DOI:
10.1039/C7RA11947J
(Paper)
RSC Adv., 2018,
8, 1361-1370
Lignin-first depolymerization of native corn stover with an unsupported MoS2 catalyst†
Received
30th October 2017
, Accepted 18th December 2017
First published on 3rd January 2018
Abstract
The lignin-first biorefinery method appears to be an attractive approach to produce phenolic chemicals. Herein, corn stover was employed for the production of phenolic monomers using an unsupported non-noble MoS2 catalyst. The yield of phenolic monomers was enhanced from 6.65% to 18.47% with MoS2 at 250 °C and about 75% lignin was degraded with more than 90% glucan reserved in the solid residues. The Fourier-Transform Infrared (FT-IR) and heteronuclear single quantum coherence-nuclear magnetic resonance (1H–13C HSQC-NMR) characterization suggested that the cleavage of the β-O-4, γ-ester and benzyl ether linkages were enhanced, promoting the delignification and the depolymerization of lignin. The catalyst performance was relatively effective with 14.30% phenolic monomer yield after the fifth run. The effects of the reaction temperature, the initial hydrogen pressure, the dosage of catalyst, and the reaction time were investigated. The model reactions were also proposed for the potential mechanism study. This work provides some basic information for the improvement of the graminaceous plant lignin-first process with a non-noble metal catalyst.
1. Introduction
Lignocellulose is the most abundant form of biomass, comprising cellulose, hemicellulose and lignin, which has been recognized as a reliable potential alternative source for fossil-based chemicals and fuels.1,2 Thus, the utilization of polysaccharides from biomass has been considered as the primary objective of the traditional lignocellulosic biorefineries to harvest the value-added chemicals.3 Compared with cellulose and hemicellulose, lignin has much lower oxygen content. Typically, it accounts for 15–30% by weight but 40% by energy of lignocellulose biomass.4 Therefore, lignin conversion is also of great significance to accomplish an economically viable biorefinery.5,6 However, lignin conversion is a challenging project due to its resistant structural feature. Lignin is the polymer of three different hydroxylated phenylpropane units (syrigyl (S), guaiacyl (G), and p-hydroxyphenyl-propane (H)). The units are linked by different ether bonds and carbon–carbon bonds, including β-O-4, β-5, β-β, and β-1 linkages, of which more than half of the linkage structures are β-O-4 linkages.4 Furthermore, lignin is cross-linked with carbohydrates in the plant cells and the middle lamella.7 The complex chemical and physical structure of lignin makes it difficult to depolymerize lignin for biochemicals and biofuels. To develop the effective depolymerization methods, the crucial tasks are to destroy the physical structure of lignin and break the chemical bonds between different units.
This is the prevalent strategy for lignin upgradation by reductive depolymerization with metal catalysts.8,9 Moreover, recent advances have been reported with different lignin sources. Noble metal catalysts, such as Ru/C, Pd/C, Pt/C, and Rh/C,10–12 and non-noble transition metal sulfide, carbide, and nitride catalysts, such as sulfided NiMo and CoMo, MoC2, and TiN,13–15 were introduced in the depolymerization of technical lignin. The active metals played an important role in the cleavage of the β-O-4 linkage through hydrogenation or hydrodeoxygenation. Moreover, technical lignin, including lignosulfonate lignin, kraft lignin, organosolv lignin, and soda lignin,16 is usually produced via pretreatments under various harsh conditions. The pretreatments dramatically lead to the structural changes by severe and irreversible condensation. In fact, the condensation, resulting from the transformation of the ether bonds into the relative stable carbon–carbon bonds during the pretreatments, makes the subsequent lignin depolymerization process harder.17 High temperature and char formation are also the common challenges in the process of lignin-to-aromatic conversion. Herein, these drawbacks have seriously restricted the depolymerization of technical lignin into value-added chemicals and biofuels under relatively mild conditions. Compared with technical lignin, native lignin has 40–60% of total intermolecular linkages in the form of β-O-4 ether bonds,16 which makes native lignin a better feedstock for lignin-to-aromatic conversion. Based on this, lignin-first as a novel concept has been put forward with native lignin to extract more valuable products from lignin.3 Lignin-first depolymerization focuses on prohibiting the lignin condensation and improving the efficiency of lignin-to-aromatic conversion.3,17 In the solvolysis process, the native lignin is extracted from biomass by organic solvents. The extracted lignin fragments are depolymerized into monomers and oligomers via a reductive pathway with a metal catalyst. In this process, the carbohydrate pulp is primarily retained in the solid residues, which can be used for producing valuable chemicals and biofuels.3,18,19 Numerous efforts have been made in the recent years. For instance, noble metal catalysts, such as Ru/C, Pd/C, Pt/C, and Rh/C, were employed for native birch.18,20,21 Furthermore, the synergistic effects of the noble metal catalysts were investigated at a relatively mild condition with an acid as a co-catalyst (such as HCl, Al(OTf)3, and metal triflates).22 To reduce the catalyst cost, the Ni-based catalysts, such as Ni/Al2O3 (ref. 23) and Ni/C,19 were introduced in the process. Based on the previous studies, the lignin-first conversion approach exhibits a high yield and selectivity for the production of phenolic monomers. The reductive metal catalyst has been proven effective for the cleavage of the lignin–carbohydrate bonds and lignin–lignin ether bonds22,23 and the acids played a significant role as the co-catalyst.24,25
Compared to lignin with a high S/G ratio from hardwoods, lignin of the graminaceous plants (e.g. corn stover) is more complex; there is a higher content of hydroxycinnamic acids, particularly p-coumaric acid (pCA) and ferulic acid (FA), which are ester-linked or ether-linked in the cell walls.26,27 However, only few studies have been conducted using lignin of graminaceous plants. For instance, Ru/C, Ni/C, and the synergistic effects with H3PO4 were studied with native corn stover as the feedstocks.24 Though the phenolic monomers were harvested, the recyclability of the catalyst was not reported. Thus, the investigation on the lignin-first process of the graminaceous plants will provide a lignin-first approach with a much wider applicability for the utilization of lignocellulose. On the whole, noble metal catalysts are high cost and the Ni-based catalysts confront with the limitation of the recyclability. Moreover, the chemocatalytic conversion of carbohydrate pulp mostly needs the involvement of acids.20,28,29 Therefore, a non-noble metal catalyst capable of hydrogenation and acid tolerance is preferred for the lignin-first process.
MoS2 is a good choice for the lignin-first reductive depolymerization. It could tolerate some common acids except strong oxidative acids and is capable of hydrogenation (HYD), hydrodesulfurization (HDS), and hydrodeoxygenation (HDO).30,31 In addition, it has been reported that Ni-modified MoS2 catalyst was effective for the cleavage of β-O-4 ether bond via a dehydroxylation–hydrogenation strategy;32 also, MoS2 has been introduced in the depolymerization of alkali lignin for bio-oil production.33 As a non-noble transition-metal sulfide catalyst, it can be easily hydrothermally synthesized by an easier method compared to the synthesis of supported NiMo or CoMo metal sulfide. The unsupported layered structure can offer more active sulphur vacancies at the edge of the slab.
In this study, the lignin-first biorefinery was further developed with the depolymerization of native corn stover lignin via a non-noble unsupported MoS2 catalyst in methanol. The effect of the reaction conditions, including the reaction temperature, the initial hydrogen pressure, the dosage of catalyst, and the reaction time, on the yield of the phenolic monomers and the liquid products were investigated in detail. In particular, FT-IR and 1H–13C HSQC-NMR techniques were utilized to perform the chemical characterization of the liquid products. The model reactions were proposed for the potential mechanism study in order to get a comprehensive understanding of the lignin-first process.
2. Methods
2.1. Materials
Corn stover was collected from Mengcheng City, Anhui Province of China. After being washed and dried at 60 °C for 48 h, the stock was cut and milled. The corn stover fragments sieved through a 30-mesh screen were collected and sealed for later use after being dried in an oven at 105 °C for 6 h. Hydrochloric acid (36–38%), sulfuric acid (95–98%), methanol, water-free ethanol, acetone, ammonium molybdate tetrahydrate, and thiourea were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Methyl sulfoxide-d6 (99.9%) for NMR characterization was purchased from Aladdin (Shanghai, China). All chemicals were used without further purification.
2.2. Synthesis and characterization of MoS2
The method for the synthesis of MoS2 was referred to that reported of Wang et al. and Li et al.33,34 Specifically, 0.614 g ammonium molybdate tetrahydrate and 0.802 g thiourea were dissolved in 80 mL double distilled water via ultrasonication and then stirred for 10 min. After the pH value was adjusted to 0.8–0.9 with hydrochloric acid under stirring, the solution was transferred into a 150 mL Teflon-lined stainless-steel autoclave and the reaction was carried out at 200 °C for 24 h. The black products were collected and washed with acetone and water, in sequence, to remove the soluble matter. Finally, the ethanol washed MoS2 was dried at 60 °C for 12 h under vacuum.
The properties of the fresh and recycled catalysts were characterized using scanning electron microscope (SEM, FEI SIRION200), X-ray diffractometer (XRD, Rigaku SmartLab), X-ray photoelectron spectrometer (XPS, Thermo-VG Scientific ESCALAB 250), Autosorb iQ Station (Quantachrome), and Autosorb-iQ-C (ASIQACIV200-2).
2.3. Determination of carbohydrates and lignin
The determination of carbohydrates and lignin were conducted according to the Laboratory Analytical Procedures (LAP) established by the National Renewable Energy Laboratory (NREL) and also according to the previous study reported by our group.35 The results are listed in Table S1.†
2.4. Depolymerization of native corn stover lignin
Typically, 0.300 g MoS2, 2.000 g corn stover together with 40 mL methanol were sealed in a 100 mL autoclave made of Zirconium-702 (Anhui Kemi Machinery Technology Co., Ltd.). The autoclave was purged with H2 for three times and pressurized to 3.0 MPa with H2. A mechanical impeller speed was set at 800 rpm. The reaction temperature was raised from room temperature to 250 °C in 40 min and held for 2 h. After the reaction was complete, the autoclave was cooled down to room temperature in water.
2.5. Analysis of the products
After the gas was released, the reaction mixture was filtered into two parts. The residue was washed with 30 mL methanol for three times before being dried at 80 °C overnight. The liquid part was reserved for monomer analysis, while the rest of the liquid products (LP) were evaporated using a rotary evaporator at 38 °C and then extracted with ethyl acetate. The evaporated ethyl acetate soluble products (EP) were defined as bio-oil.
Gas chromatography/mass spectrometry (GC-MS, Agilent 7890/5975) and gas chromatography (GC, SHIMADZU GC-2010) with flame-ionization detection were employed for qualitative and quantitative analysis of the phenolic monomers. Acetophenone was used as an internal standard. Fourier transform infrared spectrometry (FT-IR, Nicolet 8700) and nuclear magnetic resonance spectroscopy (NMR, Bruker AVANCE AV III 400) were utilized for further analysis of the chemical composition of the liquid products.
The volatile products were confirmed by GC-MS as follows: guaiacyl compounds, including 2-methoxy-4-propenylphenol, 2-methoxy-4-ethylphenol, 2-methoxy-4-propylphenol, and methyl trans-4-hydroxy-3-methoxycinnamate (methyl ferulate, MF); p-hydroxyphenyl compounds, including 4-ethylphenol, methyl trans-p-coumarate (MpC), and methyl 3-(4-hydroxyphenyl)propionate; syringyl compounds, including 2,6-dimethoxy-4-propylphenol and 2,6-dimethoxy-4-allylphenol. In addition, 2,3-dihydrobenzofuran was detected. The yield of the products was calculated using the following equations:
| | |
YSumG = MSumG/ML × 100%
| (2) |
| | |
YSumS = MSumS/ML × 100%
| (3) |
| | |
YSumH = MSumH/ML × 100%
| (4) |
| | |
YSum = MSum/ML × 100%
| (5) |
where
Mi: the mass of monomer in the liquid products;
ML: the mass of native corn stover lignin;
MSumG,
MSumS, and
MSumH: the sum mass of guaiacyl, syringyl, and
p-hydroxyphenyl phenolic monomers in the liquid products;
MSum: the sum mass of all phenolic monomers in the liquid products;
MLP: the mass of the liquid products;
MEP: the mass of the extractive products;
MSR: the mass of the solid residual corn stover (SR);
M: the mass of corn stover; the mass of the residual corn stover was determined by subtracting the mass of the catalyst from the reaction residues.
3. Results and discussion
3.1. Catalyst characterization
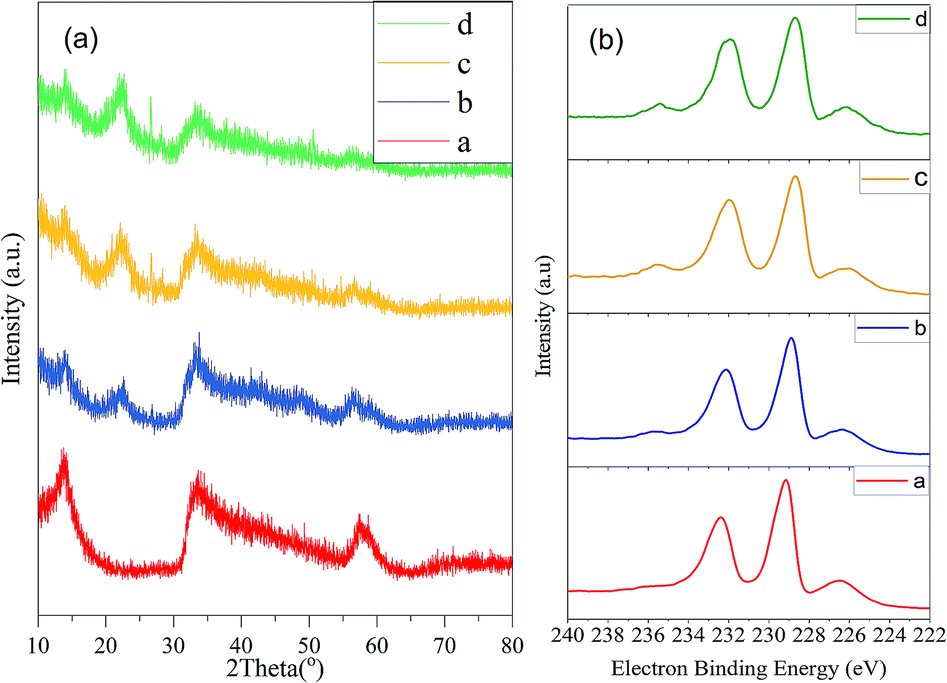
The physical properties of the fresh catalyst were evaluated. As shown in Fig. 1a the XRD 2θ peaks at 13°, 33°, and 57° are in accordance with the (001), (100), and (110) lattice planes of MoS2 (JCPDS card no. 37-1492), respectively. The broad XRD reflection of MoS2 suggested that it might have an amorphous structure and the low degree of crystallinity could provide more active sulphur vacancies, which is critical for the catalytic reactivity. In addition, the XPS (Fig. 1b) results suggested that the atomic ratio of S to Mo was 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. Thus, it can be speculated that most of the hydrothermal synthesized black powder was MoS2. In addition, the results from the nitrogen adsorption–desorption experiments revealed that MoS2 has a large Barrett–Joyner–Halenda (BJH) adsorption surface area of 68.831 m2 g−1, pore volume of 0.252 cm3 g−1, and a mesoporous pore diameter of 2.193 nm, which is in accordance with the porous flower-like layer structure observed by SEM/TEM (Fig. 2). This structure ensured the frequent access of the substance to the active sites on the edge of the layered structure, which promoted the degradation of lignin and the generation of the phenolic monomers.
:4. Thus, it can be speculated that most of the hydrothermal synthesized black powder was MoS2. In addition, the results from the nitrogen adsorption–desorption experiments revealed that MoS2 has a large Barrett–Joyner–Halenda (BJH) adsorption surface area of 68.831 m2 g−1, pore volume of 0.252 cm3 g−1, and a mesoporous pore diameter of 2.193 nm, which is in accordance with the porous flower-like layer structure observed by SEM/TEM (Fig. 2). This structure ensured the frequent access of the substance to the active sites on the edge of the layered structure, which promoted the degradation of lignin and the generation of the phenolic monomers.
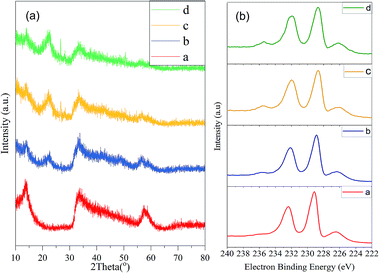 |
| | Fig. 1 XRD/XPS spectra of the MoS2: (a) XRD and (b) XPS, a: fresh MoS2, b: used once, c: used 4 times, and d: used 5 times. | |
 |
| | Fig. 2 TEM/SEM of MoS2: (a) TEM and (b) SEM. | |
3.2. Influence of the reaction conditions
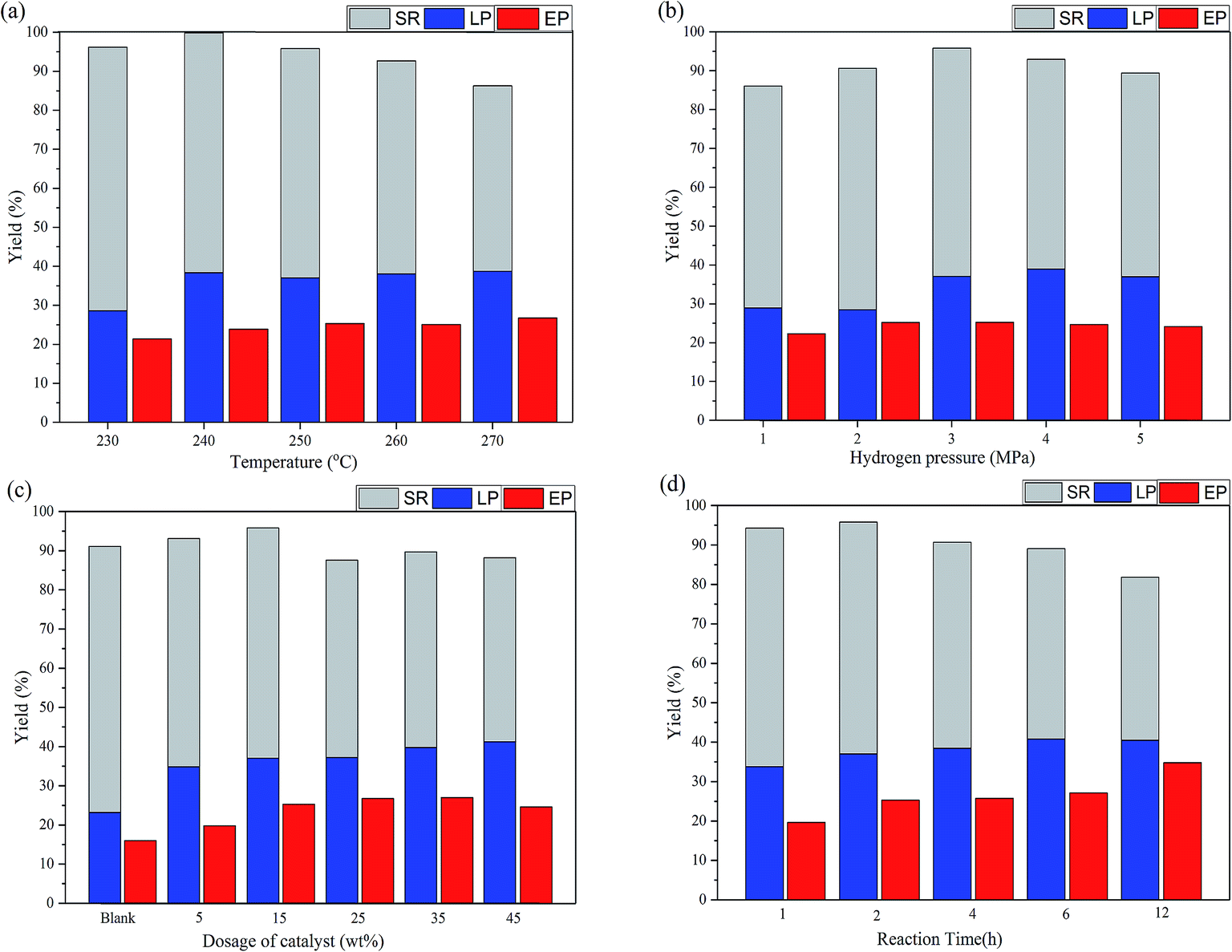
The depolymerization of native corn stover lignin was studied at various reaction conditions, including the reaction temperature, the initial hydrogen pressure, the dosage of catalyst, and the reaction time. In particular, the maximum phenolic monomer yield was 19.18% (260 °C, 3 MPa H2, 15 wt% MoS2, 2 h) and the maximum EP yield was 34.80% (250 °C, 3 MPa H2, 15 wt% MoS2). Collectively, the LP, EP, and SR yields revealed the liquefaction degree of the corn stover, the depolymerization degree of lignin, and the sugar retention of the corn stover, respectively. The phenolic monomer yield was not only dependant on the delignification of the corn stover and the depolymerization of lignin, but was also affected by the HDO and repolymerization of the monomer products, which constantly occurred in competition with the lignin-first process. Moreover, the LP yield was beyond the total lignin content in corn stover (21.10%), suggesting that not only lignin, but also the carbohydrate components were liquefied and that would be proven by the characterization of the liquid products and the chemical components analysis of the products.
The effect of the reaction temperature was significant on the lignin-first process as shown in Fig. 3 and Table 1. The LP yield was stable at around 38% at over 240 °C, indicating that more volatile gas products were released. The yield of total phenolic monomers was changed from 16.69% to 19.18% as the temperature rose from 230 °C to 260 °C and slightly down to 16.79% when the temperature increased up to 270 °C. This could be attributed to the increase of the depolymerization through the cleavage of the β-O-4 linkages in supercritical methanol as the temperature increased up to 260 °C. Moreover, the repolymerization reactions might be more dominant as the reaction temperature increased from 260 to 270 °C, resulting in the decline of the phenolic monomer yield. The decline in the yield of both guaiacols and p-hydroxyphenyl phenols was in accordance with this tendency, while the yield of syringols maintained at around 4%. This change could be ascribed to the active opening at 5-position on the aromatic ring, which made the G and H-type units prone to the repolymerization reactions at a higher temperature comparing with that for the S units.36,37
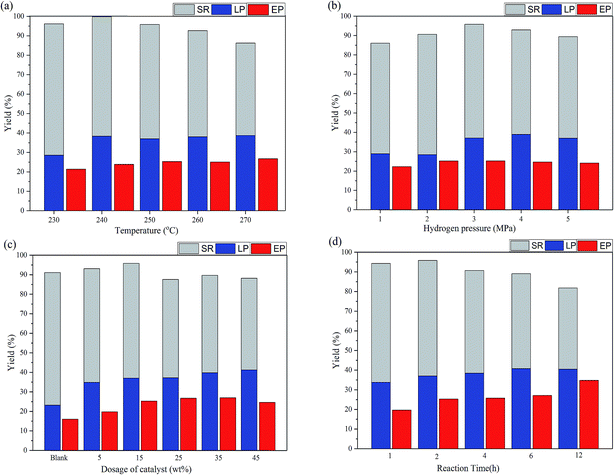 |
| | Fig. 3 Conversion of corn stover at different conditions: (a) the effect of temperature (2.0 g corn stover, 40 mL methanol, 0.3 g MoS2, 3 MPa H2, and 2 h); (b) the effect of pressure (2.0 g corn stover, 40 mL methanol, 0.3 g MoS2, 250 °C, and 2 h); (c) the effect of the dosage of the catalyst (2.0 g corn stover, 40 mL methanol, 250 °C, 3 MPa H2, and 2 h); (d) the effect of the reaction time (2.0 g corn stover, 40 mL methanol, 0.3 g MoS2, 250 °C, and 3 MPa H2) (SR: solid residual corn stover, LP: liquid products, EP: ethyl acetate soluble products). | |
Table 1 The phenolic monomer yield at different reaction temperaturesa (°C)
| Reaction temperature (°C) |
Yield of phenolic monomer (%) |
| 1b |
2c |
3d |
4e |
5f |
6g |
7h |
8i |
9j |
10k |
SumG |
SumS |
SumH |
Sum |
| Condition: 2.0 g corn stover, 40 mL methanol, 0.3 g MoS2, 3 MPa H2, and 2 h. 1: 4-Ethylphenol. 2:2,3-Dihydrobenzofuran. The calibration factor of an internal standard method was calculated by an effective carbon number (ECN) method explained in the ESI. 3: 2-Methoxy-4-ethylphenol. 4: 2-Methoxy-4-propylphenol. 5: 2-Methoxy-4-propenylphenol. 6: Methyl 3-(4-hydroxyphenyl)propionate. The calibration factor of an internal standard method was calculated by an effective carbon number (ECN) method explained in the ESI. 7: 2,6-Dimethoxy-4-allylphenol. 8: 2,6-Dimethoxy-4-propylphenol. 9: Methyl trans-p-coumarate. The calibration factor of an internal standard method was calculated by an effective carbon number (ECN) method explained in the ESI. 10: Methyl trans-4-hydroxy-3-methoxycinnamate. |
| 230 |
1.95 |
0.93 |
0.43 |
2.38 |
1.71 |
0.75 |
0.83 |
3.51 |
2.27 |
1.93 |
6.45 |
4.34 |
4.97 |
16.69 |
| 240 |
2.38 |
1.08 |
0.95 |
2.81 |
1.86 |
0.00 |
0.61 |
3.80 |
2.10 |
1.88 |
7.50 |
4.41 |
4.48 |
17.47 |
| 250 |
2.63 |
0.61 |
1.3 |
3.75 |
1.55 |
1.45 |
0.64 |
3.01 |
2.02 |
1.51 |
8.11 |
3.65 |
6.10 |
18.47 |
| 260 |
3.18 |
0.31 |
1.32 |
3.92 |
1.54 |
2.09 |
0.00 |
3.12 |
2.06 |
1.64 |
8.42 |
3.12 |
7.33 |
19.18 |
| 270 |
3.89 |
0.23 |
1.93 |
1.47 |
1.29 |
1.81 |
1.50 |
2.43 |
1.22 |
1.02 |
5.71 |
3.93 |
6.92 |
16.79 |
Therefore, the lignin fragments were constantly generated, resulting in the yield of EP to rise slowly. Moreover, the SR yield declined to 47.56%, which was less than the sum of cellulose and hemicellulose (53.04%), indicating that the degradation of the carbohydrate components occurred extensively with the increase in temperature.
Compared to that of the reaction temperature, the effect of the initial hydrogen pressure was much more significant on the lignin-first process as shown in Fig. 3 and Table 2. The liquefaction of corn stover was enhanced when the harsh reaction conditions were employed. In particular, the LP yield increased clearly and the yield of the phenolic monomers rose sharply from 9.91% to 18.47% when the initial hydrogen pressure changed from 1 MPa to 3 MPa. As the pressure was further raised to 4 or 5 MPa, the yield remained at a stable level. This suggested that a certain hydrogen pressure had a facilitation effect on the delignification and the depolymerization of lignin because in the reductive depolymerization of lignin, the hydrogenolysis and hydrodeoxygenation reactions are the hydrogen-consuming processes.8 In addition, a part of cellulose or/and hemicellulose, particularly those in the amorphous carbohydrate fractions in corn stover, might be methylated and transformed into LP or EP due to methanolysis.38 Therefore, the LP and EP yields were beyond the total lignin content in corn stover (21.10%) and the SR yield declined under the more harsh conditions.
Table 2 The phenolic monomer yield at different initial hydrogen pressuresa (MPa)
| Initial hydrogen pressure (MPa) |
Yield of phenolic monomer (%) |
| 1b |
2c |
3d |
4e |
5f |
6g |
7h |
8i |
9j |
10k |
SumG |
SumS |
SumH |
Sum |
| Condition: 2.0 g corn stover, 40 mL methanol, 0.3 g MoS2, 250 °C, and 2 h. 1: 4-Ethylphenol. 2:2,3-Dihydrobenzofuran. The calibration factor of an internal standard method was calculated by an effective carbon number (ECN) method explained in the ESI. 3: 2-Methoxy-4-ethylphenol. 4: 2-Methoxy-4-propylphenol. 5: 2-Methoxy-4-propenylphenol. 6: Methyl 3-(4-hydroxyphenyl)propionate. The calibration factor of an internal standard method was calculated by an effective carbon number (ECN) method explained in the ESI. 7: 2,6-Dimethoxy-4-allylphenol. 8: 2,6-Dimethoxy-4-propylphenol. 9: Methyl trans-p-coumarate. The calibration factor of an internal standard method was calculated by an effective carbon number (ECN) method explained in the ESI. 10: Methyl trans-4-hydroxy-3-methoxycinnamate. |
| 1 |
1.51 |
0.6 |
1.48 |
1.56 |
0.71 |
0.60 |
0.00 |
1.42 |
1.10 |
0.93 |
4.68 |
1.42 |
3.21 |
9.91 |
| 2 |
2.49 |
0.46 |
1.06 |
2.92 |
1.23 |
1.16 |
0.54 |
2.50 |
2.05 |
1.48 |
6.69 |
3.04 |
5.70 |
15.89 |
| 3 |
2.63 |
0.61 |
1.30 |
3.75 |
1.55 |
1.45 |
0.64 |
3.01 |
2.02 |
1.51 |
8.11 |
3.65 |
6.10 |
18.47 |
| 4 |
3.43 |
0.52 |
1.57 |
2.73 |
1.43 |
1.74 |
0.79 |
2.79 |
1.59 |
1.21 |
6.94 |
3.58 |
6.76 |
17.80 |
| 5 |
3.03 |
0.41 |
1.42 |
3.39 |
1.43 |
1.77 |
0.87 |
2.95 |
1.89 |
1.50 |
7.74 |
3.82 |
6.69 |
18.66 |
Together with the effects of the reaction temperature and initial hydrogen pressure, the influences of the dosage of catalyst and the reaction time were also investigated. As summarized in Table 3, the yield of the phenolic monomers was stable at around 18% with the presence of MoS2 in the reaction. The yield was three times higher than that for the blank reaction, indicating that MoS2 played a positive role in the lignin-first process. The sulfur vacancies in MoS2 could effectively adsorb oxygen in β-O-4 and other oxygen-containing linkages, thus accelerating the bond cleavage and leading to the production of monomers.32 The monomeric products were stabilized through hydrogenation,23,32 which would be proven later by NMR characterization and the analysis of the chemical components of the solid residues. The HDO might be enhanced when the reaction time was prolonged. This phenomenon could be observed from the results displayed in Table 4, with the decline of the yield of the phenolic monomers, as well as in Fig. 3, with the increasing of the EP yield. Moreover, the increase of the LP yield and the decrease of the SR yield suggested the improvement of the liquefaction and the aggravation of the degradation of the carbohydrate components.
Table 3 The phenolic monomer yield at different dosages of catalysta (wt%)
| Dosage of catalyst (wt%) |
Yield of phenolic monomer (%) |
| 1b |
2c |
3d |
4e |
5f |
6g |
7h |
8i |
9j |
10k |
SumG |
SumS |
SumH |
Sum |
| Condition: 2.0 g corn stover, 40 mL methanol, 250 °C, 3 MPa H2, and 2 h. 1: 4-Ethylphenol. 2: 2,3-Dihydrobenzofuran. The calibration factor of an internal standard method was calculated by an effective carbon number (ECN) method explained in the ESI. 3: 2-Methoxy-4-ethylphenol. 4: 2-Methoxy-4-propylphenol. 5: 2-Methoxy-4-propenylphenol. 6: Methyl 3-(4-hydroxyphenyl)propionate. The calibration factor of an internal standard method was calculated by an effective carbon number (ECN) method explained in the ESI. 7: 2,6-Dimethoxy-4-allylphenol. 8: 2,6-Dimethoxy-4-propylphenol. 9: Methyl trans-p-coumarate. The calibration factor of an internal standard method was calculated by an effective carbon number (ECN) method explained in the ESI. 10: Methyl trans-4-hydroxy-3-methoxycinnamate. |
| 0 |
0.34 |
1.13 |
0.00 |
0.26 |
0.76 |
0.00 |
0.00 |
0.00 |
2.02 |
2.14 |
3.16 |
0.00 |
2.36 |
6.65 |
| 5 |
1.12 |
1.72 |
1.71 |
2.40 |
1.33 |
0.00 |
0.00 |
2.87 |
4.22 |
2.55 |
7.99 |
2.87 |
5.34 |
17.92 |
| 15 |
2.63 |
0.61 |
1.30 |
3.75 |
1.55 |
1.45 |
0.64 |
3.01 |
2.02 |
1.51 |
8.11 |
3.65 |
6.10 |
18.47 |
| 25 |
2.58 |
0.36 |
2.57 |
2.52 |
1.35 |
1.18 |
0.94 |
3.48 |
1.40 |
1.22 |
7.66 |
4.42 |
5.16 |
17.60 |
| 35 |
2.97 |
0.00 |
2.48 |
2.40 |
1.02 |
1.79 |
1.28 |
2.82 |
0.93 |
0.73 |
6.63 |
4.10 |
5.69 |
16.42 |
| 45 |
2.86 |
0.00 |
2.92 |
1.83 |
0.94 |
2.52 |
2.00 |
3.14 |
0.84 |
0.48 |
6.17 |
5.14 |
6.22 |
17.53 |
Table 4 The phenolic monomer yield at different reaction timesa (h)
| Reaction time (h) |
Yield of phenolic monomer (%) |
| 1b |
2c |
3d |
4e |
5f |
6g |
7h |
8i |
9j |
10k |
SumG |
SumS |
SumH |
Sum |
| Condition: 2.0 g corn stover, 40 mL methanol, 0.3 g MoS2, 250 °C, and 3 MPa H2. 1: 4-Ethylphenol. 2: 2,3-Dihydrobenzofuran. The calibration factor of an internal standard method was calculated by an effective carbon number (ECN) method explained in the ESI. 3: 2-Methoxy-4-ethylphenol. 4: 2-Methoxy-4-propylphenol. 5: 2-Methoxy-4-propenylphenol. 6: Methyl 3-(4-hydroxyphenyl)propionate. The calibration factor of an internal standard method was calculated by an effective carbon number (ECN) method explained in the ESI. 7: 2,6-Dimethoxy-4-allylphenol. 8: 2,6-Dimethoxy-4-propylphenol. 9: Methyl trans-p-coumarate. The calibration factor of an internal standard method was calculated by an effective carbon number (ECN) method explained in the ESI. 10: Methyl trans-4-hydroxy-3-methoxycinnamate. |
| 1 |
1.48 |
1.30 |
1.74 |
2.74 |
1.87 |
0.00 |
0.00 |
4.00 |
2.33 |
2.07 |
8.42 |
4.00 |
3.81 |
17.53 |
| 2 |
2.63 |
0.61 |
1.30 |
3.75 |
1.55 |
1.45 |
0.64 |
3.01 |
2.02 |
1.51 |
8.11 |
3.65 |
6.10 |
18.47 |
| 4 |
2.25 |
0.00 |
2.52 |
2.02 |
1.40 |
0.87 |
0.69 |
2.99 |
1.57 |
1.39 |
7.33 |
3.68 |
4.69 |
15.70 |
| 6 |
2.45 |
0.44 |
2.82 |
1.54 |
1.19 |
0.00 |
0.00 |
2.56 |
1.24 |
1.36 |
6.91 |
2.56 |
3.69 |
13.60 |
| 12 |
3.21 |
0.00 |
2.29 |
0.99 |
0.57 |
0.97 |
0.98 |
1.33 |
0.36 |
0.34 |
4.19 |
2.31 |
4.54 |
11.04 |
3.3. Characterization of liquid products
The chemical compositions of the liquid products (250 °C, 3 MPa H2, 15 wt% MoS2, 2 h) were further characterized by FT-IR and 1H–13C HSQC NMR in order to clarify the structural transformation and get a comprehensive understanding of the lignin-first catalytic depolymerization process.
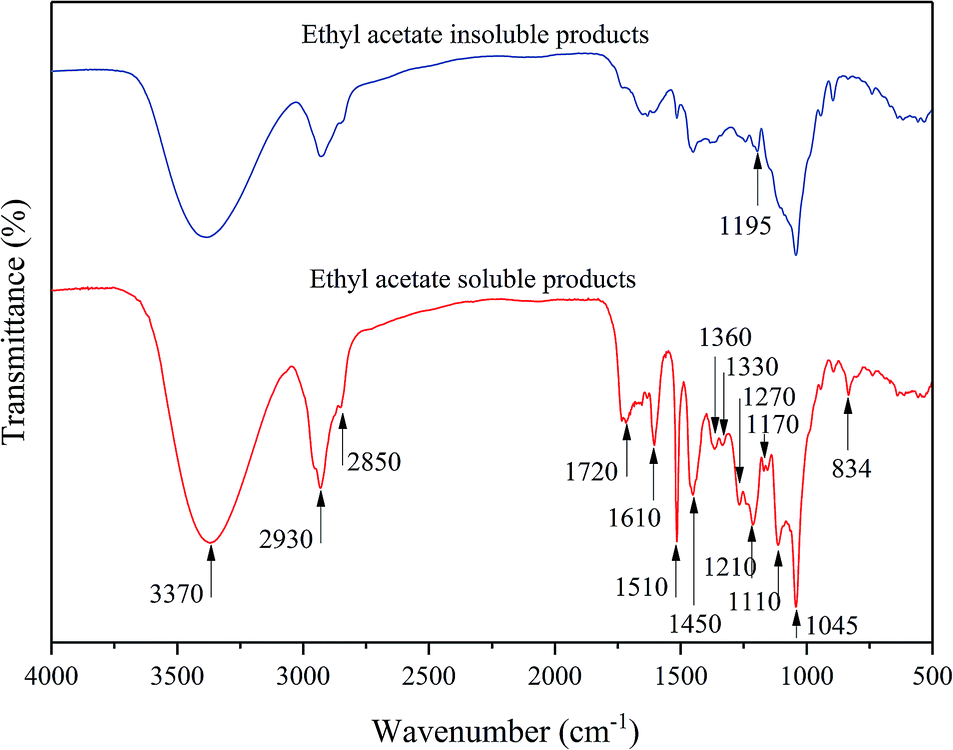
First, the EP (bio-oil) was rich in the phenolic monomers due to the abundant related functional structure observed in the FT-IR spectra shown in Fig. 4. The peak at 3370 cm−1 was attributed to the O–H stretching vibration, while the C–H symmetric and asymmetric vibrations in methyl and methylene group appeared at 2930 and 2850 cm−1, respectively.39 The 1720 cm−1 peak was assigned to the carbonyl stretching related to methyl p-coumarate. The strong absorption peak at 1610, 1510, and 1450 cm−1 in the bio-oil products suggested the existence of the benzene structure. The peak at 1360 cm−1 indicated the presence of the alkyl groups due to the sp3 C–H bending/rocking;8 the peak at 1330 cm−1 was derived from the phenolic OH (syringyl nuclei) absorptions; the guaiacyl ring breathing with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching appeared at 1270 cm−1.40 The peak at 1210 cm−1 was attributed to the asymmetric vibration of C–O–C in methylated ester. In particular, the peak at 1170 cm−1 was characterized for the ester-linked p-hydroxycinnamic acids.41 In addition, the C–O stretching in the alkoxy functional group was observed at 1110 cm−18 and the peak at 834 cm−1 was considered to be the signal of the aromatic C–H out-of-plane deformation.39 Moreover, carbohydrates were considered to be present in the ethyl acetate insoluble products because the phenolic absorptions, particularly the peaks corresponding to the benzene and alkoxyl functional structures, were weak or even faded, but O–H stretching at the β-glucosidic linkages was observed at 1195 cm−1.42 According to the FT-IR spectra, it was suggested that the liquid products of the depolymerization of native corn stover lignin were divided into two parts: one was the EP bio-oil rich of the lignin-degraded aromatic structures, including the phenolic monomers and oligomers, while the other was the ethyl acetate insoluble products mostly composed of polysaccharides. To further investigate the structure of the liquid products, 1H–13C HSQC NMR was also conducted.
O stretching appeared at 1270 cm−1.40 The peak at 1210 cm−1 was attributed to the asymmetric vibration of C–O–C in methylated ester. In particular, the peak at 1170 cm−1 was characterized for the ester-linked p-hydroxycinnamic acids.41 In addition, the C–O stretching in the alkoxy functional group was observed at 1110 cm−18 and the peak at 834 cm−1 was considered to be the signal of the aromatic C–H out-of-plane deformation.39 Moreover, carbohydrates were considered to be present in the ethyl acetate insoluble products because the phenolic absorptions, particularly the peaks corresponding to the benzene and alkoxyl functional structures, were weak or even faded, but O–H stretching at the β-glucosidic linkages was observed at 1195 cm−1.42 According to the FT-IR spectra, it was suggested that the liquid products of the depolymerization of native corn stover lignin were divided into two parts: one was the EP bio-oil rich of the lignin-degraded aromatic structures, including the phenolic monomers and oligomers, while the other was the ethyl acetate insoluble products mostly composed of polysaccharides. To further investigate the structure of the liquid products, 1H–13C HSQC NMR was also conducted.
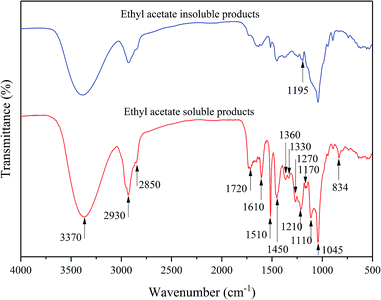 |
| | Fig. 4 FT-IR spectra of the ethyl acetate soluble and insoluble products. | |
The information of the chemical structural linkage of the liquid products was explained by the signals originated from the parts of the type-linkages of the lignin-carbohydrate complexes and the representative lignin monomer units in 1H–13C HSQC NMR (Fig. 5a and b for EP and Fig. 5c for the ethyl acetate-insoluble products). Four inter-linkages of lignin in the extractive-free corn stover (β-O-4 (53–60%), β-5 (27–29%), β-β (10–11%), and β-1 (3–7%)) were observed as reported earlier.43 First, the cleavage of the β-O-4 linkages occurred in the lignin-first process, which could be observed in the 1H–13C HSQC NMR characterization of liquid products with MoS2 catalysts, including ethyl acetate soluble products and ethyl acetate insoluble products as shown in Fig. 5a and c. Moreover, the yield of the phenolic monomers was much higher with the presence of MoS2 (Table 3, line 3) than that in the blank (Table 3, line 1). This suggested that the monomeric products were stabilized by MoS2 through hydrogenation, which was consistent with the previous study.23 For instance, the contours of the major linkages of β-O-4, including the signals of Cα–Hα (δC/δH, 72.9/4.30 ppm and 62.9/4.31 ppm for the γ-acylated lignin units), Cβ–Hβ (δC/δH, 83.6/4.40 ppm for G/S type and 86.3/4.10 for S/S type), and Cγ–Hγ (δC/δH, 60.5/3.50 ppm), disappeared after the catalytic process.42,44 Hence, it was concluded that the β-O-4 linkages of the lignin fragments in the reaction had been severely broken. Moreover, the cleavage of the G/S and S/S type β-O-4 linkages could be responsible for the partial generation of the phenolic monomers. In addition, it was reported that the hydroxycinnamic acid moieties of corn lignin, including about 18% p-coumaric acid (pCA) and 3–4% ferulic acid (FA), were deposited on the cell wall of corn stover and linked through the ester and ether linkages.27,45 The γ-ester of LCCs present the contours at δC/δH 62–64/4.0–4.25 ppm. The benzyl ether structures between the lignin units and carbohydrates showed the signals at δC/δH 80–81/4.4–4.6 ppm and 80–81/5.0–5.2 ppm.43 Both ester and ether linkages were not found in the liquid products. Therefore, the formation of MpC and MF monomers could be related to the cleavage of the ester and ether bonds. Moreover, the primary linkages retained in the liquid products were considered to be the β-5 linkages and the β-β linkages because Cβ–Hβ and Cγ–Hγ, correlated to the β-5 linkages, were distinguished at δC/δH 51.8/3.58 ppm and 61.6/3.43 ppm, respectively, and the signals for Cγ–Hγ in the β-β linkages were located at δC/δH 66.1/3.73 ppm.43 Moreover, there was only a trace of the β-1 linkages that was observed at δC/δH 79.1/3.97 ppm assigned to the Cβ–Hβ signals due to its relatively low content in the raw materials.43 Moreover, other types of the LCCs linkages and phenyl glycoside linkages were detected at δC/δH 100.4/4.51 ppm and 102.7/4.63 ppm, respectively,46 and β-D-xylopyranoside (βX) presented the contours at δC/δH 102.2/4.24 ppm, 73.6/2.98 ppm, 73.9/3.35 ppm, 75.5/3.74 ppm, and 61.6/3.43 ppm, which referred to the C1–H1, C2–H2, C3–H3, C4–H4, and C5–H5, respectively. The signals related to carbohydrates indicated that there were carbohydrates reserved in LP together with lignin oligomers. The results also confirmed the previous discussion that not only lignin but also the carbohydrate components were liquefied.
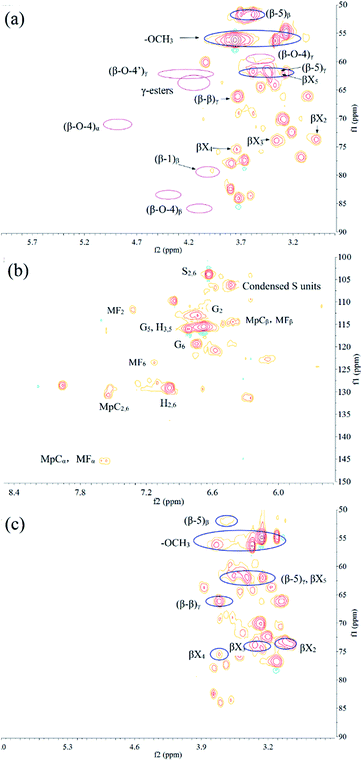 |
| | Fig. 5 1H–13C HSQC-NMR spectra of the liquid products: (a), (b) ethyl acetate soluble products, and (c) ethyl acetate insoluble products (the detailed typical structure refer to Fig. S1†). | |
In the aromatic regions of the 1H–13C HSQC NMR spectra of EP (Fig. 5b), different lignin units (G, S, and H type) of the phenolic units are clearly shown. The spectra of the ethyl acetate-insoluble products are blank, suggesting that the phenolic components were absent. C2,6–H2,6 and C3,5–H3,5 correlated to the H units at δC/δH 128.9/6.99 ppm and 115.5/6.68 ppm, respectively. In particular, MpC showed the signals for C2,6–H2,6, Cα–Hα, and Cβ–Hβ at δC/δH 130.7/7.54 ppm, 145.4/7.56 ppm, and 114.4/6.41 ppm, respectively. C2–H2 and C6–H6 in the G units were observed at δC/δH 113.2/6.73 ppm and 119.3/6.74 ppm, respectively. C2–H2 and C6–H6 correlated to MF at δC/δH 111.8/7.23 ppm and 123.5/7.13 ppm, respectively, while the signals of Cα–Hα and Cβ–Hβ in MF were overlapped with those of MpC. The S units related to C2,6–H2,6 displayed contours at δC/δH 103.7/6.63 ppm and the condensed S units of oligomers showed contours at δC/δH 106.0/6.43 ppm. The characterization of the aromatic regions supported the FT-IR spectra results, which suggested that the EP was rich in the phenolic monomers and oligomers. It was also in good agreement with the previous reports.43,44,46
3.4. Chemical components analysis of solid residues and liquid products
Typically, corn stover (2.0 g), MoS2 (0.3 g), and methanol (40 mL) were mixed and H2 was purged into the mixture at 3 MPa. The reaction was carried out at 250 °C for 2 h. The solid residues and the liquid products were further analysed. First, the determination of the solid residue suggested that, based on the corn stover feedstocks before the reaction, 90.31% glucan, 45.44% xylan, and 25.65% lignin were reserved in the residues, indicating that almost all of the glucan and about half of the xylan were reserved in the solid residues, while about 75% lignin was dissolved in methanol and depolymerized into the fragments, such as monomers and oligomers. In contrast, 76.91% glucan, 67.20% xylan, and 46.86% lignin were reserved in the solid residues at the same condition in the blank reaction. Considering the cleavage of the β-O-4 linkages observed in the 1H–13C HSQC NMR, it was concluded that the MoS2 catalyst promoted the delignification and aggravated the degradation of amorphous xylan at the same time. However, compared with the blank reaction, the reaction with MoS2 produced higher glucan content in the solid residues, which was interesting and perplexing. To figure out the loss of carbohydrates in the presence of MoS2, particularly xylan, the liquid products were extracted with water after the solvent was removed. Surprisingly, no monomeric carbohydrates were detected. Hence, EP and the ethyl acetate-insoluble products were collected and hydrolyzed by 4 wt% H2SO4 at 120 °C for 60 min and 13.41% and 17.42% xylan, respectively, were detected. These results suggested that some of the liquefied xylan could be methylated. Moreover, some of the methylated xylan with a strong molecular polarity was insoluble in ethyl acetate, while the rest might be further subjected to side reactions due to the abundance of active hydroxyl groups and high temperature. The distribution of acid hydrolyzed xylan in LP was also consistent with the contours of the β-X chemical bonds in the 1H–13C HSQC NMR spectra.
3.5. Model compounds reactions and potential mechanism
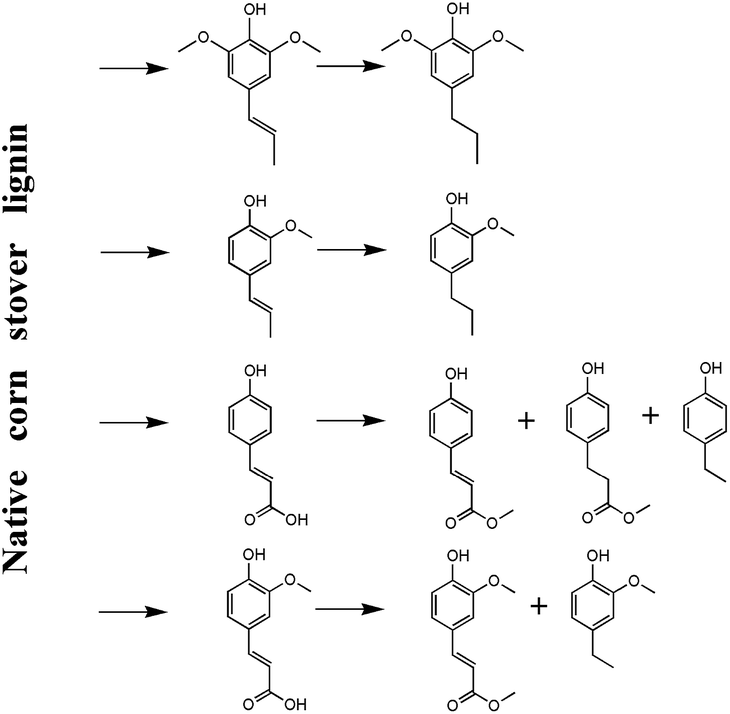
To figure out the potential mechanism of the generation of phenolic monomers, the high-content hydroxycinnamic acid moieties, including pCA and FA, in corn stover were used in the model reaction. The product yield was calculated by mole yield. First, 17.58% 4-ethylphenol and 14.85% MpC were produced after the model pCA reacted for 2 h and the mole yield respectively increased to 39.55% and decreased to 5.98% when the reaction time prolonged to 6 h. Second, 20.42% 2-methoxy-4-ethylphenol and 11.57% MF was generated when FA was used in the model reaction for 2 h. Similarly, 23.97% and 1.09% mole yield was respectively reached after 6 h, which proved the increase of the yield of 4-ethylphenol and 2-methoxy-4-ethylphenol in the lignin-first process of corn stover when the harsh reaction conditions were chosen as summarized in Tables 3 and 4.
Collectively, the potential mechanism can be explained in Fig. 6. In the lignin-first process, the native corn stover lignin can be partially dissolved in supercritical methanol together with amorphous xylan and the dissolving limitation can be promoted by a MoS2 catalyst through the cleavage of the β-O-4, γ-ester, and benzyl ether linkages, resulting in the release of pCA and FA monomer. Moreover, the dissolved lignin fragments will be further subjected to a hydrocracking reaction to be depolymerized into the phenolic monomers with MoS2. In particular, the hydroxycinnamic acid moieties (mainly pCA and FA) can further transform into 4-ethylphenol, 2-methoxy-4-ethylphenol, and methyl esters. Moreover, the 2-methoxy-4-propylphenol production can be assigned to the hydrogenation of 2-methoxy-4-propenylphenol, while the other products can be released from the cleavage of the G/S and S/S type β-O-4 linkages in native lignin.24
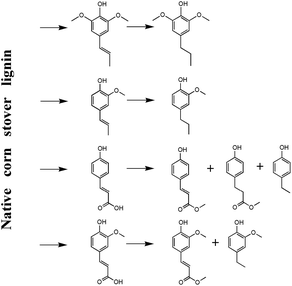 |
| | Fig. 6 The potential mechanism of the native corn stover lignin depolymerization. | |
3.6. Catalyst recycling
After the catalytic depolymerization reaction of corn stover, the residues were washed by adding water and filtered through a 100-mesh screen for several times. The filtrate containing MoS2 was collected by vacuum filtering and drying. The monomer yield decreased from 18.47% to 15.97% at the second use of the catalyst and dropped to 14.30% at the fifth reaction as summarized in Table S2.† Therefore, MoS2 was considered to be relatively effective for the depolymerization of native corn stover lignin. The fading of the (001) and (110) diffraction planes as shown in the XRD spectra (Fig. 1a) and the increase of the oxygen content detected by XPS together with the loss of the acidity of the recycled MoS2 after five times (Fig. S2†) could be responsible for the partial decline of the phenolic monomer yield.
4. Conclusion
The non-noble metal catalyst MoS2 played a positive role in the depolymerization of native corn stover lignin by lignin-first biorefinery. The yield of the phenolic monomers was enhanced from 6.65% to 18.47% with the MoS2 catalyst at 250 °C in supercritical methanol. About 75% lignin was degraded due to the cleavage of the β-O-4, γ-ester, and benzyl ether linkages with more than 90% glucan reserved in the solid residues. It has been proposed that the depolymerization of the hydroxycinnamic acid moieties primarily produces some phenolic monomers, such as 4-ethylphenol and 2-methoxy-4-ethylphenol as well as the methyl esters. The MoS2 catalyst still exhibited a relatively effective performance with 14.30% phenolic monomer yield after the fifth run.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was financially supported by the Program of National Natural Science Foundation of China (51476178 and 51536009), the National Key Technology R&D Program of China (No. 2015BAD15B06).
References
- T. E. Amidon and S. Liu, Biotechnol. Adv., 2009, 27, 542–550 CrossRef CAS PubMed
 .
. - C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed .
- T. Renders, S. Van den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 CAS .
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed .
- M. Kleinert and T. Barth, Energy Fuels, 2008, 22, 1371–1379 CrossRef CAS .
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed .
- E. Sjöström and R. Alén, Analytical methods in wood chemistry, pulping, and papermaking, Springer Science & Business Media, 2013 Search PubMed .
- R. Shu, J. Long, Y. Xu, L. Ma, Q. Zhang, T. Wang, C. Wang, Z. Yuan and Q. Wu, Bioresour. Technol., 2016, 200, 14–22 CrossRef CAS PubMed .
- J. Wang, W. Li, H. Wang, Q. Ma, S. Li, H.-m. Chang and H. Jameel, Bioresour. Technol., 2017, 243, 100–106 CrossRef CAS PubMed .
- R. Shu, J. Long, Z. Yuan, Q. Zhang, T. Wang, C. Wang and L. Ma, Bioresour. Technol., 2015, 179, 84–90 CrossRef CAS PubMed .
- J. Yang, L. Zhao, S. T. Liu, Y. Y. Wang and L. Y. Dai, Bioresour. Technol., 2016, 212, 302–310 CrossRef CAS PubMed .
- W. Xu, S. J. Miller, P. K. Agrawal and C. W. Jones, ChemSusChem, 2012, 5, 667–675 CrossRef CAS PubMed .
- L. Chen, T. I. Koranyi and E. J. M. Hensen, Chem. Commun., 2016, 52, 9375–9378 RSC .
- J. Horacek, F. Homola, I. Kubickova and D. Kubicka, Catal. Today, 2012, 179, 191–198 CrossRef CAS .
- C. R. Kumar, N. Anand, A. Kloekhorst, C. Cannilla, G. Bonura, F. Frusteri, K. Barta and H. J. Heeres, Green Chem., 2015, 17, 4921–4930 RSC .
- C. Xu and F. Ferdosian, Conversion of Lignin into Bio-Based Chemicals and Materials, Springer, 2017 Search PubMed .
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed .
- W. Schutyser, S. Van den Bosch, T. Renders, T. De Boe, S. F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Green Chem., 2015, 17, 5035–5045 RSC .
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 CAS .
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 CAS .
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L.-t. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef CAS PubMed .
- X. Huang, O. M. Morales Gonzalez, J. Zhu, T. I. Koranyi, M. D. Boot and E. J. M. Hensen, Green Chem., 2017, 19, 175–187 RSC .
- S. Van den Bosch, T. Renders, S. Kennis, S. F. Koelewijn, G. Van den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin, J. M. Thevelein, W. Schutyser and B. F. Sels, Green Chem., 2017, 19, 3313–3326 RSC .
- E. M. Anderson, R. Katahira, M. Reed, M. G. Resch, E. M. Karp, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2016, 4, 6940–6950 CrossRef CAS .
- X. Huang, X. Ouyang, B. S. Hendriks, O. M. M. Gonzalez, J. Zhu, T. I. Koranyi, M. D. Boot and E. J. M. Hensen, Faraday Discuss., 2017, 202, 141–156 RSC .
- F. Lu and J. Ralph, J. Agric. Food Chem., 1999, 47, 1988–1992 CrossRef CAS PubMed .
- R. Sun, X. F. Sun, S. Q. Wang, W. Zhu and X. Y. Wang, Ind. Crops Prod., 2002, 15, 179–188 CrossRef CAS .
- J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC .
- A. Wang and T. Zhang, Acc. Chem. Res., 2013, 46, 1377–1386 CrossRef CAS PubMed .
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed .
- B. Yoosuk, J. H. Kim, C. Song, C. Ngamcharussrivichai and P. Prasassarakich, Catal. Today, 2008, 130, 14–23 CrossRef CAS .
- C. Zhang, J. Lu, X. Zhang, K. MacArthur, M. Heggen, H. Li and F. Wang, Green Chem., 2016, 18, 6545–6555 RSC .
- N. Li, L. Wei, R. bibi, L. Chen, J. Liu, L. Zhang, Y. Zheng and J. Zhou, Fuel, 2016, 185, 532–540 CrossRef CAS .
- W. Wang, L. Li, K. Wu, G. Zhu, S. Tan, W. Li and Y. Yang, RSC Adv., 2015, 5, 61799–61807 RSC .
- Q. Liu, W. Li, Q. Ma, S. An, M. Li, H. Jameel and H.-m. Chang, Bioresour. Technol., 2016, 211, 435–442 CrossRef CAS PubMed .
- M. M. Jensen, R. B. Madsen, J. Becker, B. B. Iversen and M. Glasius, J. Anal. Appl. Pyrolysis, 2017, 126, 371–379 CrossRef CAS .
- J. Li, G. Henriksson and G. Gellerstedt, Bioresour. Technol., 2007, 98, 3061–3068 CrossRef CAS PubMed .
- Y. Ishikawa and S. Saka, Cellulose, 2001, 8, 189–195 CrossRef CAS .
- S.-N. Sun, M.-F. Li, T.-Q. Yuan, F. Xu and R.-C. Sun, Ind. Crops Prod., 2012, 37, 51–60 CrossRef CAS .
- N. Kobayashi, N. Okada, A. Hirakawa, T. Sato, J. Kobayashi, S. Hatano, Y. Itaya and S. Mori, Ind. Eng. Chem. Res., 2008, 48, 373–379 CrossRef .
- C. Jose, A. Gutiérrez, I. M. Rodríguez, D. Ibarra and A. T. Martinez, J. Anal. Appl. Pyrolysis, 2007, 79, 39–46 CrossRef .
- S. Yang, Y. Zhang, W. Yue, W. Wang, Y.-Y. Wang, T.-Q. Yuan and R.-C. Sun, Biotechnol. Biofuels, 2016, 9, 238 CrossRef PubMed .
- D. Y. Min, H. Jameel, H. M. Chang, L. Lucia, Z. G. Wang and Y. C. Jin, RSC Adv., 2014, 4, 10845–10850 RSC .
- C. Peng, Q. Chen, H. Guo, G. Hu, C. Li, J. Wen, H. Wang, T. Zhang, Z. K. Zhao, R. Sun and H. Xie, ChemCatChem, 2017, 9, 1135–1143 CrossRef CAS .
- J. Ralph, Phytochem. Rev., 2010, 9, 65–83 CrossRef CAS .
- S. Yang, T.-Q. Yuan and R.-C. Sun, ACS Sustainable Chem. Eng., 2016, 4, 1006–1015 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11947j |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *e,
Qi Zhang
*e,
Qi Zhang