
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical assembling of amine functionalized boron nitride nanotubes onto polymeric nanofiber film for improving their thermal conductivity†
Dukeun Kima,
Sumin Hab,
Hoi Kil Choid,
Jaesang Yu*d and
Yoong Ahm Kim *bc
*bc
aSMART Textile Material R&D Team, Korea High Tech Textile Research Institute, 170, Geomjun-gil, Nam-myeon, Yangju-si, Gyeonggi-do 11410, Korea
bAlan G. MacDiarmid Energy Research Institute, School of Polymer Science and Engineering, Department of Polymer Engineering, Graduate School, Chonnam National University, 77 Yongbong-ro, Buk-gu, Gwangju 61186, Republic of Korea. E-mail: yak@chonnam.ac.kr
cInstitute for Biomedical Sciences, Interdisciplinary Cluster for Cutting Edge Research, Shinshu University, 3-1-1 Asahi, Matsumoto, Nagano, Japan
dMutifunctional Structural Composites Research Centre, Institute of Advanced Composite Materials, Korea Institute of Science and Technology (KIST), 92, Chudong-ro, Bongdong-eup, Wanju-gun, Jeonbuk, 55324, Republic of Korea. E-mail: jamesyu@kist.re.kr
First published on 24th January 2018
Abstract
The interfacial effect between an organic matrix and inorganic nanofillers on the thermal conductivity of a polymer composite was systematically explored by assembling amine-functionalized boron nitride nanotubes (BNNTs) onto an electrospun polyacrylic acid/polyvinyl alcohol nanofibrous web through either physical electrostatic interactions or chemical coupling reactions. The amine functionalization of BNNTs and their integration onto the electrospun nanofiber webs were confirmed by various analytical techniques. No distinctive change in thermal stability and conductivity could be observed between the pristine- and physically assembled polymeric nanofiber films; however, the chemically assembled nanofiber film produced via the chemical coupling reaction showed large improvements in both thermal stability and thermal conductivity. Our study demonstrated that it is possible to produce a highly thermally conductive polymer nanofiber film through interfacial engineering, even if the incorporation of inorganic filler is below 1 wt%.
1. Introduction
Due to the ever-growing demand for high performance as well as miniaturization of devices utilized in heat exchangers, electronics, machines and electronic circuits, protection of such devices against heat has been recognized as one of the important issues.1–4 To this end, considerable attention has been focused on the fabrication of heat conducting polymer composites due to their good ability to protect devices against heat in various applications.5 Unfortunately, polymers have intrinsically low thermal conductivity.6 Therefore, highly thermal conductive polymer composites had been typically produced by filling polymers with highly thermal conductive fillers, such as carbon materials,7–9 ceramics10 and metals.11,12 Among them, carbon nanotubes (CNTs) have received large attention due to their high thermal conductivity (e.g., theoretical estimate, 3000–6000 W mK−1, experimental estimate, 2000–4000 W mK−1).13,14 However, CNTs are also electrically conducting. Therefore, a minor addition of CNTs to a polymer matrix brings a large change in the electrical properties of the polymeric composite, leading to adverse effects on the electrical properties of the devices utilized.5 The use of dielectric boron nitride nanotubes (BNNTs), which have a cylindrical structure similar to that of CNTs, as fillers to dissipate heat was considered due to their high thermal conductivity (e.g., theoretical estimate, 6600 W mK−1 or more) and perfect electrical insulation property.15–18Recently, several researchers attempted to prepare polymeric composites having a high fraction of BNNTs (>10 wt%) with a view to achieving high thermal conductivity.5,10,17 However, those studies reported that addition of BNNTs into polymers does not guarantee a large enhancement in thermal conductivity, because the thermal conductivity of a polymer composite depends upon various factors, such as the characteristics of filler (i.e., structural defects, the amount of loading and the orientation), the crystallinity of the polymer matrix,19,20 the interfacial interaction between filler and the polymer matrix,21–24 and dispersibility of individuated fillers in the polymer matrix.6,17,25 Accordingly, it is indicated that the degree of crystallinity of a polymer is an important parameter to achieve high thermal conductivity of the polymer composites. However, there have been no other alternative but to change the polymer.26–29 Alternatively, reducing interfacial thermal resistance between fillers and polymer matrixes is considered as a more effective factor to achieve the thermal conductivity. To produce the polymer composite with high thermal conductivity, development of a suitable interfacial engineering technique is a necessity. In this regard, we anticipated that on adding a small amount of filler, specific interactions (say, formation of chemical bonds) between fillers and polymer matrixes might help reduce the interfacial resistance, thereby resulting in a highly thermal conductive polymer composite.
To verify our anticipation, in the current work, amine-functionalized BNNTs (a-BNNTs) were synthesized through chloride-amine reaction, and nanofibrous polymeric films were produced via electrospinning using polyacrylic acid (PAA)/polyvinyl alcohol (PVA) polymers with active functional groups (e.g. carboxyl groups). The a-BNNTs were then assembled onto the surface of PAA/PVA nanofibrous polymeric film via either physical electrostatic interaction or chemical covalent bonding (Fig. S1†). On attaching BNNTs onto PAA/PVA nanofibrous film through chemical bonds, a large enhancement in thermal conductivity could be realized. Experimental results were theoretically supported by the molecular dynamics (MD) simulations. To the best of our knowledge, this is the first report which demonstrates interfacial engineering via chemical assembly of functionalized BNNTs is an efficient strategy to enhance the thermal conductivity of polymeric composites.
2. Experimental
2.1 Materials
Multiwall boron nitride nanotubes (purity 40–50% without catalysts, length: up to 200 μm) were purchased from BNNT LLC. Succinyl chloride, PAA (average Mw = 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), PVA (average Mw = 89000–98000), and N,N-dimethylformamide (DMF) were acquired from Sigma-Aldrich. Ethylene diamine (EDA) and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC) were obtained from TCI and Alfa Aesar, respectively.
000), PVA (average Mw = 89000–98000), and N,N-dimethylformamide (DMF) were acquired from Sigma-Aldrich. Ethylene diamine (EDA) and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC) were obtained from TCI and Alfa Aesar, respectively.
2.2 Preparation of amine-functionalized BNNTs
Amine-functionalized BNNTs (a-BNNTs) were synthesized via chemical reaction between chlorinated BNNT (cl-BNNTs)30,31 and EDA (Fig. S1(a)†). In brief, 100 mg BNNT, purified by acid and heating treatment, was dispersed in 5 mL succinyl chloride and the solution was refluxed at 190 °C for 120 h. The resultant powder was collected via centrifugation followed by washing repeatedly with chloroform to remove any unreacted succinyl chloride. The powder was next dispersed in 5 mL of EDA solution and stirred at 80 °C for 24 h.32 The final product was collected as white solid through centrifugation, followed by repeated washing with water.2.3 Preparation of PAA/PVA nanofibrous thin films
A schematic representation of the electro-spin synthesis of PAA/PVA nanofibrous film has been displayed in Fig. S1(b).†33,34 A homogeneous solution of PAA/PVA (mass ratio of 1:1) and a total polymer concentration of 8 wt% in H2O was prepared by stirring at 70 °C for 24 h. The polymer solution was sprayed as polymeric nanofibers with a high voltage of 19 kV applied between the needle and the collector (aluminum foil) at a feeding rate of 4 mL h−1 and 20 cm tip-to-collector distance. Through this process, 6 layers of PAA/PVA nanofibrous webs were stacked which were then hot-pressed at 80 °C to produce films. The resultant PAA/PVA nanofibrous films were further thermally treated at 145 °C for 30 min to render them water insoluble.
2.4 Assembly of a-BNNTs onto PAA/PVA nanofibrous films
The processes of assembling BNNTs onto the PAA/PVA nanofibrous thin films via physical and chemical routes have been illustrated in Fig. S1(c).†34 In the chemical route, a-BNNTs were covalently assembled onto the PAA/PVA nanofibrous thin films through imide formation between the amine groups of BNNT sidewalls and the carboxylic acid groups of PAA in the EDC aqueous solution. Specifically, 1 mg (1 wt%) of a-BNNTs was dispersed in EDC aqueous solution (32.6 M, 5 mL) by tip-sonication for 3 h. Next, the water stable (or insoluble) PAA/PVA nanofibrous thin films (3 × 0.5 cm2, 60 mg) displayed in Fig. S1(b)† were dipped into the resultant solution for 48 h, rinsed with water 3 times, and then dried under vacuum at room temperature for 24 h. In the physical absorption, the water stable nanofibrous thin films were dipped into a dispersion of a-BNNTs in aqueous solution without EDC, followed by the same process of the chemical route. The produced nanofibrous thin films with a-BNNTs by physical absorption or chemical reaction were also displayed in Fig. S1(c).†2.5 Structural characterizations
To determine the functionalization of BNNTs and their assembly on the nanofibrous films via physical absorption and chemical reaction, Fourier transform infrared spectroscopy (FT-IR, Nicolet iS10 spectrometer using ATR accessory, Thermo scientific Ltd., Japan) was used and X-ray photoelectron spectroscopy (XPS, Multilab 2000 VG Ltd., UK) was investigated with a monochromatic Al Kα line and a photo energy of 1486.6 eV, using a 180 hemispherical analyzed in a constant-resolution mode with a pass energy of 50 eV and 20 eV for whole and individual element, respectively. According to the state of products, XPS investigation was performed as powder and film state, respectively. All morphological changes of the electrospun nanofibrous thin films before and after the assembling of a-BNNTs were studied using scanning electron microscopy (SEM, S-4700, Hitachi Ltd., Japan) at an operating voltage of 15 kV after platinum-coating for 180 second (E-1030, Hitachi Ltd., Japan). Field emission transmission electron microscopy (FE-TEM, JEM-2100F, JEOL Ltd., Japan) was performed at an operating voltage of 100 kV. To prepare the TEM sample, a drop of suspensions (50 μL) produced by sonicating nanofibrous thin-films with and without a-BNNTs in ethanol for 1 h was placed on carbon coated nickel grid, followed by evaporation of the ethanol at room temperature. Thermogravimetric analysis (TGA, TGA-50, Shimadzu Ltd., Japan) was conducted at a heating rate of 10 °C min−1 under aerobic condition.2.6 Thermal conductivity measurement
Thin films were cut into 3 cm × 0.5 cm rectangle. The Angstrom's method with AC periodic scanning laser heating (ULVAC-RIKO, incorporation, Laser PIT) was used to measure the thermal diffusivity (α) of nanofibrous thin films. In this technique, a periodic square wave of heat was applied at one end of the film (near the heat source) while the other end (near the heat sink) was kept at room temperature. As a result of propagation of heat, such periodic square wave was attenuated by lowering the amplitude and experiencing a phase shift. The resulting square waves were obtained at the two thermistors located at separate points along the long axis of thin-films. The thermal diffusivity (α) was obtained from Fourier transform of such waves. From the thermal diffusivity values obtained from experiments, thermal conductivity (λ) was calculated using the following eqn (1).5| λ = α × ρ × c | (1) |
| dcomposite = vpolymer × dpolymer + vfiller × dfiller | (2) |
2.7 All atom based simulation
To estimate quantitatively the influences of the functionalization of BNNTs and their assembly on thermal conduction of the PAA/PVA nanofibrous thin films, the molecular dynamics (MD) simulations were performed by using the Material Studio software. A force field used in the simulations was COMPASS II. The total potential energy (Utotal) is expressed as eqn (3).37| Utotal = Ub + Uθ + Uϕ + Uχ + UvW + Uel + Uco | (3) |
The MD models of PAA/PVA nanofibrous thin films were constructed by packing the PAA/PVA polymer chains (n = 10). A size of the periodic boundary cell was 2.0 × 2.0 × 12.0 nm3. An energy minimization was performed with smart algorithm to relax the potential energy and the motion of atoms. The minimized models were stabilized under isothermal and isobaric conditions (NPT ensemble, 80 °C and 50 MPa). This process was performed for 2.0 ns, and each time step was 1 fs. A single-walled structure was selected to focus on the effect of the functionalization, and its chirality was (10,0). A degree of the functionalization (no. functional groups/no. BNNT atoms) was 3.0%. The functionalized BNNT model was assembled on a stabilized PAA/PVA model. The assembled model was relaxed again via stabilization process. A stabilized model had the physical (non-bonding) routes between PAA/PVA and functionalized BNNT models. A model that had chemical (covalent bonding) routes was made by performing the stabilization process after connecting the PAA/PVA chains and functional groups. The MD models for the pristine, physical assembled, and chemical assembled PAA/PVA nanofibrous thin films were denoted as “PAA-PVA”, “PAA-PVA_phys”, and “PAA-PVA_chem”, respectively. The thermal conductivity along heat flow direction was defined by Fourier's law. Müller and Plathe38 suggested the calculation method of thermal conductivity in MD model. This method determined the resulting temperature gradient by imposing a heat flux through the energy variation between cool and hot layers in an MD model. The MD model was divided into N layers to heat flow direction, and the thickness of each layer was identical. Its center layer and two end layers were defined as the cold layer and hot layer, respectively. The fastest atom in cool layer and the slowest atom of same mass in hot layer exchange the velocity vector. Then the temperature increase in hot layer and decrease in cold layer. The repetition of above process generates an energy transfer from hot layer to cold layer. It also leads to temperature gradient between cold layer and hot layer. The thermal conductivity (λ) between the cool and hot layers was expressed as eqn (4).39
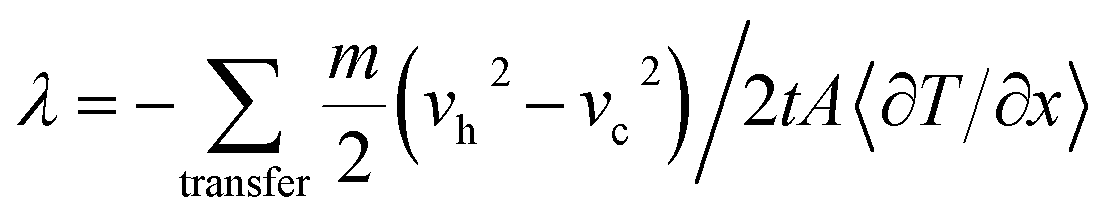 | (4) |
3. Results and discussion
To assemble BNNTs onto PAA/PVA nanofibrous thin films, amine functionalized BNNTs (a-BNNTs) were synthesized via chloride-amine reaction. FT-IR and XPS measurements were done to verify the introduction of amine groups on the surface of BNNTs. A typical FT-IR spectrum of the purified BNNT revealed two peaks at 796 and 1375 cm−1, which were assigned to A2u (B–N–B out-of-plan bending vibration mode) and E1u (in plane B–N transverse optical mode) (Fig. S2†).40 On the other hand, for the chlorinated BNNTs (cl-BNNTs), the absorption peaks at 752 cm−1, 1780 cm−1, and 2931 cm−1 were ascribed to C–Cl, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–H stretching vibration, respectively.31,41 These results indicated that the chlorination reaction between COCl group of succinyl chloride and the nitrogen atoms on the partially ionic B–N bonding of BNNTs occurred under extreme conditions. In the amine-functionalized BNNTs (a-BNNTs), the C–Cl absorption peak disappeared and new peaks which could be assigned to CONH and N–H stretching vibration at 1643 cm−1 and around 3180 cm−1, respectively, appeared. This result indicated a successful synthesis of a-BNNTs through imide reaction between cl-BNNTs and the diamine ligand. The same inference can also be drawn by comparing XPS Cl 2p spectra of cl-BNNTs and a-BNNTs. The XPS Cl 2p of cl-BNNTs displayed the C–Cl and Cl– ionic peaks (Fig. S3†). However, these peaks disappeared after imide reactions. For the N 1s spectra (Fig. S4†), the gradual shift toward high binding energy is clearly observed, possibly due to the chemical reaction between nitrogen and succinyl chloride molecule. In contrast, there is no change for the B 1s XPS spectra (Fig. S4†). Such result clearly indicated that succinyl chloride reacted with nitrogen atoms. The FT-IR and XPS data confirmed the formation of amine-functionalized BNNTs via chloride-amine reaction between cl-BNNTs and diamine ligand via imide formation.
O and C–H stretching vibration, respectively.31,41 These results indicated that the chlorination reaction between COCl group of succinyl chloride and the nitrogen atoms on the partially ionic B–N bonding of BNNTs occurred under extreme conditions. In the amine-functionalized BNNTs (a-BNNTs), the C–Cl absorption peak disappeared and new peaks which could be assigned to CONH and N–H stretching vibration at 1643 cm−1 and around 3180 cm−1, respectively, appeared. This result indicated a successful synthesis of a-BNNTs through imide reaction between cl-BNNTs and the diamine ligand. The same inference can also be drawn by comparing XPS Cl 2p spectra of cl-BNNTs and a-BNNTs. The XPS Cl 2p of cl-BNNTs displayed the C–Cl and Cl– ionic peaks (Fig. S3†). However, these peaks disappeared after imide reactions. For the N 1s spectra (Fig. S4†), the gradual shift toward high binding energy is clearly observed, possibly due to the chemical reaction between nitrogen and succinyl chloride molecule. In contrast, there is no change for the B 1s XPS spectra (Fig. S4†). Such result clearly indicated that succinyl chloride reacted with nitrogen atoms. The FT-IR and XPS data confirmed the formation of amine-functionalized BNNTs via chloride-amine reaction between cl-BNNTs and diamine ligand via imide formation.
The thermal properties of BNNTs were investigated through TGA (Fig. S5†). The pristine BNNTs showed no specific decomposition up to 800 °C. Beyond 800 °C, a slight mass gain (∼≤1 wt%) could be observed which was due to the oxidation of boron. On the other hand, the cl-BNNTs showed a continuous weight loss until 800 °C, whereas, three stages of weight loss could be observed for a-BNNTs. The first region (I) below 200 °C indicated weight loss of adsorbed residual solvents and/or impurities obtained from the functionalization process. The second region (II) between 200 and 500 °C could be attributed to the thermal decomposition of covalently grafted organic chlorination groups. Such result indicated that the percentage of covalently grafted organic chlorination groups is ca. 44.5%. The third region (III), interestingly, showed a continuous weight loss above 500 °C, and over 1000 °C, residues above 8% could be found. This was attributed to the removal of unstable BNNTs caused by chemical reaction. For the a-BNNTs products, the weight loss regions were more clearly observed. The first (I) and second (II) region were also from the adsorbed residual solvents and/or impurities in the products, and from the loss of the amine ligand from the surface of the BNNTs, respectively. The third region (III) revealed complete mass loss until reaching 1000 °C. These results can be explained that the functional groups were evenly functionalized onto the surface of chemically inert BNNTs.
a-BNNTs were assembled on the electrospun nanofibrous thin films via either physical or chemical approaches (called as PAA/PVA_phys and PAA/PVA_chem, Fig. S1†). The PAA/PVA nanofibrous thin-films before and after assembling of a-BNNTs were investigated by FT-IR analyses (Fig. 1(a)). For FT-IR spectrum of PAA/PVA nanofibrous thin-films,42 peaks at 2929, 1704, 1091 and 3407 cm−1 were observed which originated from C–H symmetric stretching, COO– asymmetric stretching from PAA, C–O asymmetric stretching from PVA and overlapped symmetric vibrations –OH and –COOH, respectively. After assembling of a-BNNTs onto the thin-films via physical approach, specific BNNT vibration peaks were observed at 802 cm−1 and 1373 cm−1. We observed downshifts for the absorption peaks of C–O, COO–, and overlapped peaks of –OH and –COOH related to PAA and PVA polymer, probably due to the physical electrostatic interaction between the amino group of BNNT and functional group of PAA/PVA nanofibers. By contrast, for the chemically assembled a-BNNTs on the PAA/PVA nanofibrous thin films, we observed a new absorption peak at 1641 cm−1 corresponding to CONH stretching vibration.38 Furthermore, the specific peaks related to PAA polymer showed a surprisingly large degree of change. For example, the COO– stretching peak was up-shifted to 1708 cm−1 in contrast to that of the physically assembled sample, indicating the presence of interaction between the carboxylic acid group of the polymer and amine group of the BNNTs. The overlapped peak of –OH and –COO at 3315 cm−1 become narrow, suggesting partial disappearance of the –COOH groups. Thus, the carboxylic acid groups of the PAA polymers participated in chemical interaction with a-BNNTs, leading to the formation of CONH bonds. To see the additional distinctive differences between the physical and chemical assemblies of a-BNNTs onto PAA/PVA nanofibrous thin films, we carried out XPS measurement and the resulting spectra were carefully analyzed by curved-fitting (Fig. 1(b) and S6†).
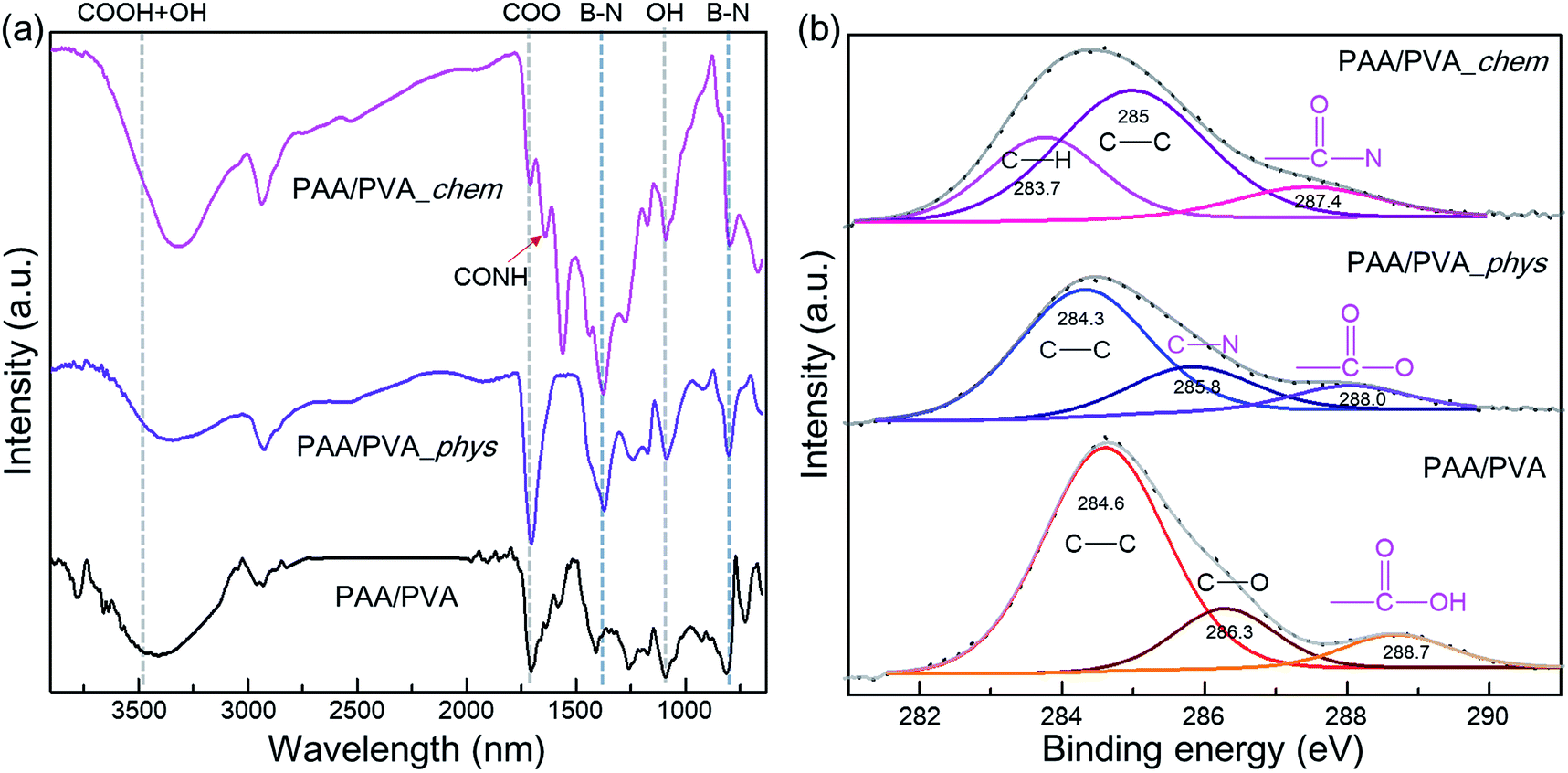 | ||
| Fig. 1 (a) FT-IR and (b) XPS C 1s spectra of pristine nanofibrous film, and the physically and chemically assembled nanofibrous films with amine functionalized BNNTs. | ||
The C 1s spectrum (Fig. 1(b)) of the pristine PAA/PVA nanofibrous thin films displayed three peaks at 284.6, 286.3, and 288.7 eV, which were ascribed to C–C, C–O, and –COOH bonding, respectively. However, the physical assembly of a-BNNTs allowed the C 1s spectrum of thin film to show new peaks of C–N and –COO– bonding at 285.8 and 288.0 eV. Such data indicated that the a-BNNTs were sufficiently adsorbed onto the thin films via weak physical electrostatic interaction. In the chemically assembled sample, we observed a new peak at 287.4, which was attributed to –CON– bonding. In addition, B 1s and N 1s spectra of physically and chemically assembled nanofibrous films (Fig. S6†) showed considerable peak changes as compared to those of the pure BNNTs, indicating strong interaction between the organic polymer and the inorganic a-BNNTs. The XPS results strongly indicated that a-BNNTs were successfully assembled onto PAA/PVA nanofibrous thin films via either physical electrostatic interaction or chemical covalent bonds.
SEM observation was performed to see the morphological changes in the PAA/PVA nanofibrous thin films before and after the assembling of a-BNNTs (Fig. 2). The SEM image of the pristine PAA/PVA nanofibrous thin film (Fig. 2(a)) showed smooth and uniform nanofibers with random orientation along with slightly pressurized and cross-linked portions caused by hot-pressure and stabilization process. The physically assembled sample (Fig. 2(b)) was very similar to that of the pristine film, except some small particles were locally attached to their surface. However, the chemically assembled film (Fig. 2(c)) displayed a rough surface with numerous small particles attached to its surface. Such difference indicated that numerous a-BNNTs were chemically assembled onto the surface of the PAA/PVA nanofibrous film with the help of EDC. From the additional TEM observation (Fig. 2(d)), we identified that the small particles were actually modified BNNTs on the surface of the nanofiber. The experimental evidence from SEM and TEM studies suggested that the BNNTs were assembled on nanofibrous thin films through either physical absorption or chemical reaction.
 | ||
| Fig. 2 Scanning electron microscopic images of (a) pristine PAA/PVA nanofibrous film, (b) physically and (c) chemically assembled nanofibrous films with functionalized BNNTs. (d) A typical transmission microscopic image of (c). | ||
The effect of the assembled a-BNNTs on the thermal stability of nanofibrous films were also investigated by TGA and DSC analyses (Fig. 3 and S7†). The weight loss of PAA/PVA nanofibrous thin-films can be categorized into 3 sections; evaporation of water and/or organic solvents (I), thermal decomposition of (II) functional groups and (III) backbone of polymers. The PAA/PVA nanofibrous thin-film showed complete decomposition at 500 °C. An enhanced decomposition temperature (600 °C) was observed for the physically assembled nanofibrous film which was due the effect of the physically incorporated BNNTs. By contrast, a dramatically increased thermal stability was observed for the chemically assembled film (more than 30% at 1000 °C). It is assumed that the chemically introduced a-BNNTs on the surface of nanofibrous polymer matrixes acted as protective agent with regard to oxygen attack. These results can be used as evidence that a-BNNTs can affect the thermal stability of PAA/PVA nanofibrous thin-films, especially if BNNTs are chemically assembled onto the polymeric thin film.
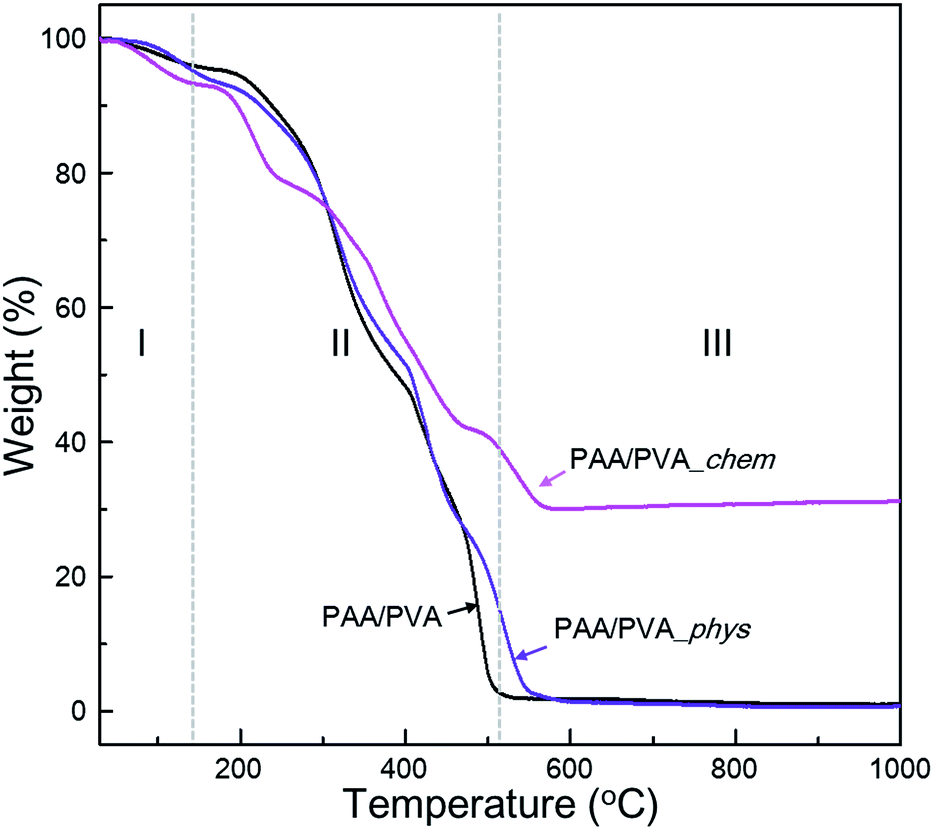 | ||
| Fig. 3 Thermogravimetric curves of pristine nanofibrous film and the physically and chemically assembled nanofibrous film with functionalized BNNTs under aerobic condition. | ||
Thermal conductivity of the nanofibrous thin films before and after assembling a-BNNTs was experimentally evaluated by measuring their thermal diffusivities. The experimentally obtained thermal diffusivities (Fig. 4(a)) and calculated thermal conductivities (Fig. 4(b)) revealed that the chemical assembling approach was more effective than the physical route. We achieved high thermal conductivity of nanofibrous film up to 0.65 W mK−1 via the chemical assembly of a-BNNTs, which is equivalent to thermal diffusivity of 9.01 × 10−7 m2 s−1. This is 1.5-fold higher than both the pristine nanofibrous film (0.42 W mK−1, 5.95 × 10−7 m2 s−1) and the physically assembled nanofibrous film with a-BNNTs (0.43 W mK−1, 5.99 × 10−7 m2 s−1). It is interesting to achieve such a high thermal conductivity from the chemically assembled nanofibrous film, considering the small amount of BNNTs incorporated (1 wt%). Therefore, it is worth mentioning that the covalent bond formation between amine-functionalized BNNTs and the –COOH of polymer matrixes reduced the interfacial thermal resistance, which resulted in the improvement of thermal conductivity for polymeric composite films.
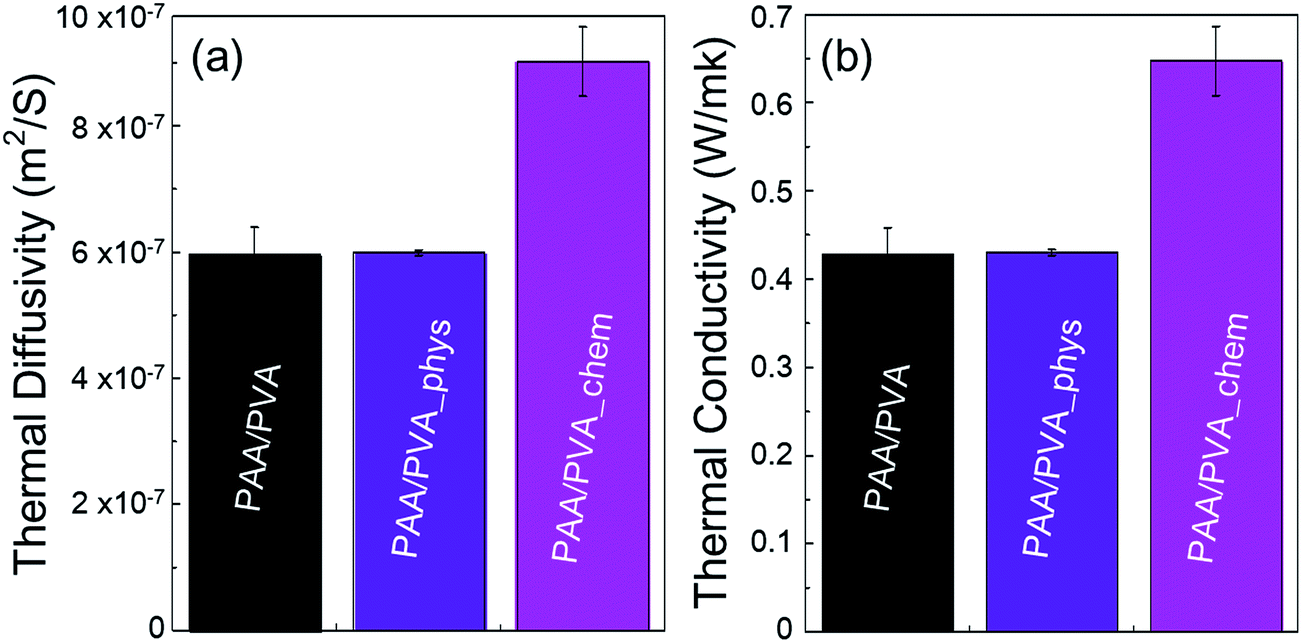 | ||
| Fig. 4 (a) Thermal diffusivity and (b) thermal conductivities of pristine nanofibrous film and the physically and chemically assembled nanofibrous films with functionalized BNNTs. | ||
Finally, the influences of the functionalization of BNNTs and their assembly was estimated quantitatively by analyzing the results of MD simulations (Fig. S8†) and compared with the experimental results. The thermal conductivities of three MD models are well matched with the experimentally measured results (Fig. 5(a)). It means that the three MD models were well constructed to simulate the characteristics of PAA/PVA nanofibrous thin-films. The specific heats and average atomic velocities obtained from MD simulation (Fig. 5(b)) revealed that the capacity of enthalpy in PAA/PVA nanofibrous thin-films was significantly improved by adding the functionalized BNNTs. A high capacity of enthalpy means that the variation of potential energies was increased by the temperature change. The potential energies with temperature show the largest variations for PAA/PVA_chem, and those of PAA/PVA and PAA/PVA_phys shows a small difference (Fig. 6(a)). These results show that the chemical assembled PAA/PVA nanofibrous thin-film had a lager heat flux than the others in the identical temperature gradient. It was the one of reason that the thermal conductivity was improved by the functionalization of BNNTs and their assembly. Another reason was the effect of interactions improved in the interphase between the PAA/PVA nanofibrous thin-film and functionalized BNNT. The interaction energies of PAA/PVA_chem were a higher than those of PAA/PVA_phys in every temperature range (Fig. 6(b)). This result means that the expansion of the conductive path and interphase area by a formation of the covalent bond improved the exchange of energy in the interphase. When the interphase had the high interaction energy, the functionalized BNNTs on PAA/PVA nanofibrous thin-films can performed the role as good conductive path. Therefore, the functionalization of BNNTs and their assembly on the nanofibrous films are the effective method to improve the conductive performance of the nanofibrous thin-film based the polymers.
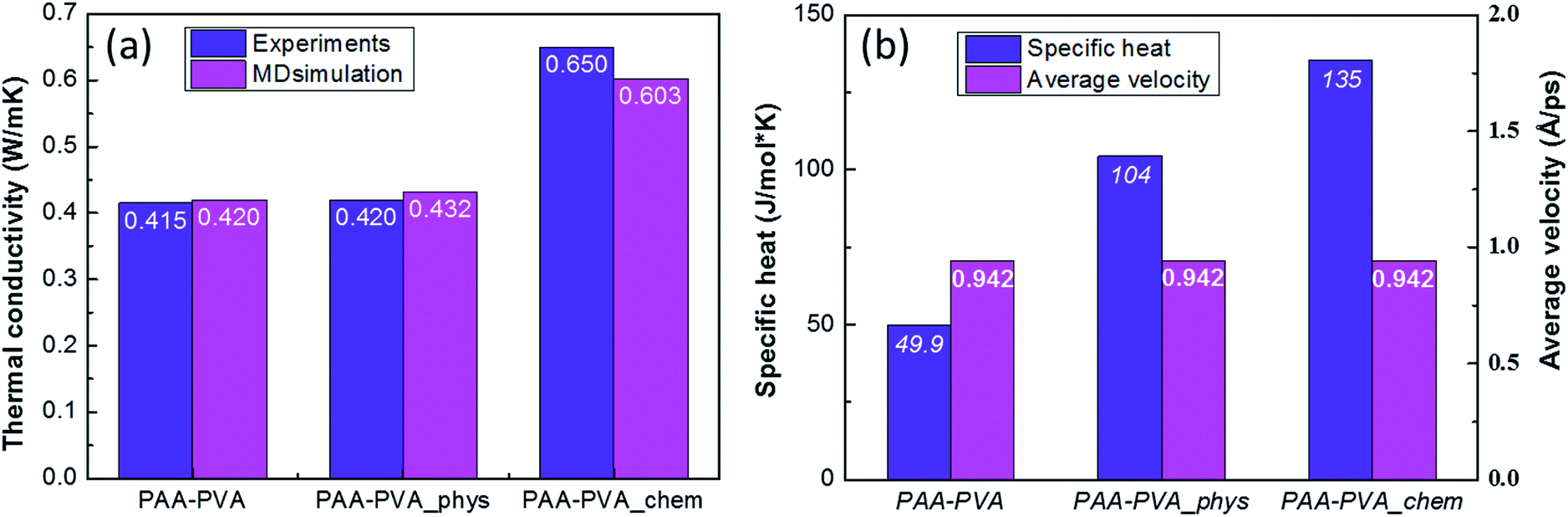 | ||
| Fig. 5 (a) Thermal conductivities, (b) specific heats, and average atomic velocities of pristine, the physically, and chemically assembled nanofibrous films obtained from the MD simulations. | ||
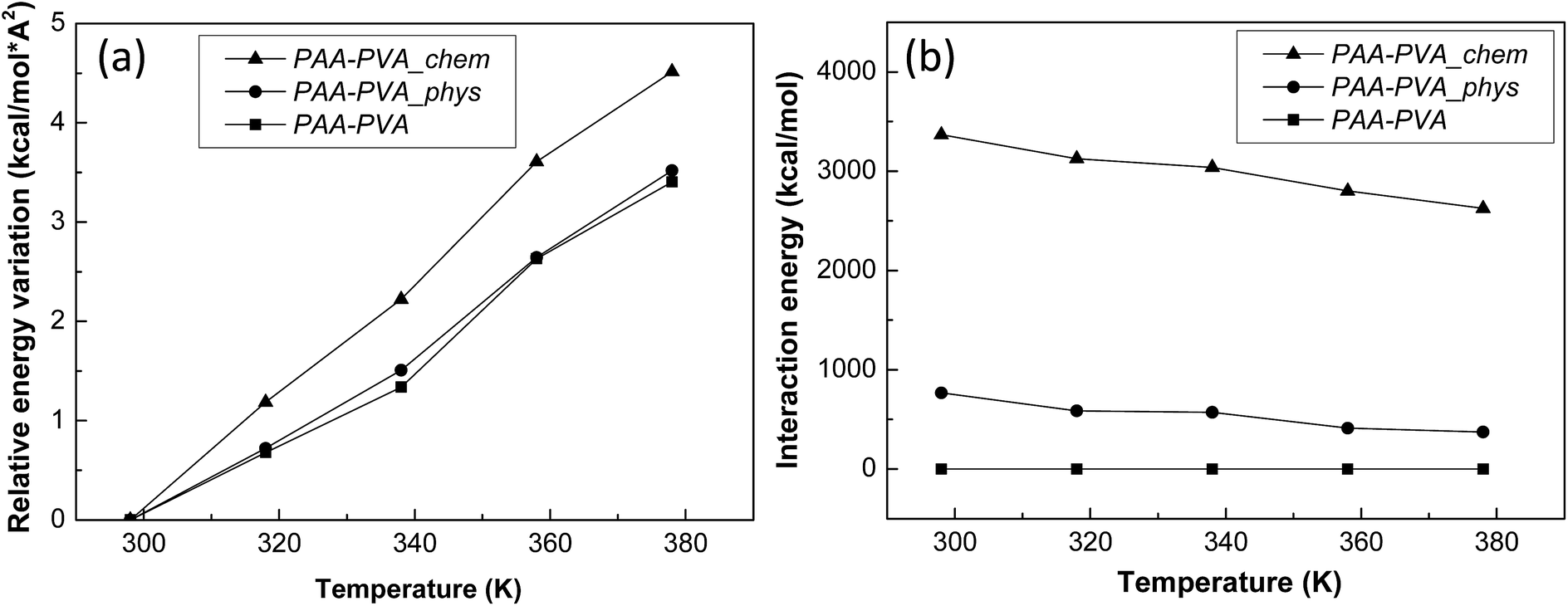 | ||
| Fig. 6 (a) Relative variations of potential energy and (b) interaction energies between the nanofibrous films and functionalized BNNTs obtained from the MD simulations. | ||
4. Conclusion
In this work, a new approach for assembling functionalized BNNTs onto electrospun polymer nanofibrous thin films was attempted to enhance thermal conductivity of the film. The amine-functionalized BNNTs, produced by chlorine-amine reaction, were assembled onto PAA/PVA nanofibrous films through either physical absorption or chemical reaction. Although a small amount of BNNTs (1 wt%) was chemically assembled onto the polymer nanofibrous film, the thermal stability of the film became very high at 1000 °C (30%). In addition, the chemically assembled nanofibrous film with a-BNNTs showed 1.5-fold higher thermal conductivity than those of the pristine and the physically assembled nanofibrous films. We clearly demonstrated that interfacial engineering between inorganic filler and organic matrixes is critical to realize high thermal conductivity of polymer composites. The chemical approach we developed here can be extended to prepare other types of polymeric composites with high thermal conductivity for applications in devices, sensing, and catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. A. K. acknowledges the financial support from the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. NRF-2017R1A2A1A17069771) and from the Nano Material Technology Development Program through the NRF funded by the Ministry of Science, ICT and Future Planning (2016M3A7B4021149). This study was also supported by Korea Institute of Science and Technology (KIST) Institutional Program and Space Core Technology Development Program (No. 2017M1A3A3A02016310) through the National Research Foundation of Korea (NRF).Notes and references
- L. Xinxia, Y. Yaping, D. Lan, G. Jie, A. Adili, X. Xiangfan and L. Baowen, J. Phys. D: Appl. Phys., 2017, 50, 104002 CrossRef.
- J. Hou, G. Li, N. Yang, L. Qin, M. E. Grami, Q. Zhang, N. Wang and X. Qu, RSC Adv., 2014, 4, 44282–44290 RSC.
- Y. Hwang, M. Kim and J. Kim, RSC Adv., 2014, 4, 17015–17021 RSC.
- C. Seran, I. Hyungu and K. Jooheon, Nanotechnology, 2012, 23, 065303 CrossRef PubMed.
- T. Terao, C. Zhi, Y. Bando, M. Mitome, C. Tang and D. Golberg, J. Phys. Chem. C, 2010, 114, 4340–4344 CAS.
- Y.-J. Xiao, W.-Y. Wang, T. Lin, X.-J. Chen, Y.-T. Zhang, J.-H. Yang, Y. Wang and Z.-W. Zhou, J. Phys. Chem. C, 2016, 120, 6344–6355 CAS.
- M. Kim, Y. Kim, S. H. Baeck and S. E. Shim, Carbon Lett., 2015, 16, 34–40 CrossRef.
- X. Huang, T. Iizuka, P. Jiang, Y. Ohki and T. Tanaka, J. Phys. Chem. C, 2012, 116, 13629–13639 CAS.
- J.-H. Kim, K. H. Kim, M.-S. Park, T.-S. Bae and Y.-S. Lee, Carbon Lett., 2016, 17, 65–69 CrossRef.
- C. Zhi, Y. Bando, T. Terao, C. Tang, H. Kuwahara and D. Golberg, Adv. Funct. Mater., 2009, 19, 1857–1862 CrossRef CAS.
- L. C. Sim, S. R. Ramanan, H. Ismail, K. N. Seetharamu and T. J. Goh, Thermochim. Acta, 2005, 430, 155–165 CrossRef CAS.
- D. Kumlutaş, İ. H. Tavman and M. Turhan Çoban, Compos. Sci. Technol., 2003, 63, 113–117 CrossRef.
- C. Yu, L. Shi, Z. Yao, D. Li and A. Majumdar, Nano Lett., 2005, 5, 1842–1846 CrossRef CAS PubMed.
- H. Huang, C. H. Liu, Y. Wu and S. Fan, Adv. Mater., 2005, 17, 1652–1656 CrossRef CAS.
- C. W. Chang, W.-Q. Han and A. Zettl, Appl. Phys. Lett., 2005, 86, 173102 CrossRef.
- D. Kim, H. Muramatsu and Y. A. Kim, Nanoscale Res. Lett., 2017, 12, 94 CrossRef PubMed.
- D. Kim, M. You, J. H. Seol, S. Ha and Y. A. Kim, J. Phys. Chem. C, 2017, 121, 7025–7029 CAS.
- D. Golberg, Y. Bando, Y. Huang, T. Terao, M. Mitome, C. Tang and C. Zhi, ACS Nano, 2010, 4, 2979–2993 CrossRef CAS PubMed.
- Z. Živcová, E. Gregorová, W. Pabst, D. S. Smith, A. Michot and C. Poulier, J. Eur. Ceram. Soc., 2009, 29, 347–353 CrossRef.
- C. Poulier, D. S. Smith and J. Absi, J. Eur. Ceram. Soc., 2007, 27, 475–478 CrossRef CAS.
- Z. Han and A. Fina, Prog. Polym. Sci., 2011, 36, 914–944 CrossRef CAS.
- C. H. Liu and S. S. Fan, Appl. Phys. Lett., 2005, 86, 123106 CrossRef.
- F. A. Nutz, P. Ruckdeschel and M. Retsch, J. Colloid Interface Sci., 2015, 457, 96–101 CrossRef CAS PubMed.
- C. W. Nan, Z. Shi and Y. Lin, Chem. Phys. Lett., 2003, 375, 666–669 CrossRef CAS.
- M. Grujicic, G. Cao and B. Gersten, Mater. Sci. Eng., B, 2004, 107, 204–216 CrossRef.
- C. L. Choy, W. H. Luk and F. C. Chen, Polymer, 1978, 19, 155–162 CrossRef CAS.
- R. Haggenmueller, C. Guthy, J. R. Lukes, J. E. Fischer and K. I. Winey, Macromolecules, 2007, 40, 2417–2421 CrossRef CAS.
- X. Xie, D. Li, T.-H. Tsai, J. Liu, P. V. Braun and D. G. Cahill, Macromolecules, 2016, 49, 972–978 CrossRef CAS.
- B. Tonpheng, J. Yu and O. Andersson, Macromolecules, 2009, 42, 9295–9301 CrossRef CAS.
- T. H. Ferreira, L. M. Hollanda, M. Lancellotti and E. M. de Sousa, J. Biomed. Mater. Res., Part A, 2015, 103, 2176–2185 CrossRef CAS PubMed.
- C. Zhi, Y. Bando, C. Tang, S. Honda, K. Sato, H. Kuwahara and D. Golberg, Angew. Chem., Int. Ed., 2005, 44, 7932–7935 CrossRef CAS PubMed.
- Z. Wu, L. Meng, L. Liu, Z. Jiang, L. Xing, D. Jiang and Y. Huang, Fibers Polym., 2014, 15, 659–663 CrossRef CAS.
- Y. Huang, H. Ma, S. Wang, M. Shen, R. Guo, X. Cao, M. Zhu and X. Shi, ACS Appl. Mater. Interfaces, 2012, 4, 3054–3061 CAS.
- D. Hu, Y. Huang, H. Liu, H. Wang, S. Wang, M. Shen, M. Zhu and X. Shi, J. Mater. Chem. A, 2014, 2, 2323–2332 CAS.
- R.-C. Zhang, D. Sun, A. Lu, S. Askari, M. Macias-Montero, P. Joseph, D. Dixon, K. Ostrikov, P. Maguire and D. Mariotti, ACS Appl. Mater. Interfaces, 2016, 8, 13567–13572 CAS.
- S. Uma and J. Philip, J. Appl. Polym. Sci., 2014, 131, 41142 CrossRef.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS.
- F. Müller-Plathe, J. Chem. Phys., 1997, 106, 6082–6085 CrossRef.
- C. Nieto-Draghi and J. B. Avalos, Mol. Phys., 2003, 101, 2303–2307 CrossRef CAS.
- D. Kim, S. Nakajima, T. Sawada, M. Iwasaki, S. Kawauchi, C. Zhi, Y. Bando, D. Golberg and T. Serizawa, Chem. Commun., 2015, 51, 7104–7107 RSC.
- H. Veisi, R. Masti, D. Kordestani, M. Safaei and O. Sahin, J. Mol. Catal. A: Chem., 2014, 385, 61–67 CrossRef CAS.
- K. F. Arndt, A. Richter, S. Ludwig, J. Zimmermann, J. Kressler, D. Kuckling and H. J. Adler, Acta Polym., 1999, 50, 383–390 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11808b |
| This journal is © The Royal Society of Chemistry 2018 |