
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTerminal aspartic acids promote the self-assembly of collagen mimic peptides into nanospheres†
Linyan Yaoa,
Manman Hea,
Dongfang Lia,
Jing Tiana,
Huanxiang Liu b and
Jianxi Xiao*a
b and
Jianxi Xiao*a
aState Key Laboratory of Applied Organic Chemistry, Key Laboratory of Nonferrous Meta Chemistry and Resources Utilization of Gansu Province, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: xiaojx@lzu.edu.cn
bSchool of Pharmacy, Lanzhou University, Lanzhou 730000, P. R. China
First published on 10th January 2018
Abstract
The development of novel strategies to construct collagen mimetic peptides capable of self-assembling into higher-order structures plays a critical role in the discovery of functional biomaterials. We herein report the construction of a novel type of amphiphile-like peptide conjugating the repetitive triple helical (GPO)m sequences characteristic of collagen with terminal hydrophilic aspartic acids. The amphiphile-like collagen mimic peptides containing a variable length of (Gly-Pro-Hyp)m sequences consistently generate well-ordered nanospherical supramolecular structures. The C-terminal aspartic acids have been revealed to play a determinant role in the appropriate self-assembly of amphiphile-like collagen mimic peptides. Their presence is a prerequisite for self-assembly, and their lengths could modulate the morphology of final assemblies. We have demonstrated for the first time that amphiphile-like collagen mimic peptides with terminal aspartic acids may provide a general and convenient strategy to create well-defined nanostructures in addition to amphiphile-like peptides utilizing β-sheet or α-helical coiled-coil motifs. The newly developed assembly strategy together with the ubiquitous natural function of collagen may lead to the generation of novel improved biomaterials.
Introduction
The multi-hierarchical self-assembly of collagen molecules into triple helices and higher-order structures is of pivotal importance in physiology and pathology.1,2 Collagen, the most abundant protein in the human body, provides a molecular scaffold for structural integrity and mechanical strength.3 Abnormalities in collagen structure are associated with a variety of connective tissue disorders such as osteogenesis imperfecta (OI), Ehlers–Danlos syndrome, and Alport syndrome.4,5 The unique triple helix structure of collagen results from its characteristic repetitive (Gly-X-Y)n amino acid sequences. The close packing of three chains requires Gly to be every third residue, while the hydrogen bonding between the amide proton of Gly and the carbonyl oxygen of a nearby X-position residue is critical in defining the triple helix conformation.6–8 Imino acids Pro and Hyp are frequently found in the X and Y positions, respectively, enhancing the stability of the triple helix.Model peptides with the repetitive (Gly-X-Y)n amino acid sequences have been constructed to investigate collagen structure and self-assembly.6,9–11 A variety of approaches such as electrostatic interactions,12–14 hydrophobic interactions,15,16 π–π stacking,17 π–cation interactions,18 triple helical nucleation19 and metal–ligand interactions20–27 have been harnessed to facilitate the self-assembly of collagen mimic peptides into a wide range of supramolecular structures including fibers, meshes, microflorettes, nanodiscs, and nanosheets. The development of novel strategies to create collagen mimic peptides capable to self-assemble into higher-order structures plays a critical role in the discovery of functional biomaterials.
Amphiphilic peptides have shown fascinating ability to spontaneously aggregate into a large variety of supramolecular self-assembled structures such as nanofibers, nanoribbons, nanotapes, nanotubes and nanovesicles.28–34 Peptide amphiphiles (PA) are often designed to mimic surfactant molecules containing a single polar head composed of hydrophilic amino acids and a longer hydrophobic tail consisting of nonpolar amino acids or alkyl chains.33 Heteropolymeric peptide amphiphiles containing collagen sequences have been constructed by Fields' and Tirrell's groups, and they have shown enhanced triple helix stability compared to the analogous peptide and they can be applied for surface modification and cell receptor binding.35,36 Recently, a peptide amphiphile constructed by conjugating a single hydrophobic alkyl chain with a collagen mimic peptide has been shown to form self-assembled nanofibers.37
Amphiphile-like peptides containing a hydrophilic head composed of one or two charged amino acids and a hydrophobic tail composed of several consecutive amino acids of increased hydrophobicity have recently been shown to spontaneously self-assemble to form well-ordered nanostructures including micelles, nanovesicles, and nanotubes.30,38,39 Peptides (A6D, V6D, V6D2 and L6D2) comprising one or two terminal aspartic acids and a tail of hydrophobic amino acids such as alanine, valine, or leucine consistently self-assembled to form nanotubes and nanovesicles.30 Peptides (G4D2, G6D2, G8D2 and G10D2) containing variable glycines as the component of the hydrophobic tail and aspartic acids as the hydrophilic head also underwent self-assembly to form ordered nanostructures.39 These studies suggested that peptides containing hydrophilic aspartic acids at one terminus and consecutive amino acids of increased hydrophobicity at the other terminus may provide a convenient approach to construct self-assembled nanostructures.
We herein report the design of amphiphile-like collagen mimic peptides consisting of only natural amino acids, which facilitate easy synthesis and high biocompatibility. The peptides contain a single hydrophilic head composed of charged aspartic acids conjugated with the repetitive triple helical (GPO)m sequences of increased hydrophobicity. We have demonstrated for the first time that these amphiphile-like collagen mimic peptides with terminal aspartic acids may provide a general and convenient strategy to construct well-defined nanostructures.
Experimental section
Peptide synthesis
Peptides were synthesized in-house by standard Fmoc solid phase synthesis method. Briefly, the peptides were assembled on 2-chlorotrityl chloride resin (substitution level = 0.88 mmol g−1 resin) at a 0.1 mmol scale. Stepwise couplings of amino acids were accomplished using a double coupling method with Fmoc-amino acids (4 eq.), DIEA (6 eq.) and activator reagents (HBTU + HOBt 0.66 mmol ml−1, 4 eq.). The reaction mixture was washed with DMF (3 × 5 ml) and DCM (3 × 5 ml) after each step of coupling, and the Fmoc protection group was removed with 20% piperidine in DMF. Test reagent (2% ethanol DMF, 2% chloranil DMF) was used to ensure the successful completion of each coupling reaction and Fmoc deprotection. The peptides were cleaved from the resin and the tBu groups were deprotected by treating the resin with TFA/TIS/H2O (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5) for 3 h. The peptides were collected by precipitation with cold Et2O and centrifugation. They were re-suspended in cold Et2O, sonicated and centrifuged again. Crude products were then dissolved in water, and lyophilized. The peptides were purified using reverse phase HPLC, and their purity was confirmed by mass spectrometry. m/z calculated 2119.3 [M + H]+ for (GPO)7D2, found 2118.9 [M + H]+; m/z calculated 1584.7 [M + H]+ for (GPO)5D2, found 1584.69 [M + H]+; m/z calculated 2653.9 [M + H]+ for (GPO)9D2, found 2653.2 [M + H]+; m/z calculated 1927.1 [M + K]+ for (GPO)7, found 1926.8 [M + K]+; m/z calculated 2502.6 [M + K]+ for (GPO)7D5, found 2504.0 [M + K]+.
:2.5:2.5) for 3 h. The peptides were collected by precipitation with cold Et2O and centrifugation. They were re-suspended in cold Et2O, sonicated and centrifuged again. Crude products were then dissolved in water, and lyophilized. The peptides were purified using reverse phase HPLC, and their purity was confirmed by mass spectrometry. m/z calculated 2119.3 [M + H]+ for (GPO)7D2, found 2118.9 [M + H]+; m/z calculated 1584.7 [M + H]+ for (GPO)5D2, found 1584.69 [M + H]+; m/z calculated 2653.9 [M + H]+ for (GPO)9D2, found 2653.2 [M + H]+; m/z calculated 1927.1 [M + K]+ for (GPO)7, found 1926.8 [M + K]+; m/z calculated 2502.6 [M + K]+ for (GPO)7D5, found 2504.0 [M + K]+.
Circular dichroism spectroscopy
CD spectra were acquired on an Aviv model 400 spectrophotometer (Applied Photophysics Ltd, England) with a Peltier temperature controller. Peptides were prepared at a concentration of 400 μM in 10 mM phosphate buffer at pH 7.0 or 200 mM glycine–HCl buffer at pH 3.0, respectively. The samples were equilibrated for at least 24 h at 4 °C prior to the CD measurements. Cells with a path length of 1 mm were used. Wavelength scans were conducted from 215 to 260 nm with a 0.5 nm increment per step and a 0.5 s averaging time. Thermal unfolding curves were measured by monitoring the amplitude of the characteristic CD peak at 225 nm, while the temperature was increased 0.3 °C min−1 from 4 °C to 70 °C with an equilibration time of 2 min at each temperature. The melting temperature (Tm) was calculated from the first derivative of the thermal unfolding curves.Dynamic light scattering
Dynamic light scattering (DLS) measurements were performed on a BI-200SM system (Brookhaven Instruments, USA) equipped with a 514 nm laser at a 90° scattering angle. Solutions of peptide (GPO)7D2 with a concentration of 0.5 mg ml−1 were prepared in 100 mM HEPES buffer (pH 7.0) at 4 °C. The peptide (GPO)7D2 solution was heated at 20 °C for 30 min to initiate the self-assembly, and then incubated at 4 °C for 60 h. DLS profiles were recorded for the peptide stored at 4 °C and that incubated at 4 °C for 60 h after the heating for 30 min at 20 °C.Transmission electron microscopy
TEM images were obtained on a JEM-2100 transmission electron microscope (JEOL, Japan). Solutions of peptides (GPO)7D2, (GPO)5D2, (GPO)9D2, (GPO)7 and (GPO)7D5 with a concentration of 0.5 mg ml−1 were prepared in 100 mM HEPES buffer, pH 7.0. The samples of peptide (GPO)7D2 were incubated at 4 °C for 60 h after heating at 20 °C for 30 min. The samples of peptide (GPO)5D2 and (GPO)7D5 were incubated at 4 °C without preheating. Peptide (GPO)9D2 was heated at 47 °C for 30 min and then incubated at 4 °C for 60 h. Peptide (GPO)7 was heated at 28 °C for 30 min and incubated at 4 °C for 60 h. A volume of 10 μl of the peptide solution was deposited on a copper grid coated with carbon film. The copper grid was negatively stained with 2.5% phosphotungstic acid (PTA). Images were recorded at 200 kV.Results and discussion
Design of collagen mimic peptides
We herein construct amphiphile-like peptides (GPO)mDn (m = 5, 7, 9; n = 0, 2, 5) incorporating hydrophilic aspartic acids at one terminus and a relatively hydrophobic tail of repetitive (GPO)m sequences at the other terminus (Fig. 1). The most widely used method to describe the hydrophobicity or hydrophilicity of a compound or peptide is the octanol–water partition coefficient logP. Here, to describe the relative hydrophobicity of two terminals of designed peptides, AlogP was calculated.40 AlogP of the amino acids or groups was calculated by summing the hydrophobic constant of each atom using Discovery Studio (version 2.5.5). The AlogP of amino acids Gly, Pro, Hyp and Asp was −1.145, −0.116, −1.207 and −2.753, respectively. A more negative value of AlogP represented a more hydrophilic amino acid, suggesting that Pro is more hydrophobic than Gly and Hyp, while all these three residues are more hydrophobic than Asp. The AlogP of amino acid groups GPO, GGG and DD was −2.511, −3.025 and −5.303, respectively, suggesting that all the residue groups GPO and GGG were more hydrophobic than DD. These results suggested that our constructed peptides (GPO)mD2 (m = 5, 7, 9) as well as previously reported peptides GnD2 (n = 4, 6, 8, 10) were amphiphile-like.
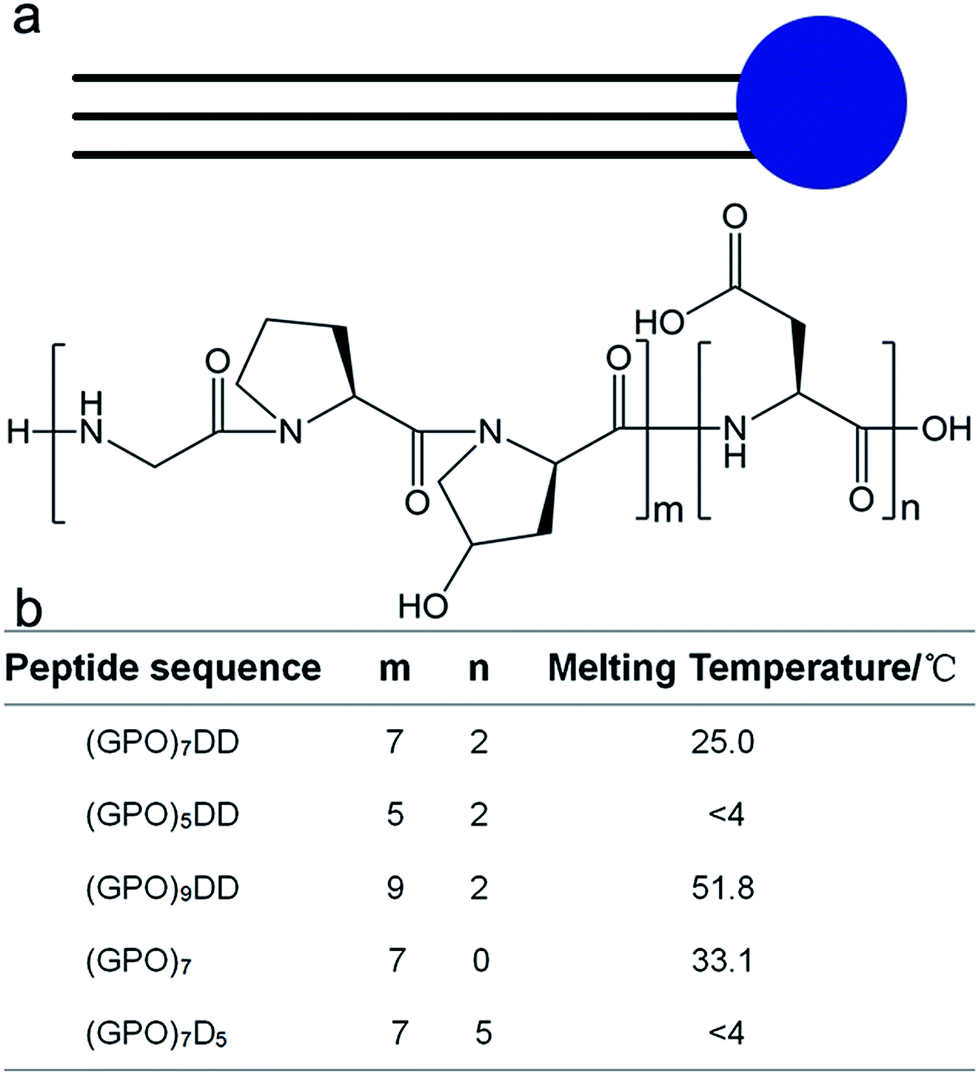 | ||
| Fig. 1 Design and thermal stability of collagen mimic peptides (GPO)mDn. The amphiphile-like peptides consist of hydrophilic aspartic acids Dn (n = 0, 2, 5) at C-terminus and repetitive triple helical (GPO)m sequences (m = 5, 7, 9) of increased hydrophobicity at N-terminus. | ||
Three peptides (GPO)7D2, (GPO)5D2, and (GPO)9D2 were constructed to investigate if the lengths of the repetitive GPO triplets affect the self-assembly of the peptides. The GPO triplet is the most abundant amino acid sequence in collagen, and is known to stabilize the triple helix structure. Two more peptides (GPO)7 and (GPO)7D5 were constructed to evaluate if the number of aspartic acids modulates the self-assembly of the peptides. All the peptides were composed of only natural amino acids, leading to high biocompatibility and easy synthesis.
Self-assembly of peptide (GPO)7DD
To mimic collagen, the constructed peptides should possess the characteristic collagen triple helix structure. The terminal negatively charged amino acids may provide peptide (GPO)7DD pH-responsive properties, and its pI was calculated as 3.6. We therefore characterized the peptide at two pHs: pH 7.0 at which the peptide was negatively charged, and pH 3.0 at which the peptide was almost neutral (Fig. 2a–f). Circular dichroism (CD) spectra of peptide (GPO)7DD exhibited a characteristic peak at 225 nm at both pH 7.0 and pH 3.0, indicating the formation of triple helix structure. Thermal unfolding studies showed that peptide (GPO)7DD formed a stable triple helix with a melting temperature (Tm) of 25.0 °C for pH 7.0 and 29.2 °C for pH 3, respectively. The CD results suggested that the peptide could form stable triple helix conformation under a wide range of pHs, while the repulsive electrostatic interactions of the terminal charged aspartic acids finely tuned the thermal stability of the peptide (Fig. 2a–f).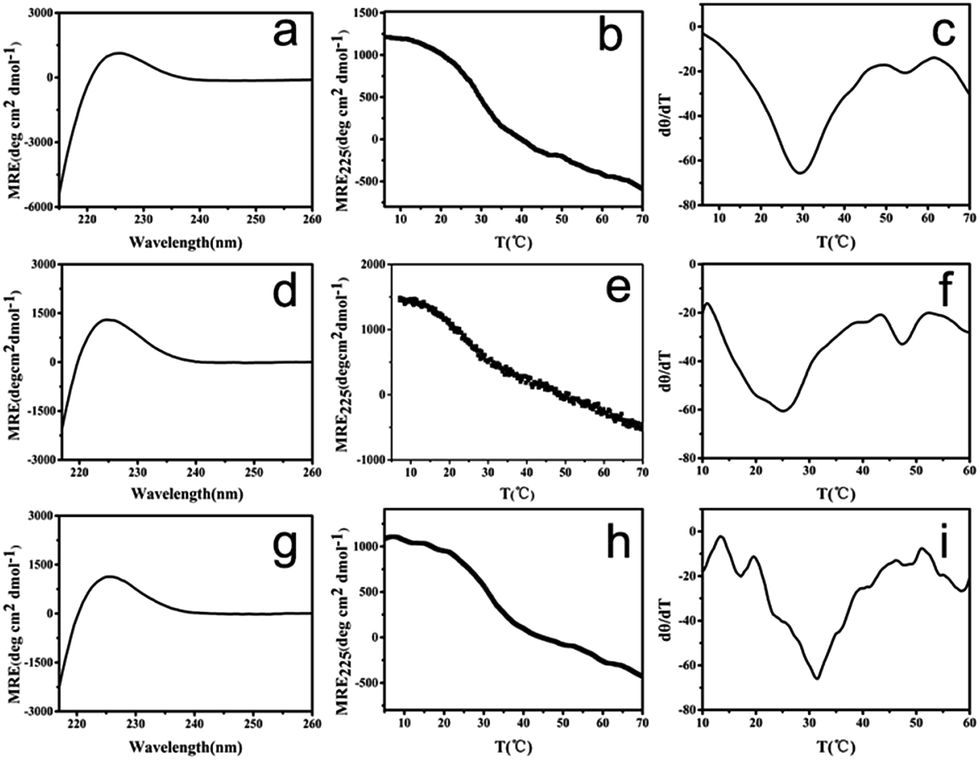 | ||
| Fig. 2 CD characterization of collagen mimic peptide (GPO)7DD. CD spectra (a), CD thermal unfolding (b) and the first derivative (d[θ]/dT) of the CD unfolding curve (c) of the unheated peptide at pH 3.0. CD spectra (d), CD thermal unfolding (e) and the first derivative (d[θ]/dT) of the CD unfolding curve (f) of the unheated peptide at pH 7.0. CD spectra (g), CD thermal unfolding (h) and the first derivative (d[θ]/dT) of the CD unfolding curve (i) of the preheated peptide at pH 7.0. | ||
The ability of peptide (GPO)7DD to self-assemble was monitored by dynamic light scattering (DLS). Peptide solutions with a concentration of 0.5 mg ml−1 were prepared, and maintained at 4 °C. The peptide solution was heated at 20 °C for 30 min to initiate the self-assembly, and incubated at 4 °C for 60 h. These conditions were optimized, as earlier studies have suggested that the temperature near Tm is a favoured condition for the self-assembly of collagen mimic peptides, since the loosening of the triple helix and the increased mobility of side chains may promote intermolecular interactions leading to supramolecular structures.41 The DLS data showed that the peptide stored at 4 °C (black) displayed a hydrodynamic radius of approximately 2 nm, suggesting no aggregate formation (Fig. S1†). The incubation at 4 °C for 60 h after preheating at 20 °C for 30 min (red) significantly increased the hydrodynamic radius to be ∼300 nm, indicating the formation of large assembled particles (Fig. S1†).
The morphology of the assembled materials formed by peptide (GPO)7DD was characterized by transmission electron microscopy (TEM). Peptide solutions of (GPO)7DD with a concentration of 0.5 mg ml−1 were prepared in 100 mM HEPES buffer, pH 7.0. TEM image of the peptide maintained at 4 °C without preheating indicated the absence of any assembled supramolecular structures (Fig. 3a). In contrast, TEM images of the peptide incubated at 4 °C for 60 h after preheating at 20 °C for 30 min resulted in relatively uniformly distributed nanospheres with an average diameter of ∼300 ± 10 nm (Fig. 3b–c). It suggested that the amphiphile-like collagen mimic peptides may provide an excellent strategy to create well-ordered nanospheres.
 | ||
| Fig. 3 TEM images of peptide(GPO)7DD maintained at 4 °C (a) or incubated at 4 °C for 60 h after preheating at 20 °C for 30 min (b and c). Peptide solutions of (GPO)7DD with a concentration of 0.5 mg ml−1 were prepared in 100 mM HEPES buffer, pH 7.0. | ||
The peptide solution incubated at 4 °C for 60 h after preheating at 20 °C for 30 min was also characterized by CD (Fig. 2g–i). The CD spectra exhibited a characteristic peak at 225 nm, indicating that the assembled peptide maintained the triple helix structure. Thermal unfolding studies further demonstrated that the assembled peptide formed a stable triple helix with a melting temperature (Tm) of 31.0 °C, which was 6 °C higher than the Tm of the free, un-assembled peptide (25.0 °C). These results indicated that the self-assembly of peptide (GPO)7DD into nanospheres stabilized the triple helix structure.
The effect of the (GPO)m sequences on the self-assembly of collagen mimic peptides
Peptides (GPO)5D2 and (GPO)9D2 were constructed to evaluate whether the lengths of the GPO triplets affect the self-assembly of the amphiphile-like collagen mimic peptides. Both peptides were prepared at a concentration of 400 μM in 10 mM phosphate buffer at pH 7.0. CD spectra of peptide (GPO)5D2 displayed a distinct peak at 225 nm at 4 °C, while its thermal unfolding studies indicated a linear decrease of the CD225 nm intensity during its melting, suggesting that peptide (GPO)5D2 may contain some triple helical species at 4 °C, but it could not form stable triple helix at higher temperatures (Fig. S2a–c†). Meanwhile, CD spectra of peptide (GPO)9D2 contained a characteristic peak at 225 nm at 4 °C, and its thermal unfolding studies indicated that peptide (GPO)9D2 formed a stable triple helix with Tm of 51.8 °C (Fig. S2d–f†).The morphology of the assembled materials formed by both peptides was characterized by transmission electron microscopy (TEM). Peptide solutions with a concentration of 0.5 mg ml−1 were prepared in 100 mM HEPES buffer, pH 7.0. Peptide (GPO)5DD was incubated at 4 °C for 60 h. Peptide(GPO)9DD was heated at 47 °C for 30 min and incubated at 4 °C for 60 h. The DLS data showed that both peptides displayed a large hydrodynamic radius after incubation, indicating the formation of assembled particles (Fig. S3†). TEM image of the assemblies of peptide (GPO)5DD revealed uniformly distributed nanospheres with an average diameter of ∼325 ± 10 nm (Fig. 4a and b), while TEM image of peptide (GPO)9DD showed nanospheres with an average diameter of ∼270 ± 10 nm (Fig. 4c and d). These results indicated that amphiphile-like collagen mimic peptides (GPO)mDD with a variable length of GPO triplets (m = 5, 7, 9) all resulted in relatively uniform nanospheres. As the lengths of the triple helix formed by peptides (GPO)5DD, (GPO)7DD and (GPO)9DD were gradually increased, the nanoparticle size of the assemblies became smaller. It implied that smaller collagen mimic peptides may be more flexible and easier to nucleate and aggregate. The lengths of the (GPO)m sequences may not affect the proper self-assembly of the peptides, while they may finely tune the size of the final assembled nanospheres.
 | ||
| Fig. 4 TEM images of amphiphile-like collagen mimic peptides (GPO)5DD (a and b) and (GPO)9DD (c and d). Peptide solutions with a concentration of 0.5 mg ml−1 were prepared in 100 mM HEPES buffer, pH 7.0. Peptide (GPO)5DD was incubated at 4 °C for 60 h. Peptide (GPO)9DD was heated at 47 °C for 30 min and incubated at 4 °C for 60 h. | ||
The effect of the aspartic acids on the self-assembly of collagen mimic peptides
Peptides (GPO)7 and (GPO)7D5 were designed to examine whether the number of aspartic acids at C-terminal affect the self-assembly of the amphiphile-like collagen mimic peptides. Both peptides with a concentration of 400 μM were prepared in 10 mM phosphate buffer, pH 7.0. CD spectra of peptide (GPO)7 showed a typical peak at 225 nm at 4 °C, and its thermal unfolding studies indicated that peptide (GPO)7 formed a stable triple helix with Tm of 33.1 °C (Fig. S4a–c†). Meanwhile, CD spectra of peptide (GPO)7D5 contained a characteristic peak at 225 nm at 4 °C, while its thermal unfolding studies showed a linear decrease of the CD225 nm intensity, suggesting that peptide (GPO)7D5 may possess some triple helical species at 4 °C, but it could not form stable triple helix at higher temperatures (Fig. S4d–f†).Peptide (GPO)7D5 was incubated at 4 °C for 60 h.
The morphology of the assembled materials formed by peptides (GPO)7 and (GPO)7D5 were characterized by transmission electron microscopy (TEM). Peptide solutions with a concentration of 0.5 mg ml−1 were prepared in 100 mM HEPES buffer, pH 7.0. Peptide (GPO)7 was heated at 28 °C for 30 min and incubated at 4 °C for 60 h, while peptide (GPO)7D5 was incubated at 4 °C for 60 h. TEM image of peptide (GPO)7 did not indicate the presence of any assemblies (Fig. 5a). In contrast, TEM image of peptide (GPO)7D5 revealed the formation of some fibrous assemblies (Fig. 5b). These results indicated that the C-terminal aspartic acids played a determinant role in the proper self-assembly of amphiphile-like collagen mimic peptides. The presence of C-terminal aspartic acids is required to initiate the self-assembly, while a proper number of aspartic acids is needed to modulate the morphology of final assemblies.
 | ||
| Fig. 5 TEM images of collagen mimic peptides (GPO)7 (a) and (GPO)7D5 (b). Peptide solutions with a concentration of 0.5 mg ml−1 were prepared in 100 mM HEPES buffer, pH = 7.0. Peptide (GPO)7 was heated at 28 °C for 30 min and incubated at 4 °C for 60 h. | ||
Conclusions
The construction of novel collagen mimetic peptides capable to self-assemble into well-defined supramolecular structures plays an essential role in the discovery of functional biomaterials. Amphiphile-like peptides have attracted extensive attention as molecular building blocks for the fabrication of higher-order structures and they have shown promising applications in the fields of bioengineering and biotechnology.42,43 A variety of supramolecular structures have been achieved utilizing amphiphile-like peptides composed of β-sheet or α-helical coiled-coil motifs.32,44,45We herein report the construction of amphiphile-like peptides consisting of the repetitive triple helical (GPO)m sequences characteristic of collagen and terminal hydrophilic aspartic acids. The amphiphile-like collagen mimic peptides consisting of (Gly-Pro-Hyp)m sequences of variable lengths consistently form well-ordered nanospherical supramolecular structures, indicating that the lengths of the (GPO)m sequences may not interfere the proper self-assembly of the peptides, while they could probably finely tune the size of the final assembled nanospheres. In contrast, the C-terminal aspartic acids played a determinant role in the appropriate self-assembly of amphiphile-like collagen mimic peptides, whose presence is a prerequisite for the self-assembly, and whose lengths could modulate the morphology of final assemblies.
We have demonstrated for the first time that amphiphile-like collagen mimic peptides with terminal aspartic acids may provide a general and convenient strategy to create well-defined nanostructures in addition to amphiphile-like peptides composed of β-sheet or α-helical coiled-coil motifs. The collagen mimic peptides are composed of only natural amino acids, which will facilitate easy synthesis as well as superior biocompatibility of the peptides for promising biological applications. The newly developed assembly strategy together with the ubiquitous natural function of collagen may lead to the generation of novel improved biomaterials.
Conflicts of interest
The authors declare no competing conflict of interests.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 21775059) and the Fundamental Research Funds for the Central Universities (Grant No. lzujbky-2016-k10).References
- J. A. Fallas, L. E. O'Leary and J. D. Hartgerink, Chem. Soc. Rev., 2010, 39, 3510–3527 RSC.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- C. M. Kielty and M. E. Grant, in Connective tissue and its heritable disorders, molecular, genetic and medical aspects, ed. P. M. Royes and B. U. Steinmann, Wiley Liss, New York, 2002, pp. 159–222 Search PubMed.
- J. Myllyharju and K. I. Kivirikko, Ann. Med., 2001, 33, 7–21 CrossRef CAS PubMed.
- J. C. Marini, A. Forlino, W. A. Cabral, A. M. Barnes, J. D. San Antonio, S. Milgrom, J. C. Hyland, J. Korkko, D. J. Prockop, A. De Paepe, P. Coucke, S. Symoens, F. H. Glorieux, P. J. Roughley, A. M. Lund, K. Kuurila-Svahn, H. Hartikka, D. H. Cohn, D. Krakow, M. Mottes, U. Schwarze, D. Chen, K. Yang, C. Kuslich, J. Troendle, R. Dalgleish and P. H. Byers, Hum. Mutat., 2007, 28, 209–221 CrossRef CAS PubMed.
- J. Xiao, X. Sun, B. Madhan, B. Brodsky and J. Baum, J. Biol. Chem., 2015, 290, 24201–24209 CrossRef CAS PubMed.
- B. Brodsky and A. V. Persikov, Adv. Protein Chem., 2005, 70, 301–339 CrossRef CAS PubMed.
- J. Xiao and J. Baum, J. Am. Chem. Soc., 2009, 131, 18194–18195 CrossRef CAS PubMed.
- B. Brodsky, G. Thiagarajan, B. Madhan and K. Kar, Biopolymers, 2008, 89, 345–353 CrossRef CAS PubMed.
- G. B. Fields, Org. Biomol. Chem., 2010, 8, 1237–1258 CAS.
- Y. Li and S. M. Yu, Curr. Opin. Chem. Biol., 2013, 17, 968–975 CrossRef CAS PubMed.
- L. E. O'Leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Nat. Chem., 2011, 3, 821 CrossRef PubMed.
- S. Rele, Y. H. Song, R. P. Apkarian, Z. Qu, V. P. Conticello and E. L. Chaikof, J. Am. Chem. Soc., 2007, 129, 14780 CrossRef CAS PubMed.
- T. Jiang, C. Xu, Y. Liu, Z. Liu, J. S. Wall, X. Zuo, T. Lian, K. Salaita, C. Ni, D. Pochan and V. P. Conticello, J. Am. Chem. Soc., 2014, 136, 4300–4308 CrossRef CAS PubMed.
- M. A. Cejas, W. A. Kinney, C. Chen, J. G. Vinter, H. R. Almond Jr, K. M. Balss, C. A. Maryanoff, U. Schmidt, M. Breslav, A. Mahan, E. Lacy and B. E. Maryanoff, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8513–8518 CrossRef CAS PubMed.
- K. McGuinness, I. J. Khan and V. Nanda, ACS Nano, 2014, 8, 12514–12523 CrossRef CAS PubMed.
- M. A. Cejas, W. A. Kinney, C. Chen, G. C. Leo, B. A. Tounge, J. G. Vinter, P. P. Joshi and B. E. Maryanoff, J. Am. Chem. Soc., 2007, 129, 2202–2203 CrossRef CAS PubMed.
- C. C. Chen, W. Hsu, T. C. Kao and J. C. Horng, Biochemistry, 2011, 50, 2381–2383 CrossRef CAS PubMed.
- B. Sarkar, L. E. O'Leary and J. D. Hartgerink, J. Am. Chem. Soc., 2014, 136, 14417–14424 CrossRef CAS PubMed.
- D. E. Przybyla, C. M. Rubert Perez, J. Gleaton, V. Nandwana and J. Chmielewski, J. Am. Chem. Soc., 2013, 135, 3418–3422 CrossRef CAS PubMed.
- M. M. Pires, D. E. Przybyla, C. M. Rubert Perez and J. Chmielewski, J. Am. Chem. Soc., 2011, 133, 14469–14471 CrossRef CAS PubMed.
- D. E. Przybyla and J. Chmielewski, J. Am. Chem. Soc., 2010, 132, 7866–7867 CrossRef CAS PubMed.
- M. M. Pires and J. Chmielewski, J. Am. Chem. Soc., 2009, 131, 2706–2712 CrossRef CAS PubMed.
- D. E. Przybyla and J. Chmielewski, J. Am. Chem. Soc., 2008, 130, 12610–12611 CrossRef CAS PubMed.
- M. M. Pires, D. E. Przybyla and J. Chmielewski, Angew. Chem., 2009, 48, 7813–7817 CrossRef CAS PubMed.
- M. He, L. Wang, J. Wu and J. Xiao, Chemistry, 2016, 22, 1914–1917 CrossRef CAS PubMed.
- M. M. Pires, J. Lee, D. Ernenwein and J. Chmielewski, Langmuir, 2012, 28, 1993–1997 CrossRef CAS PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- S. Vauthey, S. Santoso, H. Gong, N. Watson and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5355–5360 CrossRef CAS PubMed.
- H. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS PubMed.
- A. Dehsorkhi, V. Castelletto and I. W. Hamley, J. Pept. Sci., 2014, 20, 453–467 CrossRef CAS PubMed.
- S. Cavalli, F. Albericio and A. Kros, Chem. Soc. Rev., 2010, 39, 241–263 RSC.
- S. E. Paramonov, H. W. Jun and J. D. Hartgerink, J. Am. Chem. Soc., 2006, 128, 7291–7298 CrossRef CAS PubMed.
- Y. C. Yu, V. Roontga, V. A. Daragan, K. H. Mayo, M. Tirrell and G. B. Fields, Biochemistry, 1999, 38, 1659–1668 CrossRef CAS PubMed.
- G. B. Fields, J. L. Lauer, Y. Dori, P. Forns, Y. C. Yu and M. Tirrell, Biopolymers, 1998, 47, 143–151 CrossRef CAS PubMed.
- J. N. Luo and Y. W. Tong, ACS Nano, 2011, 5, 7739–7747 CrossRef CAS PubMed.
- S. Zhang, Acc. Chem. Res., 2012, 45, 2142–2150 CrossRef CAS PubMed.
- S. Santoso, W. Hwang, H. Hartman and S. Zhang, Nano Lett., 2002, 2, 687–691 CrossRef CAS.
- R. Todeschini and V. Consonni, Handbook of molecular descriptors: Methods and principles in medicinal chemistry, WILEY-VCH Verlag GmbH, 2000 Search PubMed.
- K. Kar, P. Amin, M. A. Bryan, A. V. Persikov, A. Mohs, Y. H. Wang and B. Brodsky, J. Biol. Chem., 2006, 281, 33283–33290 CrossRef CAS PubMed.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- E. R. da Silva, W. A. Alves, V. Castelletto, M. Reza, J. Ruokolainen, R. Hussain and I. W. Hamley, Chem. Commun., 2015, 51, 11634–11637 RSC.
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857–11862 CrossRef CAS PubMed.
- D. Papapostolou, E. H. Bromley, C. Bano and D. N. Woolfson, J. Am. Chem. Soc., 2008, 130, 5124–5130 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S3. See DOI: 10.1039/c7ra11855d |
| This journal is © The Royal Society of Chemistry 2018 |