DOI:
10.1039/C7RA11198C
(Paper)
RSC Adv., 2018,
8, 328-341
A combination of docking and cheminformatics approaches for the identification of inhibitors against 4′ phosphopantetheinyl transferase of Mycobacterium tuberculosis†
Received
11th October 2017
, Accepted 8th December 2017
First published on 2nd January 2018
Abstract
4′ Phosphopantetheinyl transferase (PptT) is involved in post translational modification by carrying out phosphopantetheinylation of proteins in non-ribosomal peptide synthesis and polyketide synthesis pathways of various organisms. PptT was recently shown to be crucial for the survival as well as persistence of Mycobacterium tuberculosis (M. tb) in mice models thus demonstrating it to be an attractive drug target. By employing Autodock 4.2 and Glide, we virtually screened the filtered NCI library against the active site of PptT and out of the 205 molecules tested in vitro, 13 molecules exhibited potent enzyme inhibition with IC50 ≤ 10 μg ml−1. Further evaluation of the molecules against the in vitro growth of M. tb resulted in the identification of six compounds that exhibited inhibition of both enzyme activity as well as M. tb growth. Subsequently, a cheminformatics based structure similarity approach led to the identification of 5 analogues of P-52 (IC50 – 2.25 μg ml−1 and MIC90 – 77.5 μg ml−1) with IC50 ≤ 1 μg ml−1 thereby establishing N,N-diethyl-N′-(2-methylquinolin-8-yl)propane-1,3-diamine as one of the potent inhibitory scaffolds of PptT. The inhibitors were further evaluated for their MIC90 values as well as cytotoxicity against various mammalian cell lines. PS-40 (NSC-328398), an analogue of P-52, emerged as a potent inhibitory molecule which exhibited an IC50 of 0.25 μg ml−1, MIC90 of 10 μg ml−1 and negligible cytotoxicity with a selectivity index >10 against three mammalian cell lines tested. Thus, our study identified potent inhibitory scaffolds against 4′ phosphopantetheinyl transferase of M. tb, an important drug target of M. tb.
1. Introduction
Mycobacterium tuberculosis (M. tb), the causative agent of tuberculosis, killed 1.4 million people in the year 2015 worldwide with an additional 0.4 million people that were co infected with HIV.1 These massive numbers highlight the high rate of morbidity and mortality caused by tuberculosis worldwide.1 Although, an effective chemotherapeutic regimen for drug susceptible TB and a recently discovered drug bedaquiline for multi drug resistance (MDR-TB) are available, the number of patients suffering from drug susceptible as well as drug resistant forms of M. tb is ever increasing.2,3 Thus, the need to fill the drug pipeline with as many new scaffolds with distinct mechanisms of action cannot be overemphasized in order to curtail this deadly disease.
4′ Phosphopantetheinylation is an important post translational modification prevalent in organisms ranging from bacteria to humans. It is responsible for the activation of proteins belonging to fatty acid synthases, polyketide synthases and non-ribosomal peptide synthases.4–8 In the case of mycobacteria, 4′ phosphopantetheinylation is a post translation modification that immensely contributes to the survival and virulence of the pathogen as this modification converts inactive apo forms of proteins to active holo (phosphopantetheinylated) forms of proteins involved in important pathways of M. tb. These pathways are involved in the formation of mycolic acid (a key cell wall component of mycobacteria), fatty acids, mycobactins and carboxymycobactins (siderophores). Mycobacteria encodes two types of transferases, AcpS type transferase that activates the proteins belonging to fatty acid synthase (Fas) systems4,5 and Sfp type transferase (PptT) that activates type-I polyketide synthases6 and non-ribosomal peptide synthases.7,8 By constructing knockouts in various mycobacterial and corynebacterial species, Chalut et al. demonstrated that these two proteins are non-redundant and they activate distinct subset of proteins.9 Their study also exhibited that both AcpS and PptT individually are essential for the viability of M. smegmatis.9 Subsequently, the same group generated a conditional mutant of PptT in M. tb and demonstrated that it is required for the growth and persistence of M. tb in the mice model of tuberculosis infection further emphasizing the non-redundant and crucial role of PptT in the survival and virulence of M. tb.10 Overall, these genetic studies established the important role of PptT in maintaining the physiology and virulence of M. tb inside the host. Also, various researchers biochemically demonstrated the non-redundant nature of these two proteins by demonstrating the activation of a specific set of proteins only by PptT and not by AcpS.11,12 Zimhony et al.11 showed that AcpM, an important acyl carrier protein of M. tb involved in mycolic acid synthesis, was activated by PptT and not by AcpS. Also, Jung et al. demonstrated specific activation of carrier proteins MbtL and PpsC via PptT and not via AcpS.12 Thus, various genetic and biochemical studies supported the non-redundant as well as crucial role of PptT in M. tb. Further, the elucidation of 3-dimensional crystal structures of PptT as MBP–PptT fusion protein13 as well as the native PptT14 provided insights into the key residues involved in the binding of coenzyme A. A comparison of the crystal structures of human-PptT (Hs-PptT) and M. tb PptT revealed that a distinct tunnel occupied by Ppant arm of coenzyme A is present only in M. tb PptT and not in human PptT (Hs-PptT) which can be specifically exploited for the identification of selective M. tb PptT inhibitors.14 Indeed, there have been numerous studies pertaining to the identification of inhibitors against Sfp15–17 (most widely used phosphopantetheinyl transferase from Bacillus subtilis). A number of attempts to design high throughput assays for the identification of inhibitors have also been carried out that monitor the transfer of rhodamine CoA onto a substrate Acp by fluorescence polarization,15–17 FRET17 or by native gel electrophoresis.10 However, these assays require a fluorescent tagged CoA and recently it was shown by Leblanc et al., that the active site of M. tb PptT cannot accommodate a fluorescently tagged CoA thereby posing difficulty in carrying out high throughput M. tb PptT based inhibitor identification studies.10 However, recently, M. tb AcpM was shown to be activated by the M. tb PptT by employing electrophoresis using urea-denaturing polyacrylamide gel. Also, a similar study characterized the LmPptT (leishmania homolog) by monitoring the apo to holo conversion by electrophoresis using a native gel. Thus, the crystal as well as the biochemical studies provided a platform to carry out structure based drug discovery efforts against PptT, an essential phosphopantetheinyl transferase protein of M. tb.
In the present study, we employed virtual screening approach by docking a customized small molecule NCI library containing ∼ 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 inhibitors against the active site of PptT followed by screening of top 205 inhibitors against the enzymatic activity of PptT by employing a semi-quantitative but relatively high throughput native PAGE followed by IC50 determination using a quantitative UPLC based assay which resulted in 13 molecules with IC50 ≤ 10 μg ml−1. Subsequently, all the inhibitory molecules were further evaluated for their inhibitory potential against the growth of M. tb in broth culture. The enzymatic assay as well as M. tb broth inhibition assay yielded 5 molecules with moderate IC50 and MIC90 values. Subsequently, structure similarity based search was carried out for these 5 lead molecules and 31 analogues in total were again evaluated for their inhibitory potential against the activity of enzyme. Finally, two potent inhibitory scaffolds emerged from this approach, with 5 analogues of P-52 scaffold exhibiting IC50 ≤ 1 μg ml−1 and 4 analogues of the P-102 scaffold exhibiting IC50 ≤ 15 μg ml−1. Subsequently, MIC90 against the growth of M. tb in broth culture and cytotoxicity studies against various mammalian cell lines were carried out to identify inhibitors against PptT, an important drug target of M. tb.
000 inhibitors against the active site of PptT followed by screening of top 205 inhibitors against the enzymatic activity of PptT by employing a semi-quantitative but relatively high throughput native PAGE followed by IC50 determination using a quantitative UPLC based assay which resulted in 13 molecules with IC50 ≤ 10 μg ml−1. Subsequently, all the inhibitory molecules were further evaluated for their inhibitory potential against the growth of M. tb in broth culture. The enzymatic assay as well as M. tb broth inhibition assay yielded 5 molecules with moderate IC50 and MIC90 values. Subsequently, structure similarity based search was carried out for these 5 lead molecules and 31 analogues in total were again evaluated for their inhibitory potential against the activity of enzyme. Finally, two potent inhibitory scaffolds emerged from this approach, with 5 analogues of P-52 scaffold exhibiting IC50 ≤ 1 μg ml−1 and 4 analogues of the P-102 scaffold exhibiting IC50 ≤ 15 μg ml−1. Subsequently, MIC90 against the growth of M. tb in broth culture and cytotoxicity studies against various mammalian cell lines were carried out to identify inhibitors against PptT, an important drug target of M. tb.
2. Materials and methods
2.1 Grid generation and docking studies
Open NCI database consisting of 260, 071 compounds was downloaded from the NCI website (http://cactus.nci.nih.gov/download/nci/). This database was further filtered on the basis of Lipinski rule of five and PAINS descriptor features containing molecules by using the online server FAF-Drugs server (http://fafdrugs3.mti.univ-paris-diderot.fr/) as previously described.18 The 3-dimensional crystal structure of phosphopantetheinyl transferase (PptT) was downloaded from PDB database with PDB ID-4QJK. The protein molecule was prepared by the deletion of water molecules and the addition of hydrogen atoms. Autodock 4.2 and Glide software (Schrodinger Software Package) were employed to dock this filtered library against the active site of 3-dimensional structure of phosphopantetheinyl transferase (PptT). The Autodock scoring function incorporates the van der Waal/Lennard-Jones potential term, a hydrogen bonding term, coulombic electrostatic term, torsional and solvation terms. The Glide scoring function includes lipophilic term, hydrogen bonding terms, metal–ligand interaction terms, electrostatic coulombic term, van der Waals interaction term as well as solvation terms. Thus, to cover the maximum range of possible interactions that the ligand molecules can form with the active site residues of the protein, we employed two docking softwares for obtaining the list of possible hit molecules. The active site of M. tb PptT contains a tunnel containing the Ppant arm of CoA. This portion is unique to the M. tb PptT structure as compared with human PptT 3-D structure and was exploited for carrying out the docking studies. In total, 3 grids were generated by using Autodock 4.2 and two grids by using Glide software targeting the tunnel as well as the complete active site of M. tb PptT and five separate docking lists were generated. Each docking list comprised of distinct molecules in decreasing order of free energy of binding. Ultimately, 41 distinct compounds were selected from each of the five docking lists on the basis of their docking score with the top ranking molecules given first priority. Additionally, availability of a compound in the NCI repository was also a criterion for obtaining the compounds. Thus, we obtained a total of 205 distinct compounds which comprised of 41 distinct molecules from each list. All the calculations pertaining to Autodock 4.2 were carried out on a Fujitsu RX300 S7 server which served as a master node and Fujitsu CX250 S1 servers as compute nodes (35 no. with CENT-OS version 6). All the shortlisted molecules were procured from Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
2.2 Cloning, expression and purification studies of AcpM and M. tb PptT
2.2.1 Cloning strategy of AcpM and PptT. M. tb genomic DNA was used for the PCR amplification of the genes M. tb acpM and M. tb pptT. acpM gene was PCR amplified with the forward primer 5′ gataatgctagcggatccgtgcctgtcactcaggaag 3′ with NheI and BamHI restriction sites and the reverse primer 5′ gataatgaattcaagcttatcatttttcgaactgcgggtggctccaagcgctcttggactcggcctcaagc 3′ with EcoRI and HindIII restriction sites. The PCR product and the vector pET28c were digested with NheI and HindIII and ligated resulting in the construct pET28c/AcpM. pptT gene was amplified by using the forward primer containing the sequence 5′ gataatggtaccgctagcggatccatgacggtaggcacgctg 3′ with KpnI, NheI and BamHI restriction sites and the reverse primer containing 5′ gataatctcgagaagcttattatttttcgaactgcgggtggctccaagcgcttagcacgatcgcggtcag 3′ with XhoI and HindIII restriction site. The PCR product was digested with BamHI and HindIII and ligated into the vector pMal C2X digested with the same enzymes resulting in the construct pMal/PptT. A streptactin tag at the 3′ end of the reverse primer was added for the purification.
2.2.2 M. tb AcpM purification. For carrying out the expression studies, BL21 (λDE3) cells were transformed with the construct pET28c/AcpM and the cells were grown in 3 liter 2XYT media till Abs600 nm of 0.8 to 1 at 37 °C. The expression of AcpM protein was further induced by the addition of 200 μM isopropyl-1-thio-β-D-galactopyranoside (IPTG) and 0.2% (w/v) lactose for 3 hours at 37 °C. The cells were subsequently harvested by centrifugation at 4 °C, 6340g for 10 minutes. The pellets were resuspended in 60 ml lysis buffer containing 20 mM Tris (pH-8.0), 150 mM NaCl, 5 mM imidazole, 1 mM DTT, 1 mM phenylmethylsulfonylfloride (PMSF), 2 mM β-mercaptoethanol and 10% glycerol. Cells were resuspended in the lysis buffer, sonicated and the lysate was centrifuged at 12000g for 45 minutes at 4 °C. The purification was carried out by Ni-NTA affinity chromatography and the protein was eluted with 250 mM imidazole. The purified protein was quantified by Bradford assay and the purity was analyzed by electrophoresis on a 15% polyacrylamide SDS gel. The protein is endogenously produced in three isoforms in E. coli namely apo, holo and acylated as reported in the literature.11 To purify the apo form of the protein, anion exchange chromatography was employed by using a Resource Q (6 ml) column. The Ni-NTA purified and dialyzed AcpM was loaded onto an anion exchange column (Resource Q, GE Healthcare) pre-equilibrated with 20 mM Tris (pH-8.0) and eluted with a linear gradient from 100 mM NaCl to 300 mM NaCl. The apo form was subsequently pooled and loaded onto a gel filteration column (Superdex 75, GE Healthcare) to eliminate a putative contamination of E. coli PptT.
2.2.3 M. tb PptT purification. BL21 (λDE3) cells transformed with pMal C2X/PptT were grown in 3 liter LB media till Abs600 nm −0.8 to 1.0 at 37 °C and the expression of PptT was induced by the addition of 1 mM IPTG for 16–18 hours at 16 °C. The cells were harvested by centrifugation at 4 °C, approx. 6000g for 10 minutes and the cells were resuspended in 40 ml lysis buffer (20 mM Tris, 50 mM NaCl and 10% glycerol). The resuspended cells were sonicated and centrifuged at 12000g for 45 minutes and the lysate was passed through MBP resin pre equilibrated with buffer (20 mM Tris, 50 mM NaCl and 10% glycerol). The flow through was collected and resin was washed with 20 mM Tris (pH 8.0) and 50 mM NaCl wash buffer and fractions were eluted with 20 mM Tris, 50 mM NaCl and 10 mM maltose and dialyzed with buffer 20 mM Tris and 50 mM NaCl. The dialyzed protein was concentrated with the 30 kDa amicon concentrator and stored at −80 °C till further use.
2.3 IC50 inhibition studies
To monitor the conversion of apo to holo form of AcpM by M. tb PptT, a UPLC as well as electrophoresis by a 15% native polyacrylamide gel were employed. The reaction mix of 20 μl contained 20 mM Tris (pH 8.0), 150 mM NaCl, 2.5 mM DTT, 300 μM coenzyme A and 10 mM MgCl2. 236 compounds were first screened at a fixed concentration of 100 μg ml−1 by employing native PAGE. All the compounds were dissolved in DMSO at a fixed concentration of 5 mg ml−1 and 0.4 μl of the compounds (final concentration of 100 μg ml−1) were separately incubated with 2 μM of M. tb PptT containing the reaction mix at 30 °C for 60 minutes and the reaction was subsequently initiated by the addition of 300 μM coenzyme A and 10 μM AcpM and kept at 30 °C for another 60 minutes. For the determination of IC50 values, we shortlisted the molecules exhibiting more than 30% inhibition at 100 μg ml−1 which were then subjected to dose response study from 100 μg ml−1 to 0.1 μg ml−1 by employing a more precise quantitative UPLC based assay. 1 μM of M. tb PptT was incubated with varying concentrations of compounds for 60 minutes followed by the addition of 300 μM coenzyme A and 5 μM AcpM and further incubation of 60 minutes at 30 °C in 20 μl reaction volumes. The reactions were stopped by the addition of 50 mM EDTA and 30 μl of 0.1% trifluoro acetic acid (TFA) and a fixed volume of 10 μl for each sample was injected in the Acquity Ultra Performance LC machine with a BEH C-18 column 1.7 μm (2.1 × 50 mm) column under a linear gradient of 0–80% acetonitrile in 0.1% TFA and the peak area was calculated to estimate the apo and the holo forms and fraction apo to holo conversions were calculated for IC50 determinations.
2.4 Structure similarity based searching
We downloaded the .sdf files of all the selected inhibitory molecules and subjected each one of them for search against the NCI database consisting of ∼260000 compounds by using the software Open Babel and shortlisted on the basis of the Tanimoto coefficient cut-off of 0.7 as described previously.18 We employed the Open Babel software that utilizes the FP2 fingerprint subtype of the 2-D fingerprint structural descriptor used to perform the structure based similarity search. Tanimoto coefficient was employed for the selection of the similar compounds. The Tanimoto similarity index employs 2-D path fingerprint that is used to index small molecule fragments on linear segments which comprises of 7 atoms or less. It gives the measure of degree of similarity between the two molecules. We employed a cut off of 0.7 to eliminate the less similar molecules. 31 molecules were then subsequently procured and tested for inhibition studies by using native PAGE as well as UPLC based assays.
2.5 MIC90 determination of the compounds by employing resazurin based microtiter assay
Resazurin based fluorescence assay was used to determine the MIC90 of the molecules as described earlier.18 A non-fluorescent molecule (resazurin) which is blue in colour is reduced to a fluorescent molecule, resorufin (pink in colour) in the presence of viable cells. This fluorescence is measured by using fluorescence spectrophotometer (excitation = 530 nm, emission = 590 nm) and provides quantitative measurements of the cell viability in the presence of various concentrations of the compounds. Appropriate dilutions of the compounds were made from the concentration range of 1.5 μg ml−1 to 100 μg ml−1 and were incubated with M. tb cells at starting Abs600 nm of 0.02 for 7 days in a 200 μl reaction volume in MB7H9 medium with ADC. Resazurin solution (30 μl of 0.01% solution) was added on 7th day and fluorescence was measured the next day. MIC90 is defined as that concentration of inhibitor at which 90% or more of the fluorescence is reduced.
2.6 Cytotoxicity assays
To evaluate the cytotoxic potential of the inhibitory molecules, three mammalian cell lines namely THP-1 (human monocytic macrophage cell line), HepG2 (liver hepatocellular cell line) and HEK (human embryonic kidney cell line) were incubated with varying concentrations of the compounds in their respective media (RPMI for THP-1, DMEM for HEK and HepG2 with 10% fetal bovine serum and 1X antibiotic/antimycotic mix). All the cell lines, namely, THP-1, HepG2 and HEK were procured from Cell Repository, National Centre for Cell Science (NCCS), Pune, India. Briefly, 1 × 104 cells were incubated with varying concentrations of the compounds (200 μg ml−1 to 1.5 μg ml−1) for 48 hours followed by the addition of resazurin solution (30 μl of 0.01% solution) and fluorescence was measured the next day as described above. IC50 values were calculated by Graph Pad Prism with results of two independent tests and mean ± Standard Error Mean (S.E.M) was calculated.
3. Results and discussion
3.1 Virtual screening by using Autodock and Glide softwares
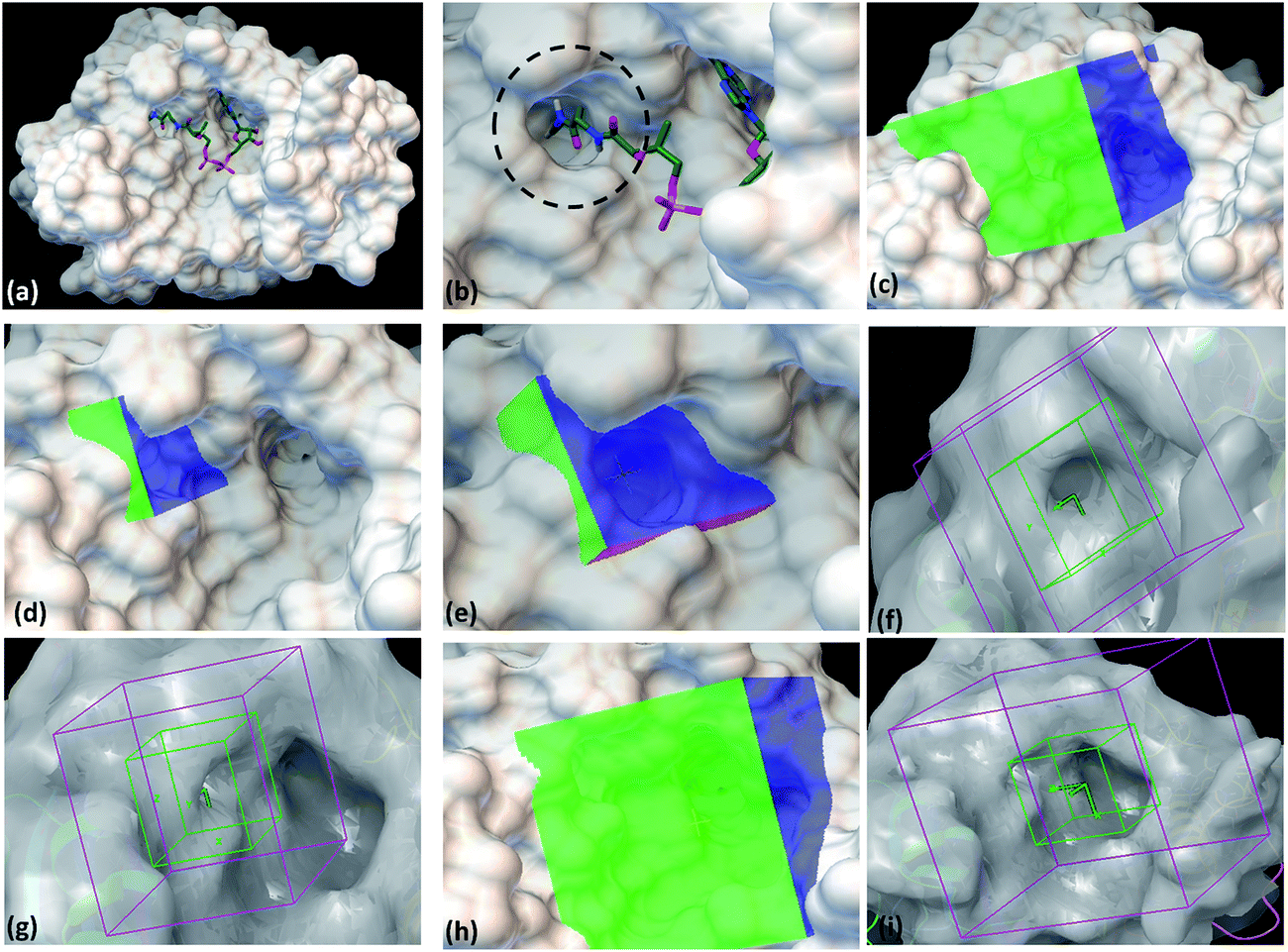
With the aim to identify inhibitors against M. tb phosphopantetheinyl transferase (PptT), we took a structure based approach by carrying out docking studies against the active site of M. tb PptT. A customized NCI library was generated by filtering the molecules using an FAF-Drugs online server on the basis of their ADME/toxicity as well as PAINS filtering as reported earlier.18–20 This filtering largely eliminated molecules exhibiting toxicity as well as PAINS like descriptor features.18,20 We docked this filtered NCI library containing ∼95000 molecules by using two softwares namely Autodock 4.2 and Glide (available in the Schrodinger Software Package). Recently, Vickery et al. determined the crystal structure of M. tb PptT and discovered a unique mode of binding of the phosphopantetheine arm of CoA with the pocket of PptT.14 This arm is buried deep inside the pocket of M. tb PptT (Fig. 1a and b). This feature is unique to M. tb PptT, as in the case of Hs-PptT (human homolog), orientation of PptT arm of CoA is shallow and is not buried deep inside the tunnel. This structural difference can be exploited to design M. tb PptT specific inhibitors. Keeping this in mind, we docked the NCI library specifically against this pocket as well as against the complete active site pocket by employing five different grids. Out of 5 grids, 3 grids were created against the pocket occupied by phosphopantetheine arm of CoA in M. tb PptT. Two of these grids were created by using Autodock 4.2, referred as AS1 (Fig. 1c) and AS2 (Fig. 1d and e) and one grid was prepared by using Glide, referred as S1 (Fig. 1f and g). These three grids covered only the phosphopantetheine pocket of PptT. The remaining two grids covering the complete active site of M. tb PptT were created, one each by Autodock (Fig. 1h) and Glide (Fig. 1i), respectively. Thus, in total, five grids were employed leading to five distinct energy based scoring lists and 41 top scoring molecules were procured from each list providing us with 205 molecules for further in vitro screening. The three separate lists obtained from docking employed by Autodock 4.2 contained free energy binding scores of less than −9 kcal mol−1 for numerous molecules. For carrying out docking studies by employing Glide, multiple conformers of the molecules were first generated by LigPrep module of Schrodinger and docked against the active site of M. tb PptT. All the conformers were docked by using first the standard precision mode followed by extra precision mode of Glide yielding numerous molecules with docking score of lower than −8 kcal mol−1. Thus, we employed two docking softwares namely Autodock 4.2 and Glide with 5 docking sites against various portions of the active site of M. tb PptT, which yielded five distinct energy based lists with numerous high affinity molecules below a good docking score of −8 kcal mol−1. Docking studies usually require a validation process to assess the ability of the docking protocol to distinguish between the known actives and inactives by the generation of decoy sets and that docking protocol which is efficiently able to distinguish this is selected ultimately. However, there is only scarce literature available regarding the known active and inactive M. tb PptT inhibitors, thus, it was difficult to carry out the validation study. Therefore, we subsequently tested 205 molecules in total so as to provide enough experimental data which can provide the basis for further development of more efficient docking protocol. The top scoring molecules were further shortlisted and procured from NCI-DTP and subsequently tested in in vitro and cytotoxicity studies for the evaluation of their inhibitory potential.
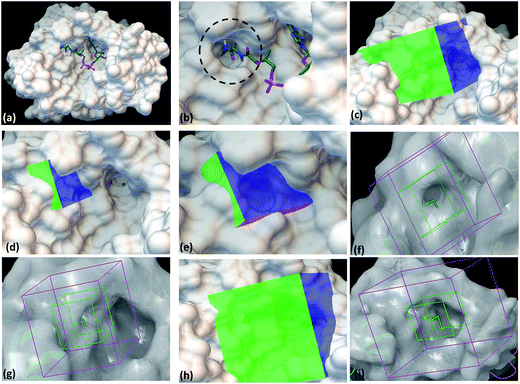 |
| | Fig. 1 Autodock 4.2 and Glide docking sites employed in the study. (a) Crystal structure of PptT bound with CoA at the active site (pdb id-4qjk), (b) zoomed in view of the cavity occupied by the Ppant arm, (c) Autodock 4.2 grid site 1 targeted against the Ppant arm of PptT, (d) Autodock 4.2 grid site 2 with grid center deep within the Ppant arm cavity, (e) zoomed in view of the Autodock grid site 2, (f) Glide grid site 1 targeted towards the Ppant arm cavity, (g) Glide grid site 1 with respect to the complete active site of PptT, (h) Autodock grid site 3 targeted towards the complete cavity of CoA, (i) Glide grid site 2 targeted towards the complete active site of PptT. In the panel (a and b), the CoA is represented in the tube model. Green represents carbon atoms backbone, red represents oxygen atoms, white represents hydrogen atoms, blue represents nitrogen atoms and yellow represents phosphate atoms. | |
3.2 Determination of inhibitory potential of molecules against M. tb PptT
Based on the docking studies, a total of 205 top scoring molecules, named as P-1 to P-205, were procured from NCI/DTP and tested against the enzymatic activity of M. tb PptT. M. tb PptT was cloned, expressed and purified as an MBP fusion protein. A number of studies have highlighted the difficulty in obtaining a soluble/active native M. tb PptT when expressed using various expression systems10–13 and thus have carried out crystallization and enzyme characterization studies by using MBP–PptT fusion protein.8,12,13 Additionally, these studies have also reported that cleavage of MBP tag results in significant precipitation of PptT protein thereby making it difficult to work with native PptT.10,12,13 Thus, we used MBP–PptT fusion protein for our activity assays and inhibition studies with M. tb AcpM as the substrate. In order to screen 205 molecules, we first characterized PptT by carrying out the time kinetics as well as Km experiment with varying concentrations of CoA by using UPLC based method for precise quantification of apo AcpM and holo AcpM peaks. It was observed that around ∼24% of apo form was converted to holo form in the presence of PptT in 60 minutes and ∼92% in 180 minute by UPLC based method (Fig. 2a). Similar conversion rate was observed by qualitative native gel electrophoreses (Fig. 2b). Kinetic parameters for CoA were determined as Km – 62 μM and Vmax – 0.12 μmoles per min and are illustrated in Fig. 2c. Subsequently, we employed relatively high throughput electrophoresis based assay by using native gel for the separation of apo and holo forms of AcpM. The molecules were screened at a fixed concentration of 100 μg ml−1 which resulted in 20 molecules which exhibited more than ∼20% inhibition of the M. tb PptT enzymatic activity (Table 1). The inhibition profile exhibited by eight of the representative compounds is depicted in Fig. 2d. Out of the 20 molecules, 12 belonged to the Autodock sites and 8 belonged to the Glide sites. Thus, our hit rate is a moderate 10% as only 12 compounds emerged to be as active amongst the 123 tested from Autodock 4.2 software and 8 out of 82 tested from Schrodinger software. However, to the best of our knowledge, this is the first in silico based study for the identification of inhibitors against M. tb PptT. We believe that the in vitro data available in this study can provide the basis for more refined docking procedures. Subsequently, these 20 molecules were then subjected to dose response studies by employing both native PAGE as well as UPLC based method. Out of the 20 molecules, 13 molecules exhibited IC50 ≤ 10 μg ml−1 (Fig. 3 and Table S1†). Two molecules namely P-137 and P-183 exhibited potent IC50 values of 1.2 μg ml−1 and 1.17 μg ml−1, respectively as determined by UPLC based assay (Fig. 4a and b) and their predicted binding mode was analyzed (Fig. 4c–f). Vickery et al. provided a detailed comparison of distinct active site residues, in terms of their identity and position that are idiosyncratic to M. tb PptT as compared to HsPptT. Residues such as Arg 48, Arg 56 and Lys 75 which varies in identity and position between M. tb PptT and HsPptT were demonstrated to be coordinating with the phosphate ion and are thus crucial for the activity of PptT.14 In concordance with this, our docking studies of P-137 predicted a few interactions such as the oxygen atoms present at 4th position attached to the oxo-hexanoic acid moiety forms key hydrogen bonds with Arg 48 and Lys 75 and oxygen atom at the 5th position of hexanoic acid forms H-bond with Arg 56 as well as key salt bridges with Arg 48 and Lys 75 and thus are predicted to inhibit the activity by disrupting the key electrostatic linkage formed by the phosphate ion with Lys 75 (Fig. 4c and d). In addition to this, a hydrogen bond was predicted between oxygen atom of the chloro benzene group with Ser 90. Thus, this provided an additional repertoire of residues apart from the phosphopantetheine arm pocket that could be important for carrying out rational modifications based on this molecule. Also, amino group of octahydronaphthalene moiety of P-183 was predicted to form H-bonding with Ala 115 and salt bridge between Asp 114 and Glu 116 and hydroxyl group at 1′ position of naphthalene moiety forming H-bond with Lys 161 (Fig. 4e and f). Thus, a few potent inhibitory molecules with IC50 ≤ 10 μg ml−1 were identified by employing UPLC based assay. The interaction of the inhibitor with various residues may provide a guide for carrying out de novo drug design or rational modification studies.
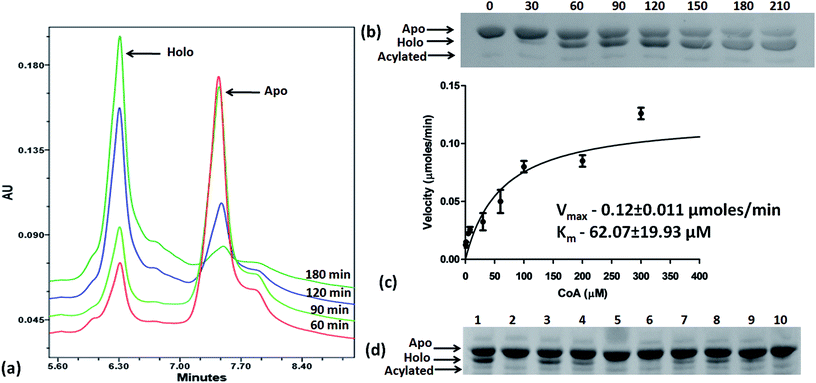 |
| | Fig. 2 Time kinetics, km experiment and native gel based screening of molecules. (a) UPLC traces of conversion of apo to holo forms of AcpM in the presence of PptT at varying time periods, (b) native gel based conversion of apo to holo forms in the presence of PptT at varying time periods, (c) km experiment of varying amounts of CoA in the presence of AcpM and PptT as measured by UPLC based assay, (d) representative figure of native PAGE based screening of molecules to evaluate their inhibitory potential against the enzymatic activity of PptT. Lanes 5, 6 and 10 denotes no visible conversion from apo to holo form of AcpM when PptT was incubated with compounds thereby depicting inhibition of PptT enzymatic activity. Lane 1 – PAC + DMSO (negative control), lane 2 – A + C, lane 3 – PAC + P-135, lane 4 – PAC + P-136, lane 5 – PAC + P-137, lane 6 – PAC + P-170, lane 7 – PAC + P-171, lane 8 – PAC + P-173, lane 9 – PAC + P-181, lane 10 – PAC + P-183. PAC stands for P-PptT, A – AcpM, C – CoA respectively, P-various P series inhibitors. | |
Table 1 Enzyme inhibition (IC50 values), whole cell inhibition as well as cytotoxicity studies of the compounds against M. tb PptT. The table depicts the IC50 against PptT activity, MIC90 as well as the IC50 values against cell lines for the final shortlisted 13 molecules which possess the ability to inhibit both enzymatic activity as well as M. tb growth in broth culture. Six out of the 205 molecules belonging to P series and 7 out of the 31 analogues belonging to PS series exhibited inhibition of both PptT and M. tb growth
| S. no. |
P/PS ID |
NCI ID |
Enzyme inhibition, IC50 values, (μg ml−1) |
MIC90, (μg ml−1) |
IC50 against THP-1, (μg ml−1) |
IC50 against HepG2, (μg ml−1) |
IC50 against HEK, (μg ml−1) |
| 1 |
P-4 |
23925 |
68.2 ± 3.9 |
17.5 ± 2.0 |
112.5 ± 5.7 |
135.0 ± 2.4 |
37.0 ± 3.6 |
| 2 |
P-52 |
53979 |
2.2 ± 1.3 |
77.5 ± 5.6 |
147.5 ± 6.8 |
>200 |
192.3 ± 5.6 |
| 3 |
P-70 |
305817 |
7.9 ± 1.4 |
24.5 ± 3.7 |
82.5 ± 2.4 |
132.7 ± 7.5 |
27.5 ± 5.5 |
| 4 |
P-102 |
95701 |
43.1 ± 1.4 |
100 |
167.5 ± 7.5 |
155.0 ± 4.7 |
175.0 ± 3.6 |
| 5 |
P-110 |
13867 |
8.5 ± 2.1 |
83.5 ± 6.2 |
>200 |
>200 |
>200 |
| 6 |
P-215 |
665278 |
7.7 ± 1.1 |
86.5 ± 8.4 |
147.5 ± 5.5 |
145.8 ± 5.4 |
167.5 ± 7.5 |
| 7 |
PS-9 |
16001 |
64.5 ± 6.2 |
15 ± 3.6 |
>200 |
>200 |
>200 |
| 8 |
PS-18 |
65385 |
66.8 ± 1.5 |
37.5 ± 4.2 |
72.5 ± 3.5 |
55.0 ± 2.7 |
85.0 ± 5.7 |
| 9 |
PS-19 |
67610 |
37.6 ± 1.6 |
40.0 ± 5.9 |
75.0 ± 2.75 |
65.0 ± 3.0 |
125.0 ± 5.5 |
| 10 |
PS-25 |
95809 |
18.0 ± 1.4 |
96.5 ± 3.1 |
>200 |
190 |
>200 |
| 11 |
PS-37 |
305815 |
29.9 ± 3.2 |
17.5 ± 4.2 |
117.5 ± 3.5 |
100.0 ± 5.8 |
190.0 ± 10.7 |
| 12 |
PS-39 |
305830 |
34.5 ± 1.5 |
20.0 ± 3.0 |
100.0 ± 4.7 |
145.0 ± 4.5 |
>200 |
| 13 |
PS-40 |
328398 |
0.2 ± 1.4 |
10.0 ± 2.5 |
117.5 ± 5.5 |
127.5 ± 3.6 |
>200 |
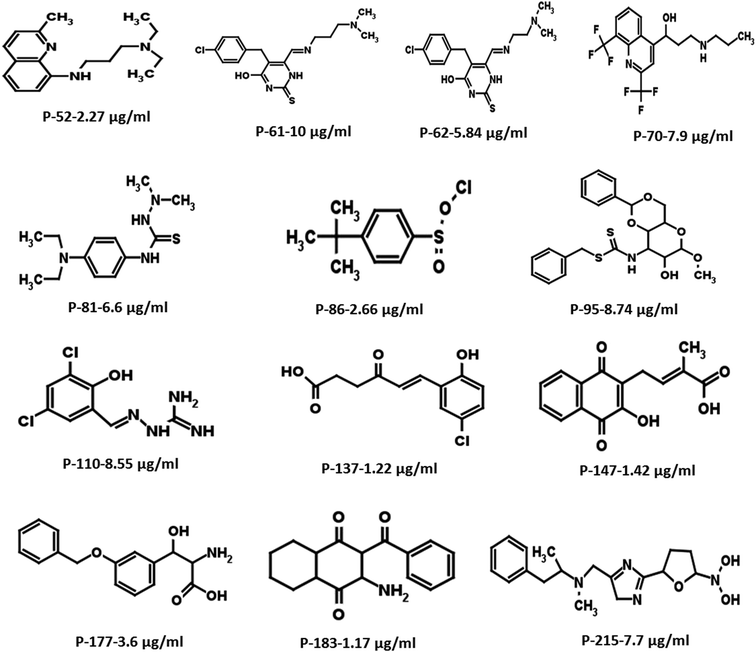 |
| | Fig. 3 Two-dimensional structures of the most potent hits with IC50 ≤ 10 μg ml−1 resulting from the initial screening of 205 molecules. | |
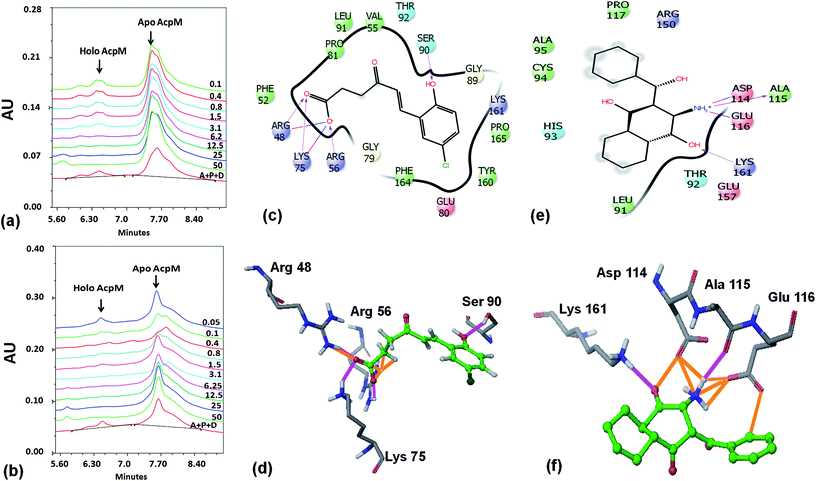 |
| | Fig. 4 UPLC traces as well as binding mode predictions of P-137 and P-183. (a) UPLC trace of various concentrations of P-137 (in μg ml−1) for IC50 calculations, (b) UPLC trace of varying concentrations of P-183 (in μg ml−1) for IC50 calculations, (c) 2-d interaction diagram of P-137 with the active site residues of PptT, (d) 3-dimensional diagram of the P-137 with residues Arg 48, Arg 56, Lys 75 and Ser 90 (e) 2-d interaction diagram of P-183 with active site residues of PptT, (f) 3-dimensional diagram of P-183 with residues Lys 161, Asp 114, Ala 115 and Glu 116. In the UPLC trace, A – AcpM, P – PptT, D – DMSO (negative control), 50–0.05 represents the respective concentrations of P-137 and P-183 in μg ml−1. In panels (d and f), the ligand molecules are shown in ball and stick model (green) and active site residues in tube model. In the ligand molecules, green represents the carbon backbone, dark green represents chlorine atom, white represents hydrogen atoms and red represents oxygen atoms. In amino acid residues, grey represents carbon backbone, blue represents nitrogen atoms, red represents oxygen and white represents hydrogen. Pink represents hydrogen bonds and orange represents salt bridges. | |
3.3 Whole cell based inhibition of M. tb in broth culture
After evaluation of the inhibitory potential of the molecules against the enzymatic activity of PptT, we then evaluated the potential of all the 20 enzyme inhibitors for their ability to inhibit the growth of M. tb in broth culture by employing a 96 well microtitre Alamar Blue based assay.18,21 Out of the 20 compounds exhibiting inhibition of enzymatic activity, 6 compounds also inhibited the growth of M. tb in broth culture. The MIC90 values of the molecules are listed in Table 1. Compounds P-52 (IC50 – 2.2 μg ml−1) and P-70 (IC50 – 7.9 μg ml−1) also inhibited the growth of M. tb at a moderate MIC90 of 77.5 μg ml−1 and MIC90 of 24.5 μg ml−1, respectively. In this study, we calculated the IC50 values as well as MIC90 of these molecules suggesting their intracellular target to be PptT, however, more detailed studies are required to validate this and the probability of off target effects cannot be completely negated. Although, few molecules such as P-137 and P-183 exhibited potent enzyme inhibition, they failed to inhibit the whole cell growth of M. tb. Few probable reasons for inability of such potent enzyme inhibitors to inhibit the whole cell growth of M. tb in broth culture may include the inability of these molecules to penetrate the cell wall of M. tb or effluxing of these molecules by various efflux pumps present at the cell wall of M. tb. Thus, we identified 6 inhibitors that exhibited IC50 ≤ 10 μg ml−1 and MIC90 in the range of 20 μg ml−1 to 100 μg ml−1 which were further taken up for lead optimization studies.
3.4 A structure similarity based search for similar analogues
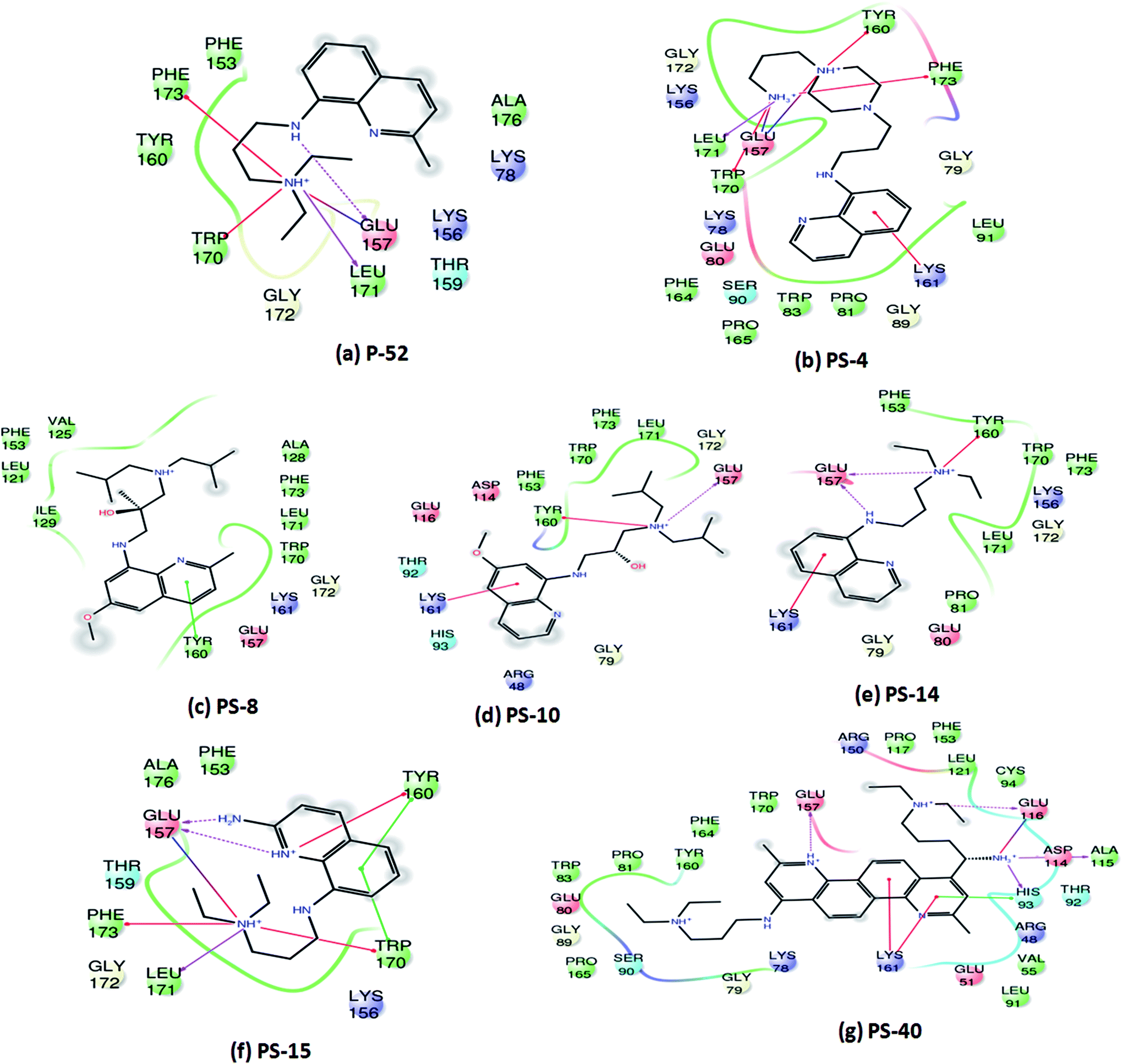
On the basis of IC50 and MIC90 studies, six lead molecules namely P-4, P-52, P-70, P-102, P-110 and P-215 were selected for further optimization studies with the aim of identification of more potent analogues. Each compound was taken as a query molecule and their analogues were searched from the complete NCI database consisting of ∼260000 compounds by using the software Open Babel. In total, 31 analogues were obtained against 5 of the 6 lead molecules taken as query molecules. No analogues of P-215 were obtained in the NCI database. Thus, 31 analogues (in total) were obtained and procured on the basis of their Tanimoto similarity coefficients with cut-off of 0.7 (prefix PS is given to all the molecules that were procured from this structure similarity based screening). All the molecules were evaluated for their enzyme inhibition potential. Out of 31 molecules evaluated, 16 exhibited enzyme inhibition with 11 molecules exhibiting IC50 below 20 μg ml−1 (Table S2†). All the 6 analogues of P-52 exhibited enzyme inhibition with five molecules having an IC50 ≤ 1 μg ml−1 with PS-40 exhibiting an IC50 – 0.25 μg ml−1 (Fig. 5a–c). Thus, P-52 bearing a diethyl-N-(2-methyl quinolin-8-y)propane-1,3-diamine emerged as a potent inhibitory scaffold. A detailed comparison of structures of P-52 (IC50 – 2.25 μg ml−1) and its analogues PS-4 (IC50 – 0.28 μg ml−1), PS-8 (IC50 – 0.64 μg ml−1), PS-10 (IC50 – 0.6 μg ml−1), PS-14 (IC50 – 0.73 μg ml−1), PS-15 (IC50 – 2.86 μg ml−1) and PS-40 (IC50 – 0.25 μg ml−1) revealed the importance of quinoline moiety attached to an amino group followed by a long carbon chain (Fig. 5d). PS-4 contains an amino propyl piperazinyl moiety attached to the quinoline moiety which may contribute to a better IC50 of PS-4. PS-8 and PS-10 contains two extra methyl groups attached to the amino group as compared to only two methyl groups attached to the amino group in P-52 lead molecule. Also, PS-8 and PS-10 contains a methoxy group attached to piperazine moiety which is absent in P-52 lead molecule. These miniscule changes could be a probable reason for their slightly better IC50 values. Removal of methyl group attached to quinoline moiety in PS-14 led to a better IC50 value as compared to P-52, however, change of methyl to amino group, as in the case of PS-15 led to a slightly poorer IC50 value. Thus, this position can also be exploited for carrying out chemistry based rational designing. Also, PS-40 is essentially two P-52 molecules attached together (Fig. 5d). This further emphasizes that a comparatively large molecular size molecule bearing a quinoline group is required for potent inhibition of PptT. These minuscule differences might explain their better IC50 values.
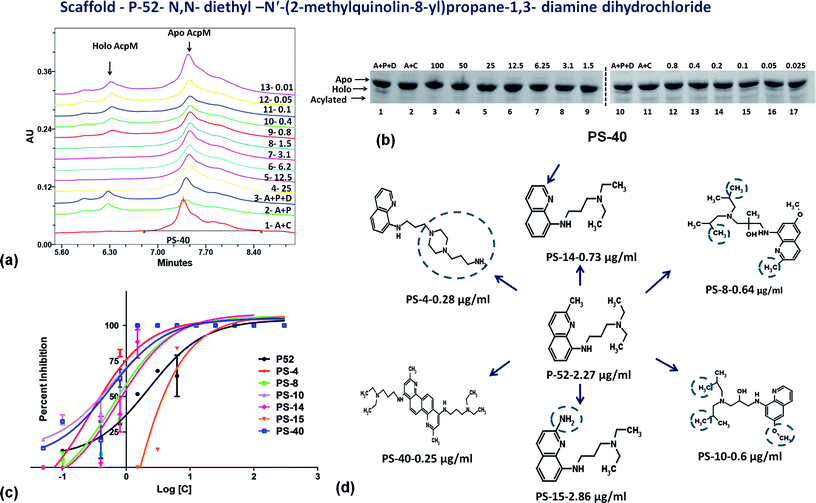 |
| | Fig. 5 Inhibition studies of the analogues of P-52. (a) UPLC trace of PS-40. In the UPLC trace, A – AcpM, P – PptT, D – DMSO (negative control), 25–0.01 represents the respective concentrations of PS-40 in μg ml−1, (b) native PAGE based screening in the presence of varying concentrations of PS-40. A – AcpM, C – CoA, P – PptT, D – DMSO, 100–0.025 represents various concentrations of PS-40 in μg ml−1, (c) IC50 curves of various analogues of P-52, (d) 2-d structure comparison of analogues of P-52. | |
Since, P-52 and its analogues were identified as the most potent inhibitory molecules in this study; detailed predicted interactions were carried out to provide an insight into their probable binding mode with the active site residues. A few important predicted interactions between these molecules and various crucial amino acid residues of PptT were identified which include hydrogen bond as well as salt bridges between Glu 157 and amino groups present in the side chain as well as the phenyl group of P-52 as well as most of its analogues such as PS-4, PS-10, PS-14, PS-15 and PS-40. This interaction might be responsible for their potent inhibitory action against the enzymatic activity of PptT (Fig. 6). Few interactions that were distinct in the analogues and not exhibited by P-52 include Lys 161 which was predicted to be important for coordination with α phosphate ion and a Π–cation interaction between the phenyl groups of quinoline moiety. These interactions were exhibited by PS-4, PS-10, PS-14 and PS-40 but not by P-52. This may explain in part, an enhanced inhibitory potential of these analogues as compared to P-52 (Fig. 6a, b, d, e and g). Amino groups of PS-40 are also predicted to form hydrogen bond with Glu 116 as well as Asp 114 and Ala 115 (Fig. 6g). The interactions corroborate with the earlier mutational experiments that demonstrated the important role of these residues in coordination with Mg2+ ion as the substitution of Asp 114 and Glu 116 led to complete abolishment and 500 fold reductions in the activity of PptT, respectively11 (Fig. 6g). These additional predicted interactions of PS-40 with important residues of PptT namely Glu 157, Glu 116 and Asp 114 might be a reason for the potent IC50 value of PS-40 (Fig. 6g). In addition, most of the molecules were also predicted to be interacting with Tyr 160, Trp 170, Leu 171 and Phe 173 thereby suggesting the role of these residues for better affinity of the inhibitory molecules (Fig. 6). Indeed, these additional residues can serve as an important resource while carrying out more rational modifications and de novo drug design.
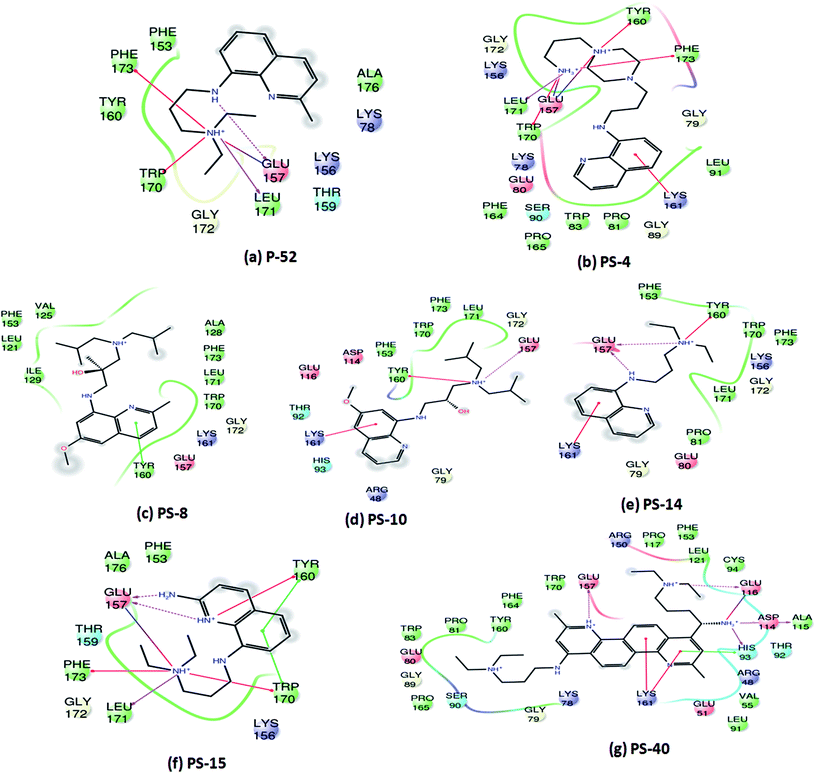 |
| | Fig. 6 Two-dimensional interaction diagrams of analogues of P-52 with various active site residues of PptT. | |
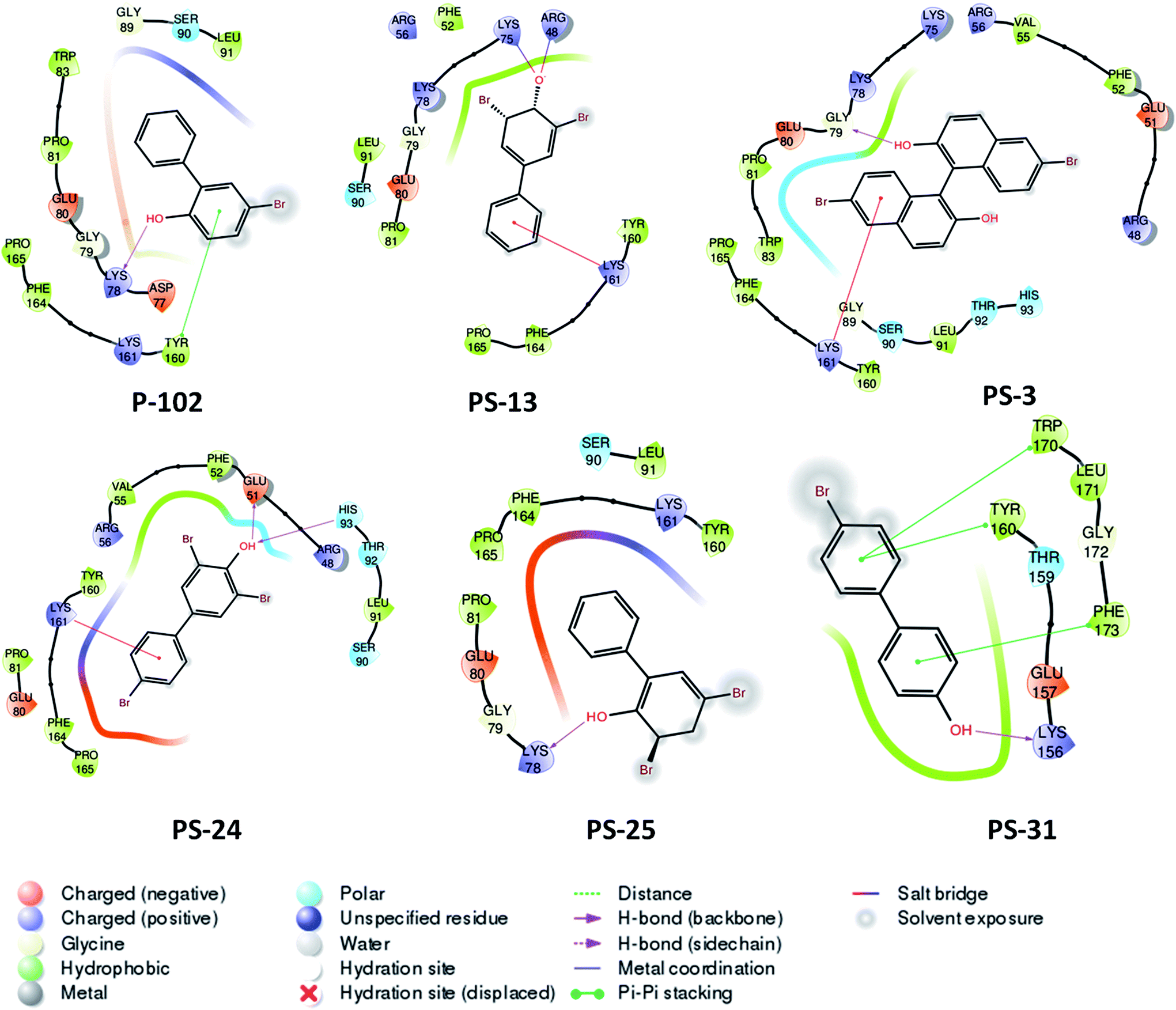
The second important scaffold that emerged from this study belongs to the molecule P-102 (IC50 – 43 μg ml−1). It contains a 5-bromobiphenyl-2-ol scaffold. All the 5 analogues of P-102 exhibited enzyme inhibition with IC50 of three molecules namely PS-3, PS-24 and PS-31 being 4 μg ml−1, 5 μg ml−1 and 7.1 μg ml−1, respectively as depicted by the UPLC traces of PS-3 (Fig. 7a) as well as the IC50 curve plot of the various analogues of P-102 (Fig. 7b). These molecules exhibited an increased inhibition of ∼10 fold, ∼8 fold and ∼6 fold, respectively, in their IC50 values from their parent molecule (Fig. 7c). A detailed structural comparison of the analogues with their parent molecule provided insights into their probable reason for a better IC50 values. A comparison of P-102 and its analogue, PS-3, revealed that the presence of an additional bromo benzyl group attached to the phenyl groups increases the potency of this scaffold (Fig. 7c). The comparison of structures of PS-24 and P-102 reveals a change in the position of the hydroxyl group from ortho to para position as well as presence of additional bromine functional groups at both the phenyl rings. These changes in comparison to P-102 could be cumulatively responsible for approximately 8 fold improvement in the IC50 values of PS-24 as compared to P-102 (Fig. 7c). A comparison between PS-24 and PS-13 suggests the importance of bromine present at the para position of benzyl ring in PS-24 which is absent in PS-13. The presence of this bromine group might corroborate with a two-fold better activity of PS-24 as compared to PS-13. Additionally, change of para position of hydroxyl position (PS-13) to ortho position (PS-25) leads to a slight decrease in the IC50 value (Fig. 7c). Thus, the detailed structural comparison of analogues with the lead molecule of P-102 might suggest further modification of these molecules by employing rational modifications.
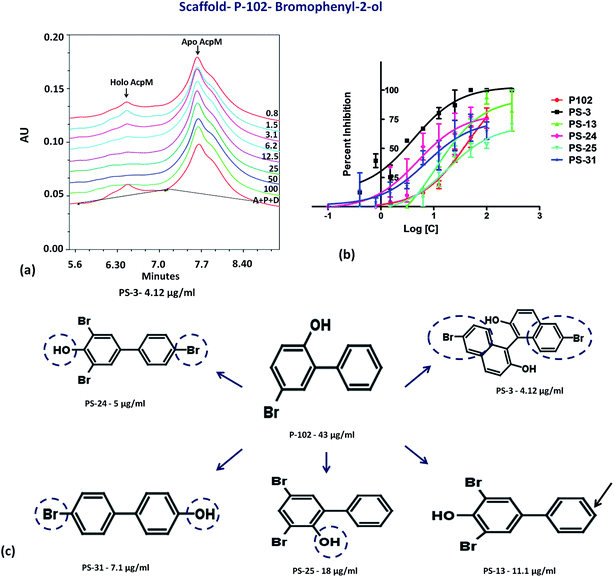 |
| | Fig. 7 Inhibition studies of the analogues of P-102. (a) UPLC trace of PptT inhibition assays in the presence of varying concentrations of PS-3. A – AcpM, P – PptT, D – DMSO, 100–0.8 depicts varying concentrations of PS-3 in μg ml−1, (b) IC50 plot of various analogues of P-102, (c) 2-d structure comparison of the analogues of P-102. | |
Probable binding poses of P-102 as well as its analogues were determined by the docking studies. The benzene moiety of P-102 as well as its analogues PS-13, PS-3, PS-31 and PS-24 forms either Π–cation or Π–Π stacking interactions with a few important residues such as Tyr 160 or Lys 161 (Fig. 8). This interaction is common between P-102 as well as most of its analogues and highlights the importance of the benzene moiety for activity against M. tb PptT. Also, additional interactions were present in few other analogues which were not observed in P-102 thereby indicating few important residues which could be important during the development of de novo drug discovery. For e.g., hydroxyl group attached to the two bromo hydroxyl benzyl moiety of PS-13 forms salt bridges with Lys 75 and Arg 48. Also, the hydroxyl moiety present in a similar position in PS-24 forms hydrogen bond with Glu 51 and His 93. The hydroxyl benzyl moiety in PS-31 forms hydrogen bond with Lys 156 and a Π–Π stacking interaction with Phe 173. Also, the benzyl group of the bromo benzyl moiety forms Π–Π stacking interactions with Tyr 160 and Trp 170. Thus, our docking studies revealed insights into the predicted mode of interactions of P-102 as well as its analogs which could help in further rational chemistry based modifications for the development of inhibitors against M. tb PptT.
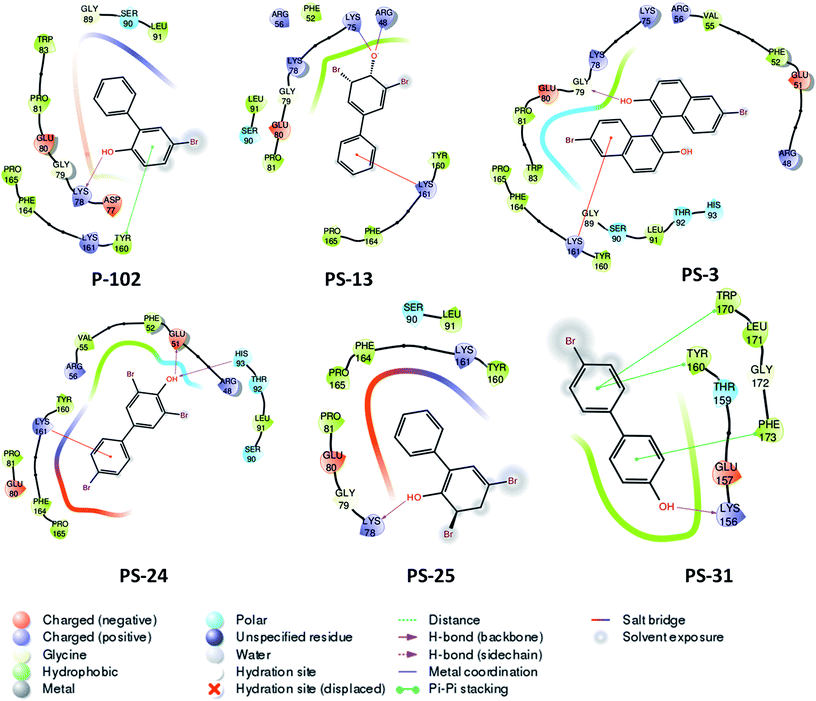 |
| | Fig. 8 Two dimensional diagram of P-102 and its analogues docked inside the active site of M. tb PptT. | |
Thus, the structure based similarity search followed by enzyme inhibition studies provided a few hot spots in the analogues of the two lead molecules that can provide a robust platform for carrying out further rational modifications by various medicinal chemistry researchers. However, the study should be considered as a proof of concept which can only serve as a platform for further detailed rational modifications studies.
3.5 Whole cell inhibition of similar analogues against the growth of M. tb in broth culture
Out of the sixteen molecules identified on the basis of structure similarity search that exhibited enzyme inhibition, seven molecules exhibited inhibition of growth of M. tb in broth culture (Table 1). Compound PS-40, the analogue of the most potent scaffold (P-52) with IC50 of 0.25 μg ml−1 exhibited a potent MIC90 of 10 μg ml−1 which was better than its parent molecule, P-52 and can serve as a starting point for further lead modification studies. Additionally, a few analogues of other lead molecules also exhibited moderate MIC90 in the range of 10 μg ml−1 to 100 μg ml−1 (Table 1). Thus, structure similarity based approach provided a few scaffolds whose lead as well as analogues exhibited enzyme as well as M. tb inhibition in vitro.
3.6 Cytotoxicity of the molecules against various mammalian cell lines
Before announcing the true promise of a molecule, it is important to evaluate its toxicity against various mammalian cell lines apart from its potential to exhibit enzyme as well M. tb growth inhibition. Since, the primary residence of M. tb in human host is macrophages, we evaluated the cytotoxicity of the molecules against THP-1 human macrophage cell line. Also, since liver and kidney are the primary sites for metabolism as well as excretion of the drug molecules, we checked the cytotoxicity of the molecules against HepG2 (human liver cancer cell line) as well as HEK (human embryonic kidney cell line). Molecules belonging to the lead molecule P-52 that exhibited moderate to potent IC50 as well MIC90 values showed negligible cytotoxicity in all the cell lines. P-52, with IC50 of 2.2 μg ml−1 and MIC90 of 77.5 μg ml−1 bear an IC50 of 150 μg ml−1 and 190 μg ml−1 against THP-1 and HEK, respectively and no cytotoxicity till 200 μg ml−1 against HepG2 (highest concentration tested) as mentioned in Table 1. PS-40, the most potent analogue belonging to this scaffold with IC50 – 0.25 μg ml−1 and MIC90 of 10 μg ml−1 also exhibited negligible cytotoxicity with IC50 of 110 μg ml−1 against THP-1, 127.5 μg ml−1 against HepG2 and no toxicity up to 200 μg ml−1 against HEK thereby exhibiting a selectivity index (S.I) of 11 against THP-1, 12.75 against HepG2 and more than 20 against HEK. Cytotoxicity of other compounds is reported in Table 1. Some of the compounds are also exhibiting lower therapeutic index and thus a probable solution could be the rational modification of these molecules by click chemistry based design and synthesis that can alleviate the cytotoxicity exhibited by these compounds. Overall, P-52 and its analogue, PS-40 exhibited the most promising cytotoxicity results thereby providing an additional support for carrying out further drug discovery efforts.
4. Conclusion
4′ Phosphopantetheinyl transferase was recently shown to be an important drug target for carrying out target based drug discovery efforts as a conditional mutant of PptT was shown to be attenuated in immune compromised as well as immune competent mice model of tuberculosis infection. Additionally, the non-redundant nature of the phosphopantetheinyl transferases shown by both genetic as well as biochemical studies further highlighted the importance of PptT as an attractive target. The recent elucidation of its 3-dimensional crystal structure also provided a platform for carrying out structure based drug discovery. In the present study, we combined virtual screening with cheminformatics technique to screen 236 inhibitors against the enzymatic activity of PptT by employing a semi-quantitative but high throughput electrophoresis using native polyacrylamide gel. Further, UPLC based assay yielded 13 molecules with IC50 ≤ 10 μg ml−1. Also, MIC90 of these inhibitors were evaluated against the growth of M. tb. Subsequently, structure similarity based search was carried out for five lead molecules that exhibited moderate IC50 as well as MIC90 values and two potent inhibitory scaffolds of P-52 and P-102 emerged from this approach, namely, N,N- diethyl-N′-(2-methyl quinolin-8-yl) propane-1,3-diamine with 5 analogues of P-52 exhibiting IC50 ≤ 1 μg ml−1 and 5-bromobiphenyl-2-ol with 3 analogues exhibiting IC50 ≤ 10 μg ml−1, respectively. Subsequently, MIC90 of these analogues as well as cytotoxicity studies against three mammalian cell lines namely THP-1, HepG2 and HEK were carried out which yielded one potent molecule, PS-40 (analogue of P-52) with IC50 of 0.25 μg ml−1 and MIC90 of 10 μg ml−1 and low cytotoxicity in various mammalian cell lines. The flowchart of the study is given in Fig. 9.
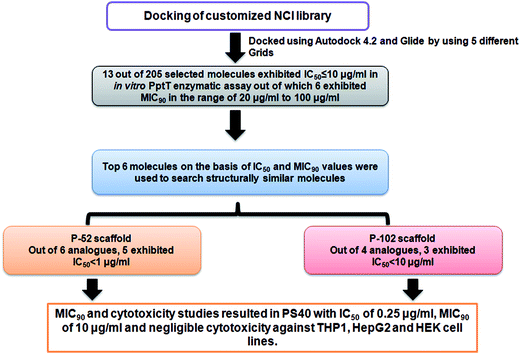 |
| | Fig. 9 Flowchart of the work carried out in this study. | |
To the best of our knowledge, no studies have carried out the identification of inhibitors against M. tb PptT as well as against the growth of M. tb in broth culture. Although, a study tested the known inhibitors of Sfp (a Bacillus subtilis homologue) against enzymatic activity of M. tb PptT, the potential of those hits to inhibit the growth of M. tb in culture was not reported.14 Thus, our study combined various in silico as well as in vitro approaches resulting in the identification of N,N- diethyl-N′-(2-methyl quinolin-8-yl)propane-1,3-diamine as a potent inhibitory scaffold of 4′ phosphopantetheinyl transferase, an important drug target of Mycobacterium tuberculosis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Drug Synthesis and Chemistry Branch, Development Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, National Institute of Health, Bethesda, MD, is thanked for supplying the compounds used in this study. Priti Singh and Tannu Priya Gosain are thanked for their technical support. DBT-supported Distributed Information Sub-Centre, Department of Biochemistry, University of Delhi South Campus, New Delhi is thanked for computational support. Dr Sangeeta at Central Instrumentation facility is acknowledged for her help with the UPLC studies. Prof. R. P. Roy is acknowledged for HPLC standardizations. A. R is thankful to Department of Science and Technology, India for the financial assistance through J. C Bose fellowship awarded to Prof. Anil K. Tyagi. This work was financially supported by Department of Biotechnology, Government of India (Grant Number-BT/01/COE/05/06-II).
References
- WHO tuberculosis report, 2016, http://apps.who.int/iris/bitstream/10665/250441/1/9789241565394-eng.pdf?ua=1.
- J. B. Nachega and R. E. Chaisson, Clin. Infect. Dis., 2003, 36, S24–S30 CrossRef CAS PubMed.
- M. C. Raviglione and I. M. Smith, N. Engl. J. Med., 2007, 356, 656–659 CrossRef CAS PubMed.
- C. T. Walsh, A. M. Gehring, P. H. Weinreb, L. E. Quadri and R. S. Flugel, Curr. Opin. Chem. Biol., 1997, 1, 309–315 CrossRef CAS PubMed.
- T. A. Keating and C. T. Walsh, Curr. Opin. Chem. Biol., 1999, 3, 598–606 CrossRef CAS PubMed.
- J. J. De Voss, K. Rutter, B. G. Schroeder and C. E. Barry 3rd, J. Bacteriol., 1999, 181, 4443–4451 CAS.
- R. H. Lambalot, A. M. Gehring, R. S. Flugel, P. Zuber, M. LaCelle, M. A. Marahiel, R. Reid, C. Khosla and C. T. Walsh, Chem. Biol., 1996, 3, 923–936 CrossRef CAS PubMed.
- L. E. Quadri, J. Sello, T. A. Keating, P. H. Weinreb and C. T. Walsh, Chem. Biol., 1998, 5, 631–645 CrossRef CAS PubMed.
- C. Chalut, L. Botella, C. de Sousa-D'Auria, C. Houssin and C. Guilhot, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8511–8516 CrossRef CAS PubMed.
- C. Leblanc, T. Prudhomme, G. Tabouret, A. Ray, S. Burbaud, S. Cabantous, L. Mourey, C. Guilhot and C. Chalut, PLoS Pathog., 2012, 8, e1003097 CAS.
- O. Zimhony, A. Schwarz, M. Raitses-Gurevich, Y. Peleg, O. Dym, S. Albeck, Y. Burstein and Z. Shakked, Biochemistry, 2015, 54, 2360–2371 CrossRef CAS PubMed.
- J. Jung, G. Bashiri, J. M. Johnston and E. N. Baker, FEBS Open Bio, 2016, 6, 1220–1226 CrossRef CAS PubMed.
- J. Jung, G. Bashiri, J. M. Johnston, A. S. Brown, D. F. Ackerley and E. N. Baker, J. Struct. Biol., 2014, 188, 274–278 CrossRef CAS PubMed.
- C. R. Vickery, N. M. Kosa, E. P. Casavant, S. Duan, J. P. Noel and M. D. Burkart, ACS Chem. Biol., 2014, 9, 1939–1944 CrossRef CAS PubMed.
- A. Yasgar, T. L. Foley, A. Jadhav, J. Inglese, M. D. Burkart and A. Simeonov, Mol. BioSyst., 2010, 6, 365–375 RSC.
- J. G. Owen, J. N. Copp and D. F. Ackerley, Biochem. J., 2011, 436, 709–717 CrossRef CAS PubMed.
- N. M. Kosa, T. L. Foley and M. D. Burkart, J. Antibiot., 2014, 67, 113–120 CrossRef CAS PubMed.
- A. Rohilla, G. Khare and A. K. Tyagi, Sci. Rep., 2017, 7, 4653 CrossRef PubMed.
- M. A. Miteva, S. Violas, M. Montes, D. Gomez, P. Tuffery and B. O. Villoutreix, Nucleic Acids Res., 2006, 34, W738–W744 CrossRef CAS PubMed.
- C. Aldrich, C. Bertozzi, G. I. Georg, L. Kiessling, C. Lindsley, D. Liotta, K. M. Merz Jr, A. Schepartz and S. Wang, ACS Med. Chem. Lett., 2017, 8, 379–382 CrossRef CAS PubMed.
- A. Martin, M. Camacho, F. Portaels and J. C. Palomino, Antimicrob. Agents Chemother., 2003, 47, 3616–3619 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11198c |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Garima Khare
a,
Garima Khare