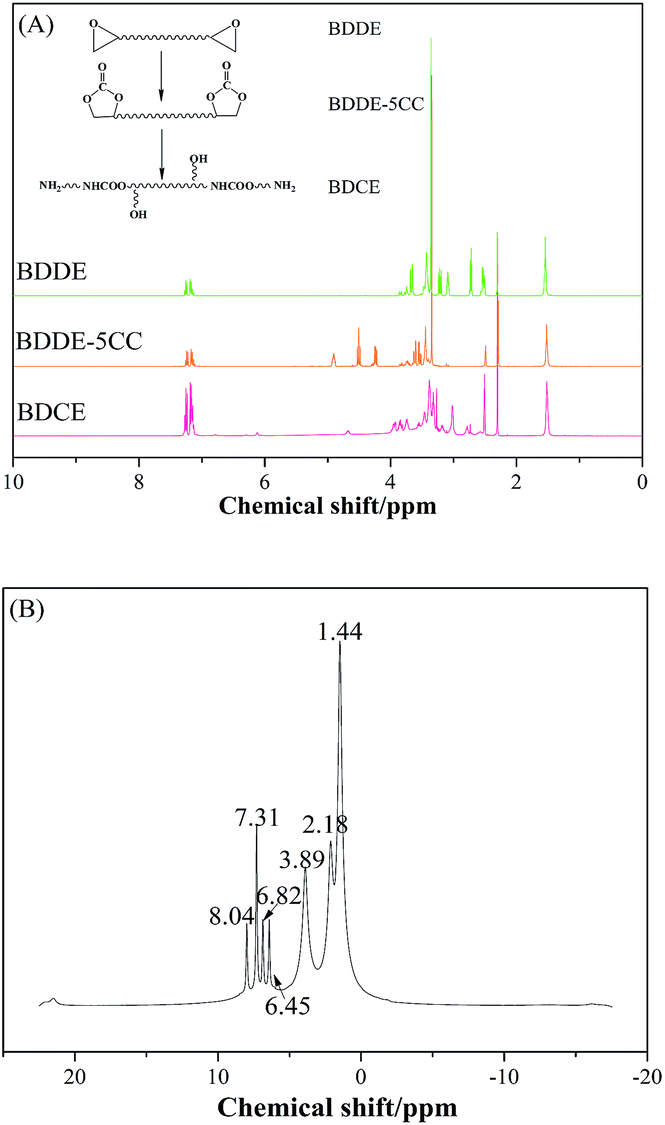
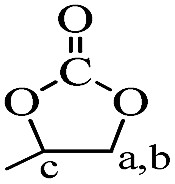
The chemical structures of raw material BDDE, the median product BDDE-5CC, final product BDCE, and the curing coating were verified by 1H NMR spectrum and are shown in Fig. 3. For BDDE, signals at 2.72, 3.08, and 3.21 ppm corresponded to protons of methylene (–CH2) and methine (–CH) in the epoxy ring. Chemical shifts at 3.43 ppm and 3.65 ppm were ascribed to the methylene in the BDDE liner chain next to the epoxy group, and shifts at 3.35 ppm and 1.53 ppm were assigned to methylene groups in the backbone. However, in comparison with BDDE, protons of methylene and methine in the cyclic carbonate group at 4.24, 4.52, and 4.91 ppm in BDCE-5CC were detected. Shifts at 3.46–3.62 ppm representing protons of methylene groups in the BDDE-5CC liner chain adjacent to the cyclic carbonate group and at 3.35 and 1.53 ppm corresponding to methylene groups in the backbone can also be observed. Additionally, chemical shifts related to epoxy groups in BDDE disappeared but new signals related to the cyclic carbonate groups appeared in BDDE-5CC. This observation, together with the IR spectrum, further proves the successful synthesis of BDDE-5CC.
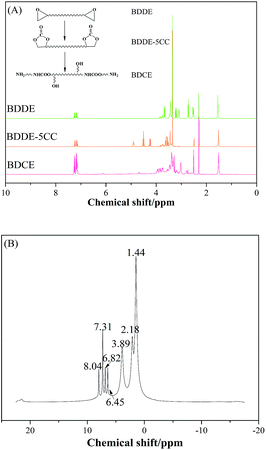 |
| | Fig. 3 1H NMR spectra of BDDE, BDDE-5CC, prepared chain extender BDCE, and coating curing by BDCE. | |
Titration results showed that the conversion of epoxy groups reached 100%, and the selectivity of cyclic carbonate was obtained through calculation from 1H NMR results of BDDE and BDDE-5CC by an internal standard method. A specified amount of BDDE, BDDE-5CC, and an internal standard solution of DMSO with toluene were weighed and tested by 1H NMR.30 Then, the related methylene peaks in BDDE, BDDE-5CC, and methyl in toluene were integrated, and the weight of BDDE-5CC was then calculated according to eqn (3),
| |
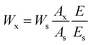 | (3) |
where
Wx is the weight of BDDE-5CC,
Ws is the weight of toluene introduced into the test system,
Ax is the integration value of the methylene peak in BDDE-5CC,
As is the integration value of methyl in toluene, and
E and
Es indicate the proton equivalent of BDDE-5CC and toluene at this chemical shift, respectively. Both of them could be calculated as dividing the molecular weight by the number of protons in the group producing the resonance peak. As calculated, the corresponding selectivity from the epoxy group to cyclic carbonates is 98.5%.
Chemical shifts and corresponding groups in the coating samples obtained from the 1H NMR spectra are listed in Table 1. From BDDE-5CC to BDCE, characteristic peaks related to the cyclic carbonate groups at 4.24, 4.52, and 4.91 ppm disappeared and peaks assigned to the urethane group appeared at 6.02 and 6.13 ppm. Additionally, signals of the hydroxyl groups at 4.61 and 4.68 ppm and peaks at 4.18 and 4.28 ppm representing the proton of methine adjacent to the hydroxyl carbon atom confirmed the isomer structures of the alcohol amine chain extender.30 As for the amino groups in BDCE, DMSO with its bibulous property could introduce water in the system and further promote rapid exchanges of active hydrogens with shape changes of peaks for amino groups. As a result, the broad peaks between 3–4 ppm ascribed to amino groups were overlapped by other groups and could not be clearly identified.
Table 1 Chemical shifts and corresponding groups in the sample structuresa
| Sample |
δ |
| 2.31; 7.16; 7.25 |
2.50 |
1.53 |
3.35 |
3.43; 3.65 |
c3.08; a2.54; b2.72 |
c4.91; a4.24; b4.52 |
6.02; 6.13 |
4.18; 4.28 |
4.61; 4.68 |
| δ = 1.53 is the methylene group in the middle of the backbone, δ = 3.35 represents the methylene group in the chain next to the ether bond, δ = 3.43, 3.65 represents the methylene group next to the epoxy/cyclic carbonate group, and δ = 4.18, 4.28 indicates the methine group next to the hydroxyl group. |
| BDDE |
PhMe |
DMSO |
–CH2– |
–CH2– |
–CH2– |
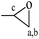 |
|
|
|
|
| BDDE-5CC |
PhMe |
DMSO |
–CH2– |
–CH2– |
–CH2–, (3.73, 3.51) |
|
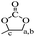 |
|
|
|
| BDCE |
PhMe |
DMSO |
–CH2– |
–CH2– |
–CH2–, (3.38, 3.65; 3.73,3.47) |
|
|
–NHCOO– |
–CH– |
–OH |
On the 1H NMR spectra of the cured coating, signals at 8.04 and 2.18 ppm were assigned to the secondary amino in the urethane group formed after the chain extension reaction between the terminal amino group and isocyanate groups and the initial hydroxyl group in the chain extender BDCE, respectively. Peaks at 7.31, 6.82, and 6.45 ppm were attributed to protons of the benzene ring in styrene. Protons corresponding to the methylene groups between the benzene ring and in the backbone of the polymer were peaks at 3.89 and 1.44 ppm. Together, the FTIR and NMR results provide a solid justification for the successful synthesis of the BDCE and PU coating. In addition, after the calculation as previously described, the calculated selectivity from cyclic carbonates to urethane is 98.9%.
3.4 Water and solvent resistance of the coatings
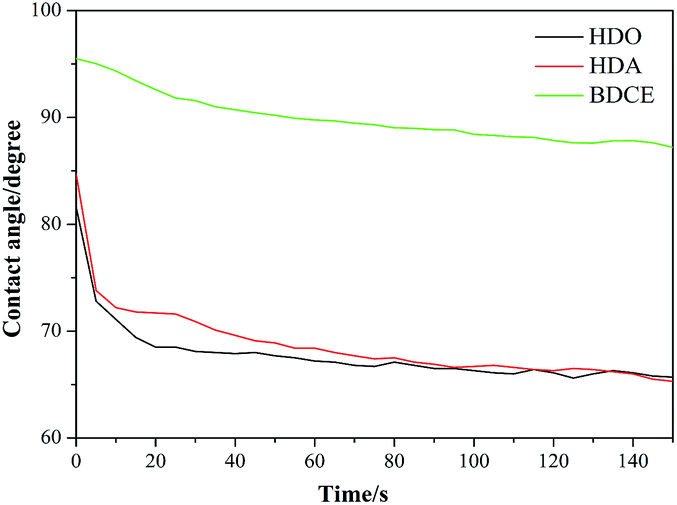
3.4.1 Contact angles of coatings. The contact angles of the coatings, as a measurement of wetting degree, are shown in Fig. 4. With the chain extender varying from BDCE to HDO, the contact angles of the coatings decreased from 95.6° to 81.6°. In comparing the HDA- and HDO-derived coatings, the relatively larger contact angle of the BDCE-derived coating might be attributed to the additional hydrogen bonds formed in the coating. The hydrogen atoms in hydroxyl groups could create physical force in the form of hydrogen bonds that occur with electronegative oxygen and nitrogen atoms in urea groups and carbamate groups. Large quantities of hydrogen bonds will increase the cohesion and integration among the polyurethane molecules, thus preventing the solvent molecules from penetrating, and then the coating will exhibit a large contact angle. Moreover, in the BDCE-based system, the 7-member ring structures that form between carbonyl oxygen and hydroxyl hydrogen might also provide contributions to the larger contact angles. The contact angle of BDCE was 95.6°, which is an indication of a hydrophobic coating, good water resistance, and decreased water wetting.
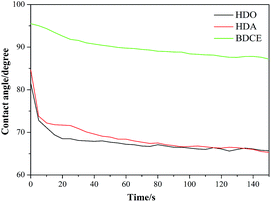 |
| | Fig. 4 Figure of contact angles of coatings with different chain extenders changed with time. | |
It is likely that the contact angles of each chain extender decreased with time prolongation, partial deformation of the coating surface, and reconstruction of the polymer surface in order to minimize the interface energy. In the BDCE-based coating, the existence of hydrogen bonds restricted the movement of the groups, and the 7-member ring as a hydrophobic structure increased the steric hindrance, resulting in a small reduction of the contact angles with time. However, in contrast to BDCE, less hydrogen bonds formed in the HDA and HDO coatings due to a lack of free hydroxyl groups, and contact angles decreased more obviously in a larger range. Moreover, for the HDA-derived coating, closer polarity between urea groups and water facilitated the spread of water, leading to the greatest degree of contact angle reduction. From another perspective, group cohesion and contribution of groups to molecular force played a key role in the performance of materials. Furthermore, the microcrystallinity and degree of phase separation of polyurethane was closely related to the cohesive energy of the groups on the molecular chain. Table 2 shows that the cohesive energy of the carbamate group was lower than that of the urea group. Correspondingly, more urea groups in the coating could lead to a more stable structure and stronger water resistance and was in accordance with the law of descent in contact angles.
Table 2 Cohesion of different groups33
| Groups |
Cohesion/kJ mol−1 |
Groups |
Cohesion/kJ mol−1 |
| –CH2– |
2.84 |
–CO– |
11.12 |
| –CH3 |
7.11 |
–COO– |
12.1 |
| –Ph |
16.3 |
–NHCO– |
35.5 |
| –OH |
24.2 |
–NHCOO– |
36.4 |
| –O– |
4.18 |
–NHCONH– |
>36.5 |
| –COOH |
23.4 |
|
|
3.4.2 Resistance to water and solvents. Anticorrosive properties are significant for practical applications of coatings. In this work, the resistance of the prepared coatings to acid, alkali, and solvents with different polarity were evaluated by an immersion method as shown in Table 3. The coating synthesized from BDCE shows excellent resistance to water and other solvents and no blistering or falling off was observed, and these positive attributes should be the result of the unique structure as previously mentioned. Moreover, the alkali resistance was better than that of commonly used coatings.31,32 After several days of immersion, all coatings faded. This might be caused by polar groups, such as urethane and hydroxyl, which enhanced the compatibility between the coatings and solvents, and changed the evenness of the coating surface. Coatings derived from BDCE exhibited better solvent resistance than others.
Table 3 Resistance of coatings with different chain extenders to different types of solventsa
| Sample |
Water resistance |
Solvent resistance |
Acid resistance (in 5% HCl) |
Alkali resistance (in 5% NaOH) |
Results for solvent double rub test |
| Acetone |
Methylbenzene |
| I – no effect, II – loss of gloss, III – blistering, IV – fell off or film removed, A – small amount of substrate was exposed, B – a fairly large amount of substrate was exposed, C – a large area of substrate was exposed. |
| PTMG + MDI + BDCE |
I |
II |
II |
I |
I |
A |
| PTMG + MDI + HDO |
II |
III |
III |
III |
III |
C |
| PTMG + MDI + HDA |
II |
III |
IV |
III |
III |
C |
3.4.3 Resistance to salt solution. Salt solution resistance is also an important factor for the performance of coatings. To investigate the resistance of the BDCE-based coating to salt, a partially coated tin plate was immersed in 10 wt% NaCl solution for 24 h, and the results are shown in Fig. 5. Due to the protection of the coating, there was no rust on the coated region, and there was only a small amount of fading of the coating. However, it was noted that the uncoated region was covered with rust. Such a result indicated good resistance of the BCDE-derived coating to the salt solution. It was also in agreement with the measured fairly larger contact angle, because of the stronger hydrophobicity of the coating.
 |
| | Fig. 5 Image of tin plate coated or uncoated after immersion in NaCl solution. | |
3.5 Mechanical properties of the coatings
Flexibility, impact resistance, pencil hardness, and adhesive force to the substrate were measured to evaluate the mechanical properties of the coating, and the results are shown in Table 4 and Fig. 6. Table 4 shows that all the coatings possessed excellent adhesive force to the substrate as well as flexibility, occupying the top ranking of the standard, which could also be seen from Fig. 6, where Fig. 6(A)–(C) show the results after tests for impact resistance, adhesive force grade, and flexibility, respectively. After the flexibility tests, the coating surfaces were observed with a 4× magnifying glass, and no cobwebbing cracks or peeling or other damages were found. Furthermore, during the adherence test, the BDCE coating firmly adhered to the substrate and integrated each part of the film in an orderly manner, and every part of the coating endured this test without defect and without detachment from the substrate.
Table 4 Mechanical properties of coatings with different chain extender
| Sample |
Flexibility/mm |
Impact resistance/50 cm |
Adhesive force/grade |
Pencil hardness |
| PTMG + MDI + BDCE |
0.5 |
No crack |
1 |
3H |
| PTMG + MDI + HDO |
0.5 |
No crack |
1 |
2H |
| PTMG + MDI + HDA |
0.5 |
With crack |
1 |
3H |
 |
| | Fig. 6 Images of coated plates recorded after mechanical tests were performed on different substrates. Results are shown after (A) the impact resistance test, (B) adhesive force grade test, and (C) flexibility test. | |
It is evident that there was a difference in impact resistance and pencil hardness between BDCE and HDA. Coatings prepared from BDCE and HDO showed no rupture to any noticeable degree and did not influence the adherence to the substrate after the tin plate was subjected to extreme impact damage from either side. However, for HDA, obvious cracks were observed at the impact site, and the coating in this area was peeled off. The small difference in pencil hardness was reduced from 3H to 2H. Differences in impact resistance might be ascribed to the existence of an additional urea group in the HDA coating system that provided increased hardness and brittleness. However, comparing BDCE with HDA, although all had urea groups in the coatings, the BDCE-based coating possessed a longer chain and side chains in the structure, which could play a role in stress dispersion and bearing stress, and therefore, lead to good resistance to impact. From another perspective, from the microstructures, a greater effect of hydrogen bonding in the BDCE system might lead to a stronger degree of microphase separation and thereby further improve the mechanical properties. In the HDO system, hard segments were mainly composed of urethane groups, of which the strength was weaker than the urea bond, and therefore, the coating had lower pencil hardness.
3.6 Thermal properties of coatings
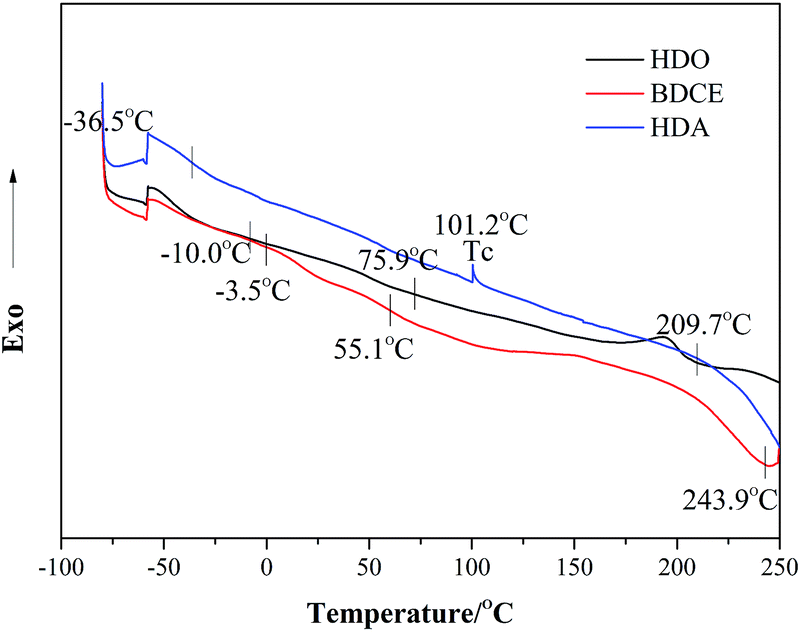
DSC was used to determine the glass transition temperature (Tg) and the pyrogenation temperature of the PU coatings. Fig. 7 shows the DSC thermogram of the PU coatings prepared from different chain extenders. The numerical values corresponding to the thermal transition are tabulated in Table 5. Coatings made from HDA and BDCE exhibited two Tg temperatures; however, the coating from HDO exhibited only one Tg. Furthermore, a crystallization transition was detected for the HDA system.
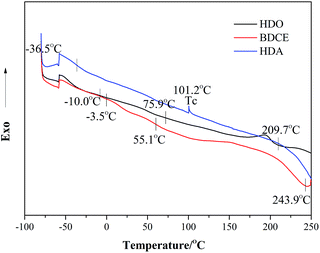 |
| | Fig. 7 DSC thermograms of coatings prepared from various chain extenders. | |
Table 5 Thermal characteristics of different chain extenders from the DSC results
| Sample |
Tg (°C) |
Tc (°C) |
T0 (°C) |
| PTMG + MDI + HDO |
−10.0 |
101.2 |
209.7 |
| PTMG + MDI + HDA |
−36.5, 75.9 |
— |
— |
| PTMG + MDI + BDCE |
−3.5, 55.1 |
— |
243.9 |
Steps at −36.5–3.5 °C were the glass transition temperatures of the soft segment, all of which were higher than the glass transition temperature of pure soft segments of PTMG at −74 °C. However, there were two Tg values for chain extender BDCE and HDA, representing the Tg temperature of the soft and hard segments in the coating structure. This occurred because of the formation of urea bonds, which resulted in the existence of urea groups that generally exhibited stronger polarity and rigidity and led to a higher degree of phase separation between the hard and soft segments. Then, the Tg-dependency could be observed in the system. Moreover, in the BDCE-based coating, the intramolecular hydrogen bonding previously described might also make a contribution to the higher extent of microphase separation. Additionally, due to the higher phase separation in HDA, a higher soft segment content might result in a lower Tg temperature closer to PTMG than that of HDO. For BDCE, the corresponding Tg was higher than that of HDO, and it might be ascribed to the higher hard segment content caused by the alcohol amine chain extender, which restricted the segmental motion of the block and the elevation of Tg.
With the increase in temperature hereafter, when the temperature was higher than the glass transition temperature and lower than the viscous flow decomposition temperature, blocks of polymers could move in an orderly manner after being heated and forming regular structures, and therefore, in the HDA coating, an apparent crystallization peak can be found at 101.2 °C.34 The endothermic peaks from 209.7 °C to 243.9 °C represent the melting of the coatings. Samples prepared from HDA and BDCE showed higher melting temperatures and heat absorption than HDO due to the difficulty in the pyrolysis of urea as compared to carbamate groups. However, in this area, no obvious endothermic peak exists for HDA because of the larger purity of hard segments caused by the secondary crystallization, resulting in a higher decomposition temperature for the polymers.
3.7 TEM of the coatings
As direct evidence of the microstructure, the microphase separation can be observed by TEM, which is a common method to study the interstructure of materials. A typical image of the microstructure of BDCE-derived coating is presented in Fig. 8 together with HDA- and HDO-based coatings. Imparities in the viscoelastic and mechanical properties exhibited by these coatings could be generally ascribed to the microphase separation in structure, arising from the thermodynamic incompatibility of the different polymer segments as previously mentioned. The resulting domain separation could lead to clustering of the same segments, which was affected by composition, molecular weight, and structures of the segments. The dark domains were presumed to be the hard urethane and urea microphase due to the aggregation of electrons, and the light areas were ascribed to the soft segments.35,36 As presented in Fig. 8, dark and light regions were clearly observed in BDCE-based coatings and in HDA and HDO systems, and the hard segments were relatively evenly distributed in the soft segments. In addition, areas of the dark portion of the samples in the images could be ordered from BCDE to HDA and then to HDO, implying that the BCDE-derived sample displayed the highest microphase separation degree among the tested coatings. This might be ascribed to the higher content of hard segments and stronger hydrogen bonding caused by the free hydroxyl groups in the BCDE-derived coating, which resulted in enhanced incompatibility between hard and soft segments. Generally, the enlarged microphase separation led to better viscoelastic and mechanical properties of PU materials.
 |
| | Fig. 8 TEM images of various types of polyurethane coatings obtained from BDCE, HDA, and HDO. | |
3.8 Storage stability
The storage stability of the synthesized polyurethane coatings was investigated by setting the sealed samples statically at environmental temperature for three months. Fig. 9 shows images of the samples. As a good solvent for polyurethane materials, all the coatings were well dispersed in DMF. The coating solution obtained from BDCE with a slight yellow color hardly changed in appearance and viscosity, indicating the best storage stability. This stability might be related to the stronger interaction between the polymer and DMF due to the strong polarity of urea groups in the BDCE system and a stable structure caused by strong hydrogen bonding. Additionally, the other two coatings with the same solid content exhibited relatively poor storage stability. During the preparation process, a light milky white color was observed for the HDA-derived sample, which changed to a deep milky white and precipitated on the bottom of the bottle after 3 months. For the HDO-derived sample, the same phenomenon was also observed after 3 months of storage.
 |
| | Fig. 9 Storage stability of different polyurethane coatings. | |
4. Conclusions
A five-membered carbonate compound was synthesized from CO2 and 1,4-butanediol diglycidyl ether with a conversion of 100% in the epoxy group and selectivity of 98.5%. The carbonate was applied to the preparation of a novel alcohol amine, and was further applied as a chain extender to the manufacture of polyurethane coatings. Free hydroxyl groups in the coating played an important role in the properties of the coating. The hydroxyl groups provided strengthened hydrogen bonding in PU coatings, especially the intermolecular hydrogen bond with a 7-member ring structure, which improved the cohesion and penetration resistance of the coating, thus endowing the coating with promising performance in adhesion, flexibility, impact resistance, anticorrosiveness, and thermal stability.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully thank the financial support from the “Major Science & Technology Project of Shanxi Province” (No. MD2014-10, 201603D312007 and 2015021043) and “Youth Scientific Funds of Shanxi Province” (No. 2015021043).
References
- X. Zhong, J. Lin, Z. Wang, C. Xiao, H. Yang, J. Wang and X. Wu, Prog. Org. Coat., 2016, 99, 216–222 CrossRef CAS.
- O. Bayer, H. Rinke, W. Siefken, L. Orthner and H. Schild, DE. Pat.728,981, 1937.
- D. Randall and S. Lee, The polyurethanes book, John Wiley & Sons Inc, 2002 Search PubMed.
- B. K. Kim and J. C. Lee, J. Polym. Sci., Part A: Polym. Chem., 2015, 34, 1095–1104 CrossRef.
- H. Xin, Y. D. Shen and X. R. Li, Polym. Bull., 2011, 67, 1849–1863 CrossRef CAS.
- R. S. Chen, C. J. Chang and Y. H. Chang, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3482–3490 CrossRef CAS.
- O. Jaudouin, J. J. Robin, J. M. Lopez-Cuesta, D. Perrin and C. Imbert, Polym. Int., 2012, 61, 495–510 CrossRef CAS.
- C. Zhang, X. Zhang, J. Dai and C. Bai, Prog. Org. Coat., 2008, 63, 238–244 CrossRef CAS.
- M. S. Shin, Y. H. Lee, M. M. Rahman and H. D. Kim, Polymer, 2013, 54, 4873–4882 CrossRef CAS.
- J. B. Dai, X. Y. Zhang, J. Chao and C. Y. Bai, J. Coat. Technol. Res., 2007, 4, 283–288 CrossRef CAS.
- T. Teruo, K. Masao and Y. Masaaki, Macromol. Chem. Phys., 2003, 191, 625–632 Search PubMed.
- X. Zhao, J. Ding and L. Ye, J. Fluorine Chem., 2014, 159, 38–47 CrossRef CAS.
- F. Çelebi, L. Aras, G. Gündüz and I. M. Akhmedov, J. Coat. Technol., 2003, 75, 65–71 CrossRef.
- A. Kadam, M. Pawar, V. Thamke and O. Yemul, Prog. Org. Coat., 2017, 107, 43–47 CrossRef CAS.
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- M. Akbarian, M. E. Olya, M. Mahdavian and M. Ataeefard, Prog. Org. Coat., 2014, 77, 1233–1240 CrossRef CAS.
- A. Noreen, K. M. Zia, M. Zuber, S. Tabasum and A. F. Zahoor, Prog. Org. Coat., 2016, 91, 25–32 CrossRef CAS.
- A. Kadam, M. Pawar, V. Thamke and O. Yemul, Prog. Org. Coat., 2017, 107, 43–47 CrossRef CAS.
- Y. González-García, S. González and R. M. Souto, Corros. Sci., 2007, 49, 3514–3526 CrossRef.
- P. Francois, P. Vaudaux, N. Nurdin, H. J. Mathieu and P. Descouts, Biomaterials, 1996, 17, 667–678 CrossRef CAS PubMed.
- H. Blattmann and R. Mülhaupt, Green Chem., 2016, 18, 2406–2415 RSC.
- G. Liu, G. Wu, J. Chen, S. Huo, C. Jin and Z. Kong, Polym. Degrad. Stab., 2015, 121, 247–252 CrossRef CAS.
- O. L. Figovsky, US. Pat., 6120905, 2000.
- O. L. Figovsky, Mendeleev Chem. J., 1988, 33, 31–36 Search PubMed.
- GB/T 1731-93, Determination of Flexibility of Film.
- GB/T 6739-2006, Paints and Varnishes-Determination of Film Hardness by Pencil Test.
- GB/T 1732-93, Determination of Impact Resistance of Film.
- GB/T 1720-89, Determination of Adhesion
of Film.
- H. T. Wang, B. L. Bai, F. Q. Bai, D. M. Pang, X. Ran and C. X. Zhao, Liq. Cryst., 2011, 38, 767–774 CrossRef CAS.
- A. Cornille, J. Serres, G. Michaud, F. Simon, S. Fouquay, B. Boutevin and S. Caillol, Eur. Polym. J., 2016, 75, 175–189 CrossRef CAS.
- A. Noreen, K. M. Zia, M. Zuber, S. Tabasum and A. F. Zahoor, Prog. Org. Coat., 2016, 91, 25–32 CrossRef CAS.
- R. Pathak, M. Kathalewar, K. Wazarkar and A. Sabnis, Prog. Org. Coat., 2015, 89, 160–169 CrossRef CAS.
- G. Oertel, Polyurethane Handbook: Chemistry, Raw Materials, Processing, Application, Properties, 1985 Search PubMed.
- C. T. Li, The 18th Biennial Conference of China Polyurethane Industry Association, 2016, pp. 425–428 Search PubMed.
- J. A. Koutsky, N. V. Hien and S. L. Cooper, J. Polym. Sci., Part B: Polym. Lett., 1970, 8, 353–359 CrossRef CAS.
- S. Horiuchi, T. Kajita and T. Tachibana, J. Appl. Polym. Sci., 1999, 74, 1762–1772 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ac,
Maoqing Kanga,
Feng Wanga,
Yuhua Zhaoa and
Qifeng Lia
*ac,
Maoqing Kanga,
Feng Wanga,
Yuhua Zhaoa and
Qifeng Lia