
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDopamine@Nanodiamond as novel reinforcing nanofillers for polyimide with enhanced thermal, mechanical and wear resistance performance†
Songlv Qinab,
Mingjun Cuib,
Shihui Qiub,
Haichao Zhao *b,
Liping Wang*b and
Afang Zhanga
*b,
Liping Wang*b and
Afang Zhanga
aCollege of Materials Science and Engineering, Shanghai University, Nanchen Road 333, Shanghai, 200444, China. E-mail: zhaohaichao@nimte.ac.cn; wangliping@nimte.ac.cn; Tel: +86-057-486325713
bKey Laboratory of Marine Materials and Related Technologies, Zhejiang Key Laboratory of Marine Materials and Protective Technologies, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, China 315201
First published on 18th January 2018
Abstract
In this study, to achieve a homogeneous dispersion of nanodiamond (ND) in a polyimide (PI) matrix and a strong interfacial adhesion between ND and the PI matrix, a biomimetic nondestructive dopamine chemistry was employed for surface modification of ND. FTIR and Raman spectroscopy studies revealed that self-polymerization of dopamine could produce thinner polydopamine (PDA) layers on the ND surface via spontaneous oxidation and the intermolecular cross-linking reaction of PDA molecules. The structure and morphology of PDA–ND were studied by FTIR, SEM, and Raman spectroscopy, which verified the π–π interactions between PDA and ND. The facile dispersion of PDA–ND in a polyamic acid prepolymer made it possible to obtain PI/ND composites with no obvious ND aggregation. The effect of PDA–ND nanoparticles on the thermal, mechanical and tribological properties of the resulting PI/PDA–ND composites were evaluated, and the results showed that the incorporation of PDA–ND could increase the hardness, tensile strength, storage modulus, as well as the wear resistance properties. PI/PDA–ND composites prepared in this study showed that PDA–ND is a promising nanoreinforcing filler for PI composites.
1. Introduction
Carbon-based nanomaterials (i.e., fullerene, nanodiamond, carbon nanotubes, graphene) combine unique physical and chemical properties, making them ideal candidates for the fabrication of composite materials with a wide range of applications, such as energy storage, in sensors, in displays, as catalysts, nanomedicines, gas separation and water purification.1–3 Despite numerous advances in the development of carbon-based composites, there are still many issues to be faced regarding the synthesis, scaled-up production, and purification of carbon-based nanomaterials.4–6 In particular, carbonaceous materials tend to aggregate in the polymer matrix due to their strong π–π stacking and van der Waals interactions, which will seriously damage the performance of nanomaterials.7,8 Therefore, the homogeneous dispersion of carbonaceous materials throughout the matrix without destroying its integrity and good interfacial bonding with the polymer matrix are essential for their effective utilization in composite applications. Covalent functionalization of carbonaceous materials is an effective approach to increase their processability and manipulate the interfacial properties of composites.9 Although several reactions have been successfully developed, such as oxidation,10 Friedel–Crafts acylation,11 diazo coupling,12 the Diels–Alder reaction,13 the oxygen radical reaction,14 and alkylation,15 most of these chemical processes rely on harsh reaction conditions, like strong acids, oxidants, high temperature, and the exclusion of oxygen and water. As a consequence, it is highly desirable to develop novel synthetic routes that are very efficient, simple, scalable and versatile for the functionalization of carbon-based materials.Recently, a biomimetic dopamine self-polymerization inspired by mussel protein has been devised to modify a variety of substrates, leading to multifunctional surfaces for diverse applications.16 In comparison with conventional chemical modification methods, mussel-inspired dopamine chemistry features a versatile ability for use with almost any organic or inorganic substrates under moderate conditions, enabling its use as a modifier for nanofillers to achieve strong interfacial adhesion and good dispersion in polymeric nanocomposites.17–19 By taking advantage of the self-polymerization of dopamine, several polydopamine (PDA)-coated carbonaceous composite materials with multifunctionality and high mechanical performance have been successfully fabricated.20,21 For instance, Fei et al. prepared PDA-coated carbon nanotubes of predefined thickness by adjusting the reaction time, temperature and pH. The resulting polydopamine coating could act as a platform for further tailoring of additional functions and electroless metal deposition.22 Cui et al. reported the fabrication of cross-linked graphene oxide (GO) sheets using dopamine as a crosslinker via an evaporation-induced assembly process. The prepared artificial-nacre-based composites integrated high tensile strength, excellent toughness and high electrical conductivity.23 Recently, dopamine has also been used as both reducing agent for graphene oxide (GO) and for noncovalent functionalization of the resulting graphene, providing a versatile platform for further functionalization24,25 or as a reactive component for the preparation of polymer/graphene composites with enhanced physical properties.26–28 Although polydopamine-modified carbonaceous materials could enhance the mechanical properties of polymers, the tribological properties of the resulting polymer composites have rarely been studied, particularly polyimide with a high coefficient of friction and poor wear resistance. Given the ultra-high hardness and wear resistance of nanodiamond, we expected that the tribological properties will be improved with the addition of nanodiamond.
Polyimides (PIs) are known as a class of aromatic polymers showing excellent mechanical properties, high thermal stability, and chemical and radiation resistance, all of which are of critical importance for their practical applications in microelectronics, in membranes, as fibers, and in the aerospace industry.29–31 It is of considerable interest to impart polyimide with improved or novel properties by incorporation of carbon-based materials like fullerene,32–34 carbon nanotubes,35,36 graphene,37–40 and nanodiamond (ND).41–43
In this study, we present the preparation of carbon-based polyimide nanocomposites using polydopamine-coated nanodiamond (PDA–ND) as reinforcing nanofillers, as shown in Scheme 1. Self-polymerization with dopamine chemistry was first employed to functionalize nanodiamond, with the aim of addressing the aggregation issue of ND and to achieve a uniform distribution in the polyamic acid prepolymer. PI/ND nanocomposites were then prepared by in situ polycondensation of 4,4′-oxydianiline (ODA), pyromellitic dianhydride (PMDA), and various amounts of PDA–ND in dimethylacetamide under N2 at room temperature, followed by thermal imidization at an elevated temperature. The dispersity of ND and PDA–ND in the polyamic acid prepolymer solution was analyzed. The structure and morphology of PDA–ND were studied by FTIR, SEM, and Raman spectroscopy, which verified the π–π interactions between PDA and ND. Microstructure analyses using XRD and SEM were performed to investigate the morphology of ND in the polyimide matrix. The thermal, mechanical and tribological properties of PI/PDA–ND nanocomposites as a function of ND content were studied in detail.
 | ||
| Scheme 1 Schematic illustration of in situ preparation of PI/PDA–ND composite films. | ||
2. Experimental section
2.1 Materials
Dopamine hydrochloride, tris(hydroxymethyl)aminomethane, pyromelliticdianhydride (PMDA), 4,4′-oxidianiline (ODA) and dimethylacetamide (DMAc) were supplied by the Aladdin Industrial Corporation. HCl solution was purchased from Sinopharm chemical Reagent Co. Ltd. Nanodiamond was provided by Beijing DK Nanotechnology Co. Ltd.2.2 Preparation of nanodiamond with dopamine (PDA–ND)
A typical procedure for the modification of nanodiamond with dopamine is as follows. Dopamine hydrochloride (0.8 g) was dissolved in 400 mL of tris(hydroxymethyl)aminomethane (pH = 8.5) in a 1000 mL round bottom flask. Then 2 g of nanodiamond was added to the flask in an ultrasonic bath for 3 h. After that, the mixture solution was stirred at 60 °C for 24 h. Finally, the reaction solution was washed with plenty of deionized water to remove the residual dopamine, centrifuged and dried overnight. Subsequently, gray dopamine-modified nanodiamond (PDA–ND) was obtained.2.3 Preparation of PAA/PDA–ND suspension with various mass fractions of PDA–ND
Various quantities of PDA–ND (0.01 g, 0.02 g, 0.03 g, 0.04 g) were dispersed in dry DMAc (28 mL). Then ODA (2.00 g, 0.01 mol) was added after sonication of the mixtures for 1 h at room temperature. When the ODA was completely dissolved in the DMAc, PMDA (2.18 g, 0.01 mol) was added to the mixture separately and stirred for 24 h at room temperature under a flow of N2, yielding a PAA/nanodiamond hybrid suspension with various PDA–ND loadings (0.25 wt%, 0.5 wt%, 0.75 wt%, 1 wt%). Further increases in the PDA–ND content in the PI matrix led to serious cracking during thermal imidation. This may be the reason why a high content of nanodiamond cannot achieve a high molecular weight of PI (Fig. S1†). Pure PAA in DMAc solution was prepared in a similar procedure without the addition of PDA–ND.2.4 Synthesis of polyimide/nanodiamond composite films by thermal imidization
PI/PDA–ND composite films and neat PI were prepared by solution casting and a subsequent thermal imidization method. The prepared PAA/nanodiamond suspension or neat PAA solution were cast on 2 cm × 2 cm sheet steel (Q235) and glass substrates; then the precursor was degassed in a vacuum oven at 100 °C for 1 h to remove excessive solvent. For the thermal imidization reactions, the above specimens were placed in an air circulation oven at 150, 200, 250 and 300 °C for 1 h to evaporate all the volatiles and achieve thermal imidization. The thickness of the PDA–ND/PI nanocomposite films is ca. 0.13 mm.2.5 Instruments and characterization
Intelligent Fourier infrared (FTIR) spectra of ND and PDA–ND were obtained by a FTIR (NICOLET 6700) spectrometer. The FTIR spectrum was recorded between 500 and 4000 cm−1. The chemical composition of the samples was obtained by X-ray photoelectron spectrum measurement (XPS), which was carried out on a multi-functional spectrometer (AXIS ULTRADLD) using Al Kα radiation. The specimens of ND and PDA–ND were characterized by a confocal micro Raman spectrometer (Renishaw inVia Reflex). The microstructures of ND and PDA–ND were performed with TEM (Tecnai F20). Particle size analysis of ND and PDA–ND was performed with dynamic light scattering (Zetasizer Nano ZS). X-ray diffraction (XRD) patterns were acquired by a D8 ADVANCE using Cu Kα radiation (λ = 0.154 nm) at an accelerating voltage of 40 kV and current of 40 mA. Atomic force micrographs (AFM) were conducted using a commercial instrument (Dimension 3100) in the tapping mode. The fracture morphology of the PI/PDA–ND composite films and the microstructure of ND and PDA–ND were observed by scanning electron microscopy (HITACHI S4800). The worn surface and wear debris of the PI/PDA–ND composite coatings were observed by scanning electron microscopy (FEIQuanta FEG250). Thermogravimetric analysis (TGA) of PI/PDA–ND composite films was carried out with Diamond TG/DTA under air flow at a heating rate of 10 °C min−1.A tensile test of the PI/PDA–ND composite film samples was performed on a 5567 universal material testing machine with a test speed of about 0.5 mm min−1. At least five specimens were used of each sample during the test. Dynamic mechanical analysis (DMA) was studied on a DMAQ800 analyzer. The Vickers hardness of the PI/PDA–ND composites was tested on a microhardness tester (HV-1000) under a load of 100 N.
The tribological properties of the samples were evaluated using a UMT-3. The friction mode is reciprocating friction. And the diameter of the steel ball is 3 mm. Evaluation of the tribological behavior was conducted at a 2 Hz sliding frequency under a constant load of 3 N with a test duration of 30 min. Before each test, the steel balls were cleaned with acetone followed by drying. All the tests were carried out at about 24 °C and a relative humidity of 40–45%. The wear rate and depths of the specimens after the tribological test were measured using a surface profiler (D-100, KLA, Tencor).
3. Results and discussions
3.1 Preparation and characterization of the poly(amic acid)/ND hybrid suspension
It is a challenge to steadily disperse ND in a polymer matrix to achieve their optimal properties. Owing to its high aspect ratio and strong interlayer van der Waals interactions, ND tends to aggregate in organic solvents or in a polymer matrix. As shown in Fig. 1a, pristine ND (marked as A) were partly aggregated in DMAc, while it was obvious that PDA-functionalized ND could be stably dispersed in DMAc solvent (marked as B), making it possible to prepare a homogenous PAA/ND suspension via in situ polymerization of ODA, PMDA, and various amounts of PDA–ND (marked as C). The morphology and particle size distribution of pristine ND and the resulting PDA-coated ND were examined by SEM, TEM and dynamic light scattering (DLS), respectively. Typical SEM and TEM images showed that primary ND particles exhibited a typical irregular shape (Fig. 1b1) and part aggregated morphology (Fig. 1b2), and the average size of the ND was around 183.5 nm with a narrow size distribution. No obvious aggregation in the morphology of PDA–ND could be seen after the dopamine treatment (Fig. 1c1–2), and the average size of PDA–ND was measured to be 196.4 nm, implying the formation of a PDA layer on the ND surface. | ||
| Fig. 1 Digital images of (a) ND in DMAc, PDA–ND in DMAc, PAA/PDA–ND in DMAc; SEM images of pristine ND (b1) and PDA–ND (c1); typical TEM images of ND (b2) ND and PDA–ND (c2); size distribution of pristine ND (b3) and PDA–ND (c3). | ||
The chemical functionality of PDA–ND and the resulting poly(amic acid)/ND suspension were characterized by FTIR, Raman spectroscopy and XPS, respectively. Fig. 2a shows the FTIR spectrum of pristine ND and PDA–ND particles. As shown in Fig. 2a, the characteristic peaks of pristine ND were a broad absorption band at 3421 cm−1 due to OH functional groups or/and absorbed H2O, a 1771 cm−1 band due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch, and a band at 1623 cm−1 due to the COOH group, suggesting the presence of hydroxy and carbonyl species. The presence of PDA layers on the ND surface was confirmed by intense peaks at 1778 cm−1 due to the presence of quinone groups, at 1531 cm−1 due to aromatic amines, and at 1387 cm−1 owing to the CNC stretching modes of the indole rings of PDA. All of which supported the successful formation of PDA layers on the ND particle surface.44
O stretch, and a band at 1623 cm−1 due to the COOH group, suggesting the presence of hydroxy and carbonyl species. The presence of PDA layers on the ND surface was confirmed by intense peaks at 1778 cm−1 due to the presence of quinone groups, at 1531 cm−1 due to aromatic amines, and at 1387 cm−1 owing to the CNC stretching modes of the indole rings of PDA. All of which supported the successful formation of PDA layers on the ND particle surface.44
 | ||
| Fig. 2 (a) FTIR spectra of ND and PDA–ND; (b) Raman spectra of ND and PDA–ND. | ||
For the Raman spectrum of ND and PDA–ND shown in Fig. 2b, pristine ND exhibits a typical crystal diamond peak at 1323 cm−1. On the other hand, there is another wider peak at 1545 cm−1, which corresponds to the sp2 structure characteristic peak of graphite.6 It could be because some graphite was left behind in the process of nanodiamond graphitization. It is apparent that the sp2 structure characteristic peak of ND shifted from 1545 cm−1 to 1555 cm−1 for the PDA–ND particles, indicating charge transfer interactions between ND and the aromatic polydopamine.
XPS spectra provided additional evidence for the formation of PDA layers on the ND surface. As shown in the wide-scan XPS spectra of ND and PDA–ND in Fig. 3a and c, we can observe sharp peaks corresponding to carbon and oxygen on the ND and PDA–ND, while there is an additional weak N 1s peak at around 397.5 eV, elucidating the existence of nitrogen from polydopamine (Fig. 3d). The C 1s spectra of ND can be resolved into four peaks, including the most intense peak at 284.6 eV (Fig. 3b), which expresses the peak of bulk diamond composed of sp3 carbon and an alkyl group. And the graphitic carbon species of sp2 carbon at 283.5, the peak of ethers at 285.9 eV, and the peak of carbonyl groups at 287.6 eV agreed well with the FTIR and Raman spectra of ND.45 After the formation of PDA on the ND surface, the C/O atomic ratio changed from 4.10 (ND) to 6.22, as can be seen in the tables in Fig. 3a and c.
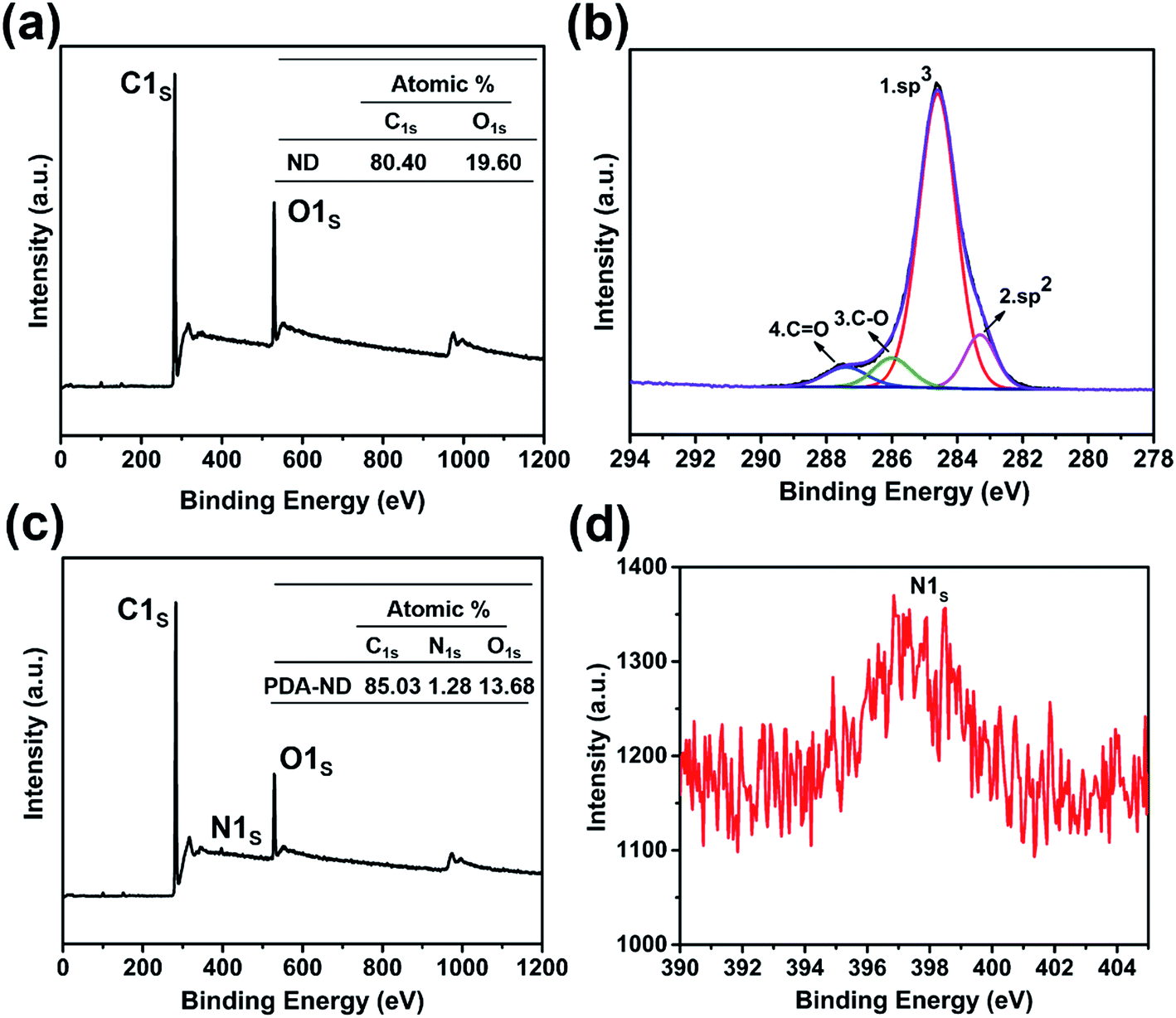 | ||
| Fig. 3 XPS spectra of ND and PDA–ND: (a) wide-scan spectrum of ND; (b) C 1s core-level spectrum of ND; (c) wide-scan spectrum of PDA–ND; (d) N 1s core-level spectrum of PDA–ND. | ||
A series of characterizations proved that PDA functionalized ND successfully. Density functional theory (DFT) was performed to calculate the interaction between ND and PDA, and the van der Waals forces of ND. And we found that the interaction between ND and PDA is slightly smaller than the van der Waals forces of ND (Fig. S2†).
3.2 Preparation and characterization PI/ND nanocomposites
XRD was employed to characterize the structures of ND, PDA–ND, pure PI and PI/PDA–ND nanocomposites. Fig. 4 shows that the XRD pattern of ND presents two sharp diffraction peaks at approximately 2θ = 44.0° and 75.4°, which indicated that the ND particles possess a three-dimensional diamond structure.45 The interlayer spacing shows little difference between ND and PDA–ND, while the intensity of the diffraction peak for PDA–ND is weakened compared to that of ND, revealing that the polydopamine was coated in the ND particles to further increase the structural heterogeneity.20 To study the dispersion state of PDA–ND in the polymer matrix, the XRD patterns were also plotted for pure PI and PI/PDA–ND composites. The pure PI shows a broad peak at around 2θ = 18.9°, revealing its amorphous nature. After PDA–ND was incorporated into the PI matrix, the XRD patterns of the PI/PDA–ND composites are almost the same as those for pure PI. The absence of the characteristic diffraction peak of ND may clearly demonstrate that PDA–ND is fully dispersed in the polyimide matrix.41 | ||
| Fig. 4 X-ray diffraction patterns of ND, PDA–ND, neat PI and PI/PDA–ND composite films. | ||
Fig. 5 presents the surface morphologies of neat PI and PI/PDA–ND nanocomposites obtained by AFM. The surface roughness (Ra) of neat PI is 4.86 nm and the surface roughness of polyimide/PDA–ND nanocomposites ranges from 7.28 to 14.9 nm, exhibiting a slight rise in surface roughness with the incorporation of PDA–ND into the PI matrix. The surface morphologies of the composite showed the highest surface roughness with a PDA–ND loading up to 1%.40
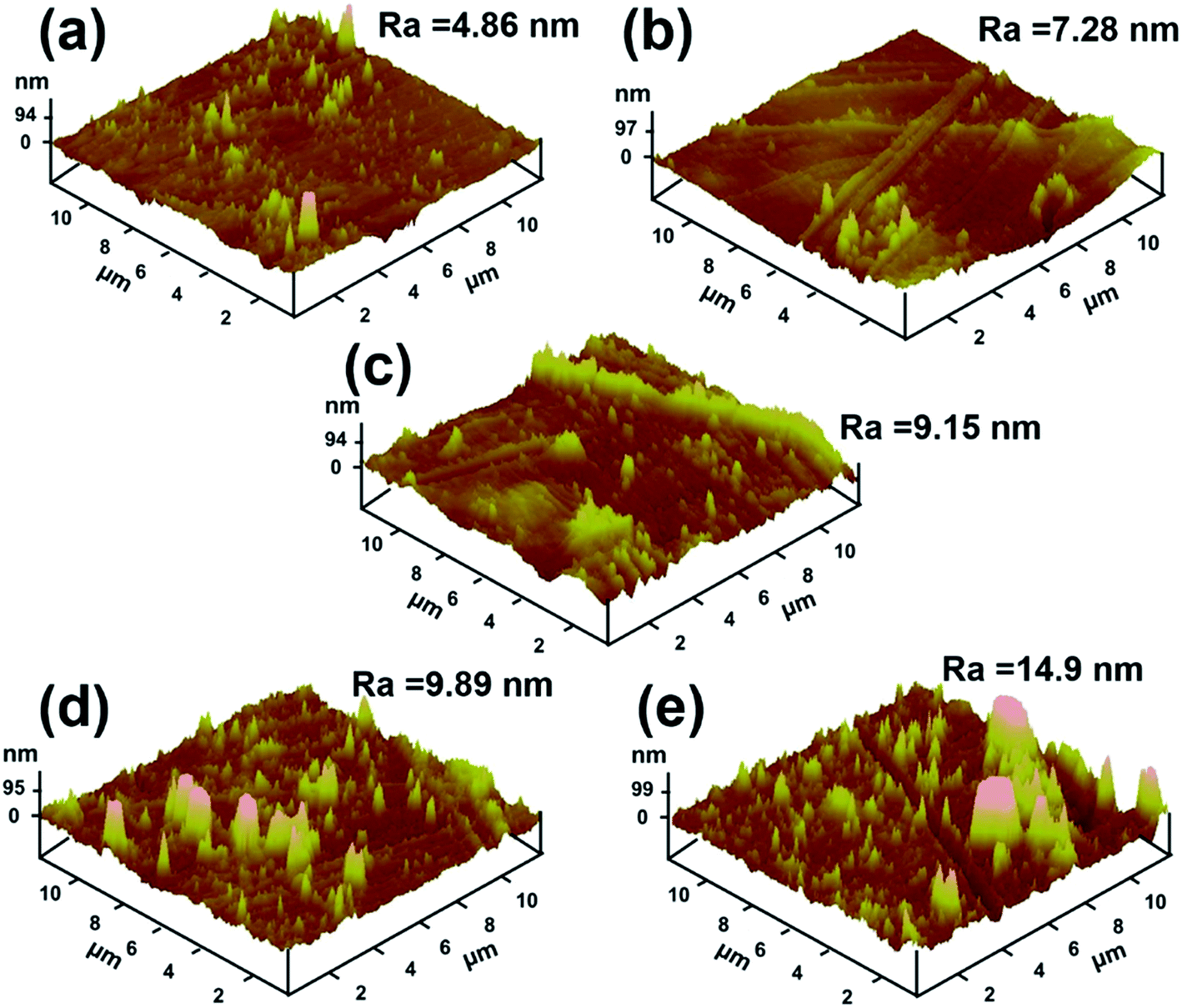 | ||
| Fig. 5 AFM surface morphologies of (a) neat PI, (b) 0.25% PI/PDA–ND, (c) 0.5% PI/PDA–ND, (d) 0.75% PI/PDA–ND, (e) 1% PI/PDA–ND. | ||
3.3 Mechanical properties
Typical stress–strain curves of neat PI and PI/PDA–ND composites are shown in Fig. 6a. Both the neat PI and the nanocomposites exhibited the typical characteristics of ductile fracture. The corresponding tensile strength, modulus, and elongation at break are summarized in Fig. 6b. Neat PI exhibited a tensile strength of 98 MPa, and a tensile modulus of 2.2 GPa. As expected, the PI/PDA–ND composites exhibit improved tensile strength and tensile modulus with the addition of PDA–ND. With the incorporation of a small quantity of 0.75 wt% PDA–ND, the tensile strength was significantly increased by 45% (142 MPa), implying more absorbed energy and greater fracture resistibility. In addition, the tensile modulus of PI/PDA–ND composites with 0.75 wt% PDA–ND was increased to 3.5 GPa, representing the best mechanical performance among the prepared specimens. On the other hand, we compared the tensile property of PDA–ND/PI and PDA–GO/PI composites. The tensile strength and the tensile modulus of PDA–ND/PI composites are obviously higher than those of the PDA–GO/PI composites (Fig. S3†). The strong interfacial adhesion and fine dispersion of ND in the PI matrix led to efficient load transfer from the PI matrix to PDA–ND and decreased the stress concentration during the stretching process.46,47 And the hydrogen bonding between PDA and polyimide improved the compatibility between ND and the polyimide matrix (Fig. S4†).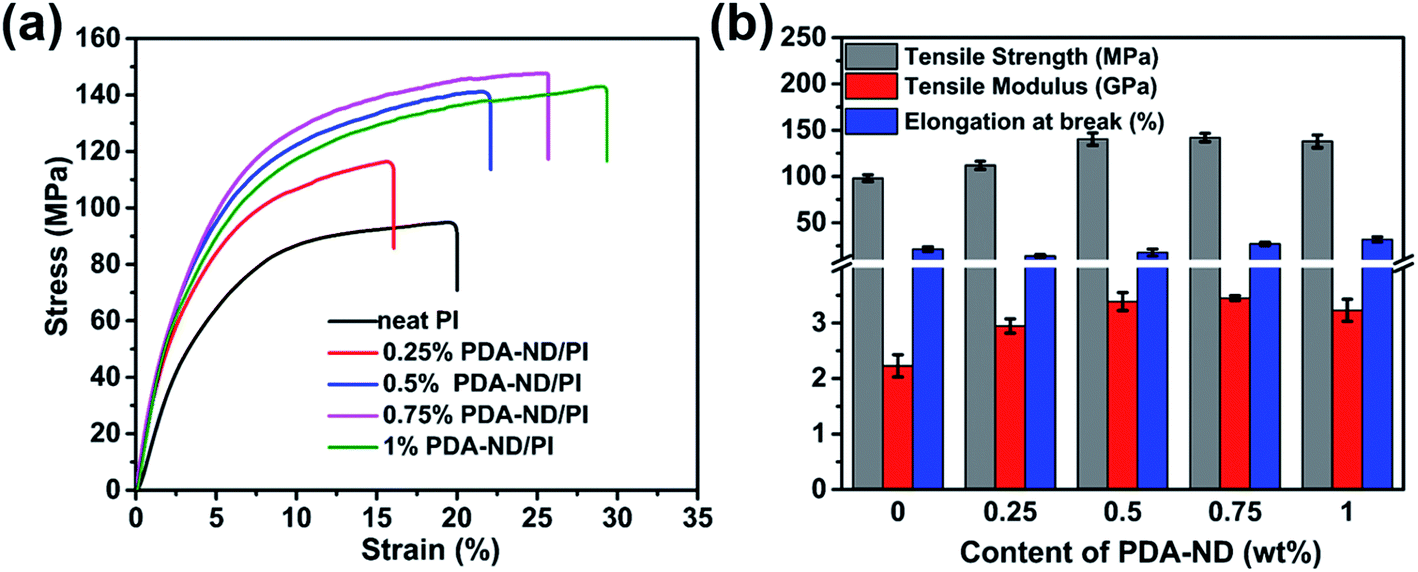 | ||
| Fig. 6 Tensile properties of neat PI and PI/PDA–ND composites: (a) typical stress–strain curves of neat PI and PI/PDA–ND composites; (b) tensile strength, tensile modulus and elongation at break of neat PI and PI/PDA–ND composites. | ||
Fig. 7a–e shows SEM images of the tensile-fractured surfaces of neat PI and the PI/PDA–ND composite films. The neat PI exhibits a relatively smooth and flat fracture surface, which implies that the crack propagates rapidly and little energy is absorbed during tensile fracture. However, the PI/PDA–ND composite films exhibited obvious rough and wrinkled morphologies. Moreover, a few crack initiation points were observed simultaneously for the nanocomposites. The much-deformed morphology of PI/PDA–ND composites revealed the strong interfacial adhesion and compatibility between ND and the PI matrix.42
 | ||
| Fig. 7 SEM images of (a) neat PI, (b) 0.25% PI/PDA–ND, (c) 0.5% PI/PDA–ND, (d) 0.75% PI/PDA–ND, (e) 1% PI/PDA–ND. | ||
DMA experiments were also undertaken to investigate the reinforcement effect of nanodiamond on the polyimide nanocomposites. Fig. 8 shows the effects of the PDA–ND mass fraction on the dynamic storage modulus, E′, of polyimide. The E′ values of all the specimens were temperature dependent and decreased gradually with an increase in temperature. The E′ of neat PI at ambient temperature is 1072 MPa, while the polyimide/PDA–ND nanocomposites exhibited an E′ of over 1188 MPa. It is evident that the E′ value (1973 MPa) of polyimide/PDA–ND nanocomposites with 0.25 wt% PDA–ND was much higher than that of neat polyimide and other composites, indicating that the rigidity of the composites reached its maximum. This prominent increase in E′ is due to the strong interfacial adhesion and good dispersion of PDA–ND in the PI matrix. However, with the increase in PDA–ND content, polydopamine molecules might be incorporated into the structure of polyimide, and increase the flexibility of polyimide/PDA–ND nanocomposites, resulting in a decrease in E′. The compiled DMA curves of loss modulus (E′′) versus temperature of neat PI and PI/PDA–ND nanocomposites are displayed in Fig. 8b. The E′′ of polyimide/PDA–ND nanocomposites also increased with the amount of PDA–ND. In addition, the loss modulus of PI/PDA–ND nanocomposites displayed a maximum at around 367 °C, owing to the backbone motion of PI molecules and was defined as α relaxation or the primary glass transition.48 This significant enhancement in storage modulus is indicative of a uniform dispersion of PDA–ND in the polyimide resin, because the PDA–ND could trap and inhibit the movement of PI molecular chains and the external stress was transferred to the much stiffer PDA–ND.49 Fig. 8c expresses the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ peaks of the damping spectra of polyimide and polyimide/nanodiamond nanocomposites, corresponding to their glass transition temperatures in the range of 385 to 397 °C. The PI/PDA–ND nanocomposites exhibited lower Tg compared with neat PI (397 °C). The reason is probably that the additional bonding between polydopamine and PI improved the flexibility of polyimide.40,50
δ peaks of the damping spectra of polyimide and polyimide/nanodiamond nanocomposites, corresponding to their glass transition temperatures in the range of 385 to 397 °C. The PI/PDA–ND nanocomposites exhibited lower Tg compared with neat PI (397 °C). The reason is probably that the additional bonding between polydopamine and PI improved the flexibility of polyimide.40,50
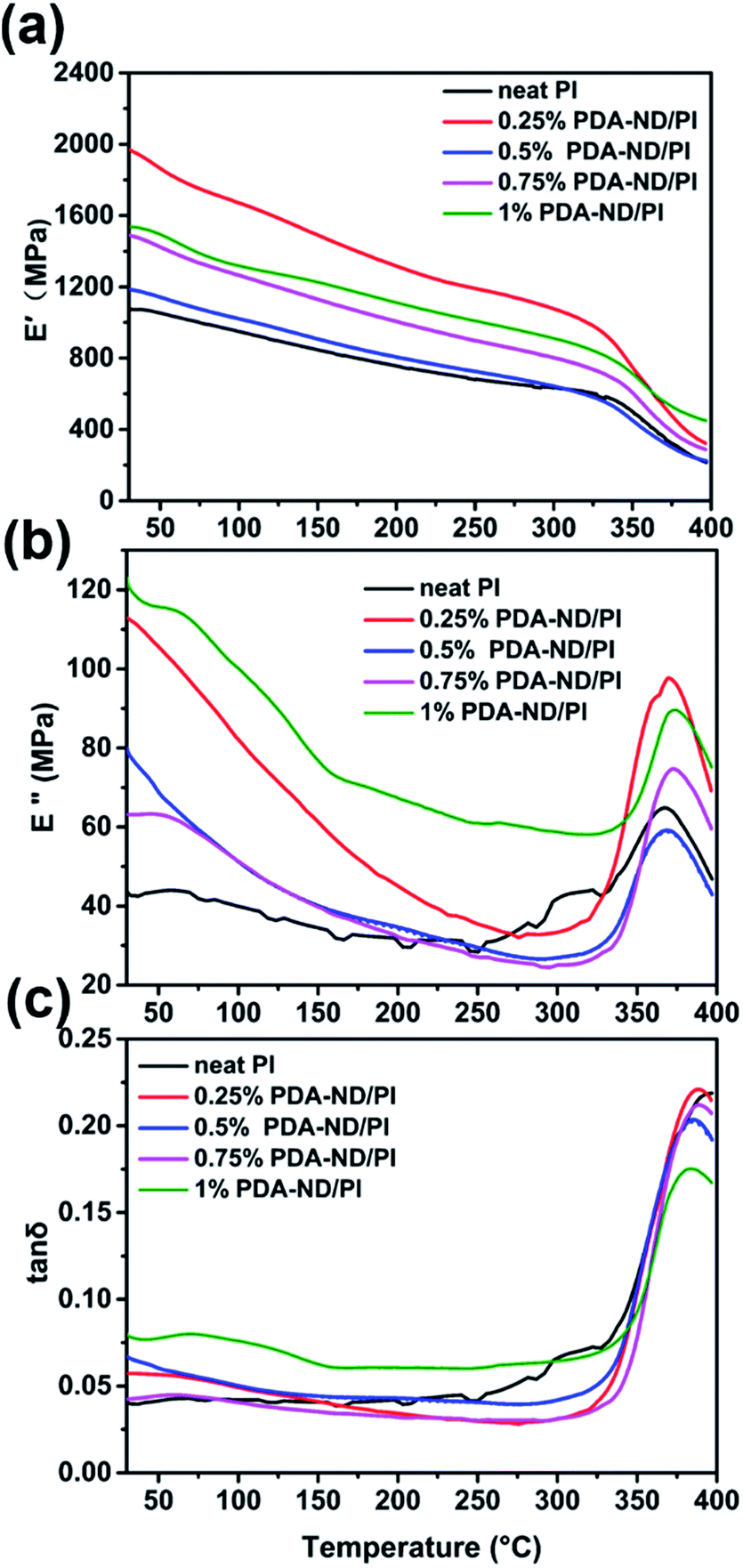 | ||
| Fig. 8 DMA results of polyimide and polyimide/nanodiamond nanocomposites as a function of nanodiamond content; (a) storage modulus E′, (b) loss modulus E′′, and (c) tanδ as a function of temperature. | ||
It is expected that the hardness of polyimide will be increased with addition of nanodiamond due to the high modulus of ND and the fine dispersion of ND in the polyimide matrix. Fig. 9 shows the Vickers hardness of the PI/PDA–ND nanocomposites with different mass fractions of PDA–ND. When a load of 100 N was applied in the Vickers hardness test, the hardness of neat PI was 18.44 HV. As the mass fraction of PDA–ND was 0.25%, the hardness slightly increased by ca. 0.52 HV compared with that of pure PI. After that, the Vickers hardness of the PI/PDA–ND nanocomposites increased obviously with the increase in PDA–ND. It went up to 20.89 HV at a 1% PDA–ND load, at which point the hardness reach its maximum. This significant increase in hardness for PI/PDA–ND composites can be attributed to the existence of well-dispersed ND and the strong interface adhesion between ND and the PI matrix. We also compared the hardness of PDA–GO/PI and PDA–ND/PI composites. As shown in Fig. S5,† the Vickers hardness of a 1 wt% PI/PDA–ND composite was 20.9 HV, and the Vickers hardness of a 1 wt% PI/PDA–GO composite was 19.9 HV. The hardness of the two composite materials was similar due to the high modulus of GO and ND.
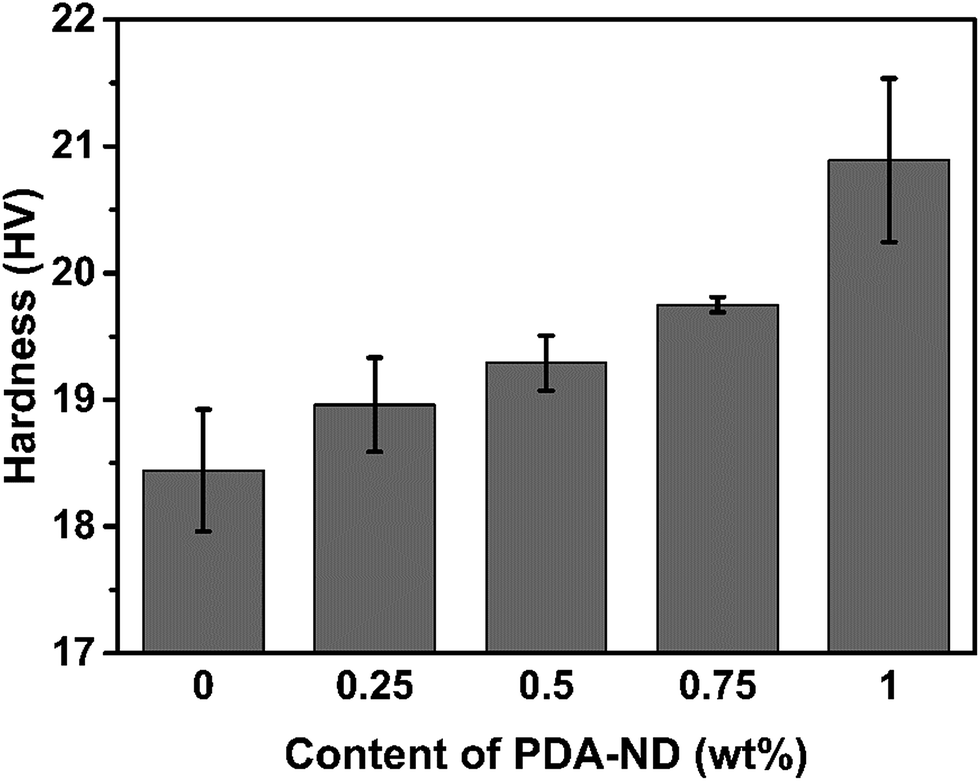 | ||
| Fig. 9 The hardness of neat PI and PI/PDA–ND nanocomposites with various mass fractions of nanodiamond. | ||
3.4 Thermal properties
To study the effects of nanodiamond on the thermal stability of polyimide, thermogravimetric (TGA) analysis was conducted under air in the temperature range of 25 to 800 °C. The TGA results of pure PI and PI/PDA–ND nanocomposites are shown in Fig. 10 and Table 1. There is almost no weight loss below 400 °C for any of the specimens, and the onset degradation temperature (Td) of pure PI was found to be 565 °C. As a result, it can be seen that the Td of the PI/PDA–ND nanocomposites increased significantly with a low PDA–ND loading (571 °C for 0.25 wt% PDA–AD, 574 °C for 0.5 wt% PDA–AD, 573 °C for 0.75 wt% PDA–AD, 576 °C for 1 wt% PDA–AD). PI/PDA–ND nanocomposites exhibited higher thermal stability under air atmosphere, owing to the incorporation of thermoduric ND nanofillers.51 The thermal stability parameters, including temperature of 50% degradation (Td50), could also be obtained from an enlarged drawing of the partial curves of Fig. 10; see Table 1. The thermal decomposition temperature at Td50 slightly decreased with the mass fractions of 0.25–1% of PDA–ND compared with that of neat PI. It is considered that the easily decomposed carboxyl or hydroxyl groups on ND might accelerate the degradation of PI. Despite the slight decrease in Td50, PI/PDA–ND nanocomposites retain superior thermal stability to satisfy their practical applications.42 | ||
| Fig. 10 TGA curves of polyimide and nanodiamond/polyimide nanocomposites as a function of temperature. | ||
| ND loading | TGA data | |
|---|---|---|
| Td/°C | Td50/°C | |
| a Td: onset degradation temperature; Td50: temperature at 50% weight loss. | ||
| 0% | 565.45 ± 0.25 | 642.72 ± 1.20 |
| 0.25% | 571.42 ± 0.43 | 632.64 ± 3.92 |
| 0.5% | 574.25 ± 0.59 | 633.30 ± 3.30 |
| 0.75% | 573.17 ± 1.21 | 638.65 ± 3.50 |
| 1% | 575.67 ± 0.60 | 641.86 ± 0.30 |
3.5 Friction and wear tests
Coefficients of friction (COF) testing was carried out under dry conditions at a 2 Hz sliding frequency and an applied load of 3 N, and the results of COF against time as a function of ND content are shown in Fig. 11a. It was shown that the mean COF slightly increased and then decreased with the addition of PDA–ND. Neat PI has a mean COF value of 0.41, while the mean COF of the PI/PDA–ND composites was higher than that of neat PI, ranging from 0.47 to 0.52 (Fig. 11b). The reason for the increase in the mean COF for the PI/PDA–ND nanocomposites can be explained on the basis of the super stiffness of nanodiamond, and the difference in mean COF for PI/PDA–ND composites is a combined result of the changes of PI chains with PDA and the different contents of ND. The wear rate tests of pure PI and PI/PDA–ND nanocomposites were determined by measuring the depth of the worn surface. Fig. 11c–d displays the variation in wear depth and wear rate as a function of PDA–ND content. The wear depth and wear rate sharply decreased with the incorporation of PDA–ND. In particular, the wear rate of the composite with 0.5 wt% PDA–ND was measured to be 0.84 × 10−5 mm3 N−1 m−1, which was reduced by 76% in comparison with that of neat PI (3.49 × 10−5 mm3 N−1 m−1). The incorporation of ND could significantly enhance the stiffness of the nanocomposites and form ND transfer films. Consequently, it could reduce the fragmentation and distortion of the PI nanocomposites and result in an obvious decrease in the wear depth and wear rate.40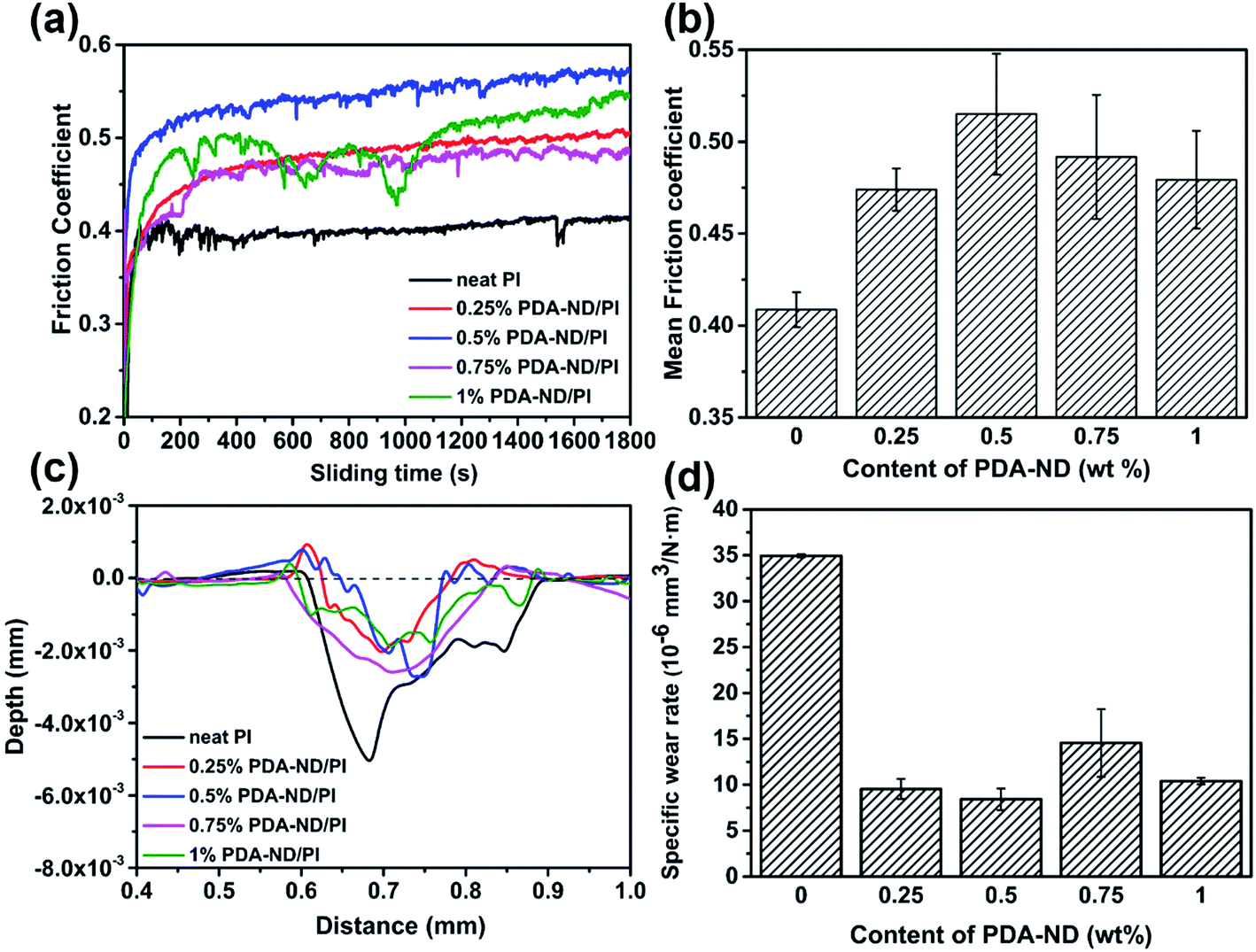 | ||
| Fig. 11 Friction coefficients (a) and (b); wear depth (c) and wear rate (d) of neat PI and PI/PDA–ND nanocomposites at a 2 Hz sliding frequency and an applied load of 3 N. | ||
The worn surface of pure PI and the resulting PI/PDA–ND nanocomposites were analyzed using SEM. As shown in Fig. 12a and e, the worn surface of neat PI showed a relatively flat morphology, displaying signs of adhesive wear.52 However, the worn surface of the PI/PDA–ND nanocomposites containing 0.25%, 0.5% and 1% ND exhibited cataphract morphologies, as shown in Fig. 12b–d and f–h. The typical fatigue deformations and super rigidity characterized by the tensile and DMA tests implied that the type of wear changed from adhesive wear to fatigue wear.38,53 The SEM images of wear debris of neat PI and PI/PDA–ND nanocomposites are given in Fig. 12i–l. The debris comes from the cracked surface at the points weakened by the adhesive force. It was observed that the wear debris of neat PI (Fig. 12i) and its nanocomposite exhibited flake-like morphology. However, the worn surface of the PI/PDA–ND nanocomposite was much smoother than that of neat PI, indicating that the rigid nanocomposites were more difficult to peel off during the sliding process.
 | ||
| Fig. 12 SEM images for the worn surface of specimens: (a) (e) neat PI, (b) (f) 0.25% PI/PDA–ND, (c) (g) 0.5% PI/PDA–ND, (d) (h) 1% PI/PDA–ND, and for the wear debris of specimens: (i) neat PI, (j) 0.25% PI/PDA–ND, (k) 0.5% PI/PDA–ND, (l) 1% PI/PDA–ND (load: 3 N, sliding frequency: 2 Hz). | ||
4. Conclusions
In summary, novel PI/PDA–ND nanocomposites were prepared by taking advantage of polydopamine chemistry to ensure the facile dispersion of ND and the good compatibility between ND particles and the PI matrix. The successful formation of PDA layers on the ND surface was verified by FTIR, Raman and XPS spectroscopy. The effect of PDA–ND on the thermal, mechanical and tribological properties of PDA–ND/PI composites were evaluated as a function of ND load. The slight decrease in the Tg of the PI/PDA–ND nanocomposites was probably due to the change in flexibility of the polymer chains by chemical bonding between PDA and PI. Obvious improvements in the tensile strength and tensile modulus were observed with incorporation of 0.25–1 wt% of PDA–ND. In addition, the incorporation of PDA–ND could distinctly increase the hardness, storage modulus and wear resisting properties. Considering their superior mechanical properties and tribological performance, the novel PI/PDA–ND nanocomposites prepared in this study are expected to extend the applications of PI as composites and wear resistance materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledged financial support provided by Strategic Leading Science & Technology Program (XDA13040600), “One Hundred Talented People” of the Chinese Academy of Sciences (Y60707WR04) and the National Key Basic Research Program (No. 2014CB643302).References
- Y. Wang, Y. Y. Shao, D. W. Matson, J. H. Li and Y. H. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed.
- C. de las Casas and W. Li, J. Power Sources, 2012, 208, 74–85 CrossRef CAS.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2011, 7, 11–23 CrossRef PubMed.
- R. S. Edwards and K. S. Coleman, Nanoscale, 2013, 5, 38–51 RSC.
- A. G. Rinzler, J. Liu, H. Dai, P. Nikolaev, C. B. Huffman, F. J. Rodriguez-Macias, P. J. Boul, A. H. Lu, D. Heymann, D. T. Colbert, R. S. Lee, J. E. Fischer, A. M. Rao, P. C. Eklund and R. E. Smalley, Appl. Phys. A: Mater. Sci. Process., 1998, 67, 29–37 CrossRef CAS.
- S. P. Hong, T. H. Kim and S. W. Lee, Carbon, 2017, 116, 640–647 CrossRef CAS.
- A. Shakun, J. Vuorinen, M. Hoikkanen, M. Poikelispää and A. Das, Composites, Part A, 2014, 64, 49–69 CrossRef CAS.
- A. Krüger, F. Kataoka, M. Ozawa, T. Fujino, Y. Suzuki, A. E. Aleksenskii, A. Y. Vul’ and E. Ōsawa, Carbon, 2005, 43, 1722–1730 CrossRef.
- R. A. Bueno, J. I. Martinez, R. F. Luccas, N. R. del Arbol, C. Munuera, I. Palacio, F. J. Palomares, K. Lauwaet, S. Thakur, J. M. Baranowski, W. Strupinski, M. F. Lopez, F. Mompean, M. Garcia-Hernandez and J. A. Martin-Gago, Nat. Commun., 2017, 8, 15306 CrossRef CAS PubMed.
- K. A. Wepasnick, B. A. Smith, K. E. Schrote, H. K. Wilson, S. R. Diegelmann and D. H. Fairbrother, Carbon, 2011, 49, 24–36 CrossRef CAS.
- S. Isikli, M. Lecea, M. Ribagorda, M. C. Carreño and R. Díaz, Carbon, 2014, 66, 654–661 CrossRef CAS.
- K. Balasubramanian and M. Burghard, Small, 2005, 1, 180–192 CrossRef CAS PubMed.
- S. Munirasu, J. Albuerne, A. Boschetti-de-Fierro and V. Abetz, Macromol. Rapid Commun., 2010, 31, 574–579 CrossRef CAS PubMed.
- A. Krueger, Chem.–Eur. J., 2008, 14, 1382–1390 CrossRef CAS PubMed.
- A. Abo-Hamad, M. Hayyan, M. A. AlSaadi, M. E. S. Mirghani and M. A. Hashim, Chem. Eng. J., 2017, 311, 326–339 CrossRef CAS.
- C. Zhang, Y. Lv, W. Z. Qin, A. He and Z. K. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 14437–14444 CAS.
- S. L. Phua, L. Yang, C. L. Toh, D. Guoqiang, S. K. Lau, A. Dasari and X. Lu, ACS Appl. Mater. Interfaces, 2013, 5, 1302–1309 CAS.
- H. Hu, B. Yu, Q. Ye, Y. Gu and F. Zhou, Carbon, 2010, 48, 2347–2353 CrossRef CAS.
- L. Han, X. Lu, K. Liu, K. Wang, L. Fang, L. T. Weng, H. Zhang, Y. Tang, F. Ren, C. Zhao, G. Sun, R. Liang and Z. Li, ACS Nano, 2017, 11, 2561–2574 CrossRef CAS PubMed.
- X. L. Hu, R. R. Qi, J. Zhu, J. Q. Lu, Y. Luo, J. Y. Jin and P. K. Jiang, J. Appl. Polym. Sci., 2014, 131, 39754 CrossRef.
- W. Li, T. Shang, W. Yang, H. Yang, S. Lin, X. Jia, Q. Cai and X. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 13037–13050 CAS.
- B. Fei, B. Qian, Z. Yang, R. Wang, W. C. Liu, C. L. Mak and J. H. Xin, Carbon, 2008, 46, 1795–1797 CrossRef CAS.
- W. Cui, M. Z. Li, J. Y. Liu, B. Wang, C. Zhang, L. Jiang and Q. F. Cheng, ACS Nano, 2014, 8, 9511–9517 CrossRef CAS PubMed.
- L. Q. Xu, W. J. Yang, K.-G. Neoh, E.-T. Kang and G. D. Fu, Macromolecules, 2010, 43, 8336–8339 CrossRef CAS.
- I. Kaminska, M. R. Das, Y. Coffinier, J. Niedziolka-Jonsson, J. Sobczak, P. Woisel, J. Lyskawa, M. Opallo, R. Boukherroub and S. Szunerits, ACS Appl. Mater. Interfaces, 2012, 4, 1016–1020 CAS.
- S. H. Hwang, D. Kang, R. S. Ruoff, H. S. Shin and Y. B. Park, ACS Nano, 2014, 8, 6739–6747 CrossRef CAS PubMed.
- N. Ning, Q. Ma, S. Liu, M. Tian, L. Zhang and T. Nishi, ACS Appl. Mater. Interfaces, 2015, 7, 10755–10762 CAS.
- J. Wu, W. Tu, Y. Zhang, B. Guo, S. Li, Y. Zhang, Y. Wang and M. Pan, Powder Technol., 2017, 311, 200–205 CrossRef CAS.
- Y. Y. Qin, Q. Y. Peng, Y. J. Ding, Z. S. Lin, C. H. Wang, Y. Li, J. J. Li, Y. Yuan, X. D. He and Y. B. Li, ACS Nano, 2015, 9, 8933–8941 CrossRef CAS PubMed.
- Q. Wu, L. Pan, H. Wang, W. Deng, G. Yang and X. Liu, Polym. J., 2015, 48, 169–175 CrossRef.
- F. Rahmani, S. Nouranian, X. Li and A. Al-Ostaz, ACS Appl. Mater. Interfaces, 2017, 9, 12802–12811 CAS.
- N. V. Kamanina, S. V. Serov, N. A. Shurpo, S. V. Likhomanova, D. N. Timonin, P. V. Kuzhakov, N. N. Rozhkova, I. V. Kityk, K. J. Plucinski and D. P. Uskokovic, J. Mater. Sci.: Mater. Electron., 2012, 23, 1538–1542 CrossRef CAS.
- N. V. Kamanina, L. N. Kaporskii and B. V. Kotov, Opt. Commun., 1998, 152, 280–282 CrossRef CAS.
- J. Subocz, A. Valozhyn and M. Zenker, Rev. Adv. Mater. Sci., 2007, 14, 193–196 Search PubMed.
- S. Li, Y. Feng, Y. Li, W. Feng and K. Yoshino, Carbon, 2016, 109, 131–140 CrossRef CAS.
- W. Xu, Y. Ding, S. Jiang, J. Zhu, W. Ye, Y. Shen and H. Hou, Eur. Polym. J., 2014, 59, 129–135 CrossRef CAS.
- X. Ye, X. Liu, Z. Yang, Z. Wang, H. Wang, J. Wang and S. Yang, Composites, Part A, 2016, 81, 282–288 CrossRef CAS.
- T. Huang, Y. Xin, T. Li, S. Nutt, C. Su, H. Chen, P. Liu and Z. Lai, ACS Appl. Mater. Interfaces, 2013, 5, 4878–4891 CAS.
- T. Huang, R. Lu, C. Su, H. Wang, Z. Guo, P. Liu, Z. Huang, H. Chen and T. Li, ACS Appl. Mater. Interfaces, 2012, 4, 2699–2708 CAS.
- S. Qin, C. Chen, M. Cui, A. Zhang, H. Zhao and L. Wang, RSC Adv., 2017, 7, 3003–3011 RSC.
- Q. Zhang, K. Naito, Y. Tanaka and Y. Kagawa, Macromol. Rapid Commun., 2007, 28, 2069–2073 CrossRef CAS.
- Y. Xiaoyan, Q. Xiongwei, K. Naito and Z. Qingxin, J. Reinf. Plast. Compos., 2011, 30, 661–670 CrossRef.
- Q. Zhang, K. Naito, Y. Tanaka and Y. Kagawa, Macromolecules, 2008, 41, 536–538 CrossRef CAS.
- R. A. Zangmeister, T. A. Morris and M. J. Tarlov, Langmuir, 2013, 29, 8619–8628 CrossRef CAS PubMed.
- S. Morimune, M. Kotera, T. Nishino, K. Goto and K. Hata, Macromolecules, 2011, 44, 4415–4421 CrossRef CAS.
- O.-K. Park, J.-Y. Hwang, M. Goh, J. H. Lee, B.-C. Ku and N.-H. You, Macromolecules, 2013, 46, 3505–3511 CrossRef CAS.
- W. H. Liao, S. Y. Yang, J. Y. Wang, H. W. Tien, S. T. Hsiao, Y. S. Wang, S. M. Li, C. C. Ma and Y. F. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 869–877 CAS.
- H. L. Tyan, Y. C. Liu and K. H. Wei, Chem. Mater., 1999, 11, 1942–1947 CrossRef CAS.
- I. H. Tseng, Y.-F. Liao, J.-C. Chiang and M.-H. Tsai, Mater. Chem. Phys., 2012, 136, 247–253 CrossRef CAS.
- W. P. Serena Saw and M. Mariatti, J. Mater. Sci.: Mater. Electron., 2011, 23, 817–824 CrossRef.
- A. Beyler-Çiğil, E. Çakmakçı and M. Vezir Kahraman, Polym. Compos., 2016, 37, 2285–2292 CrossRef.
- H. Cai, F. Yan and Q. Xue, Materials Science and Engineering: A, 2004, 364, 94–100 CrossRef.
- K. H. Hu, X. G. Hu, J. Wang, Y. F. Xu and C. L. Han, Tribol. Lett., 2012, 47, 79–90 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10688b |
| This journal is © The Royal Society of Chemistry 2018 |