
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMapping molecular binding by means of conformational dynamics measurements†
Noelle M. do Nascimentoa,
Augusto Juste-Dolza,
Paulo R. Bueno b,
Isidro Monzóc,
Roberto Tejeroc,
José L. Lopez-Paza,
Angel Maquieiraa,
Sergi Morais*a and
David Gimenez-Romero*c
b,
Isidro Monzóc,
Roberto Tejeroc,
José L. Lopez-Paza,
Angel Maquieiraa,
Sergi Morais*a and
David Gimenez-Romero*c
aInstituto Interuniversitario de Investigación de Reconocimiento Molecular y Desarrollo Tecnológico, Departamento de Química, Universitat Politècnica de València, Camino de Vera, s/n Valencia, 46022, Spain
bInstituto de Química, Univ. Estadual Paulista (UNESP), Departamento de Físico-Química, Nanobionics Research Group, Araraquara, São Paulo, Brazil Web: http://www.nanobionics.pro.br
cDepartamento de Química-Física, Universitat de València, C/Dr Moliner 50, 46100 Burjassot, Spain. E-mail: giroda@uv.es
First published on 3rd January 2018
Abstract
Protein–protein interactions are key in virtually all biological processes. The study of these interactions and the interfaces that mediate them play a key role in the understanding of biological function. In particular, the observation of protein–protein interactions in their dynamic environment is technically difficult. Here two surface analysis techniques, dual polarization interferometry and quartz crystal microbalance with dissipation monitoring, were paired for real-time mapping of the conformational dynamics of protein–protein interactions. Our approach monitors this dynamics in real time and in situ, which is a great advancement within technological platforms for drug discovery. Results agree with the experimental observations of the interaction between the TRIM21α protein and circulating autoantibodies via a bridging bipolar mechanism. This work provides a new chip-based method to monitor conformational dynamics of protein–protein interactions, which is amenable to miniaturized high-throughput determination.
Introduction
Proteins are vital components of living organisms, and are responsible for nearly every task of cellular life. In fact the human genome codes for around 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 proteins, of which some 400000 have not yet been isolated.1 By considering at least three interactions per protein, there may be around 1200000 protein–protein interactions (PPIs). PPIs are key in virtually all biological processes and are crucial for comprehending the pathogenesis of numerous human diseases due to the close ties between function and structure.2 Furthermore, the targeted modulation of PPIs is one of the most promising approaches in drug discovery.3,4
000 proteins, of which some 400000 have not yet been isolated.1 By considering at least three interactions per protein, there may be around 1200000 protein–protein interactions (PPIs). PPIs are key in virtually all biological processes and are crucial for comprehending the pathogenesis of numerous human diseases due to the close ties between function and structure.2 Furthermore, the targeted modulation of PPIs is one of the most promising approaches in drug discovery.3,4
Computational studies have been the commonly applied approach to predict the protein orientation in PPIs.5 However, these tools produce questionable results given the difficulty of approaching in vivo conditions. As a result, a variety of experimental approaches have been developed to help studies on PPIs, such as X-ray-triggered spontaneous crystallisation,6 fluorescence spectroscopy7 and molecular electronics.8
Most protein studies usually employ dynamic conditions, which show only the limits of conventional experimental tools on real-time orientation measurements. Currently, available information on orientation is obtained from X-ray diffraction, nuclear magnetic resonance (NMR) or atomic force microscopy research conducted under fixed orientation conditions.9 Conversely, the biological relevance of an interaction lies in its structural dynamics. Hence in order to address the protein physics, huge efforts are being made to establish alternative techniques to detect the protein orientation in real-time.10 Intense UV femtosecond lasers allow protein dynamic pictures to be captured by 2D UV spectroscopy.11 Cryo-electron microscopy is also gaining popularity in structural biology because it allows proteins to be observed in their native environment.12 However despite some progress, no method yet encompasses all the characteristics needed to monitor in situ protein orientation and to explore reaction intermediates.
The protein array, a solid phase assay to study PPIs, is an attractive tool in proteomics for diagnostics and therapeutic purposes as well as for basic research. Currently, the quartz crystal microbalance with dissipation monitoring (QCM-D) detection system, which requires immobilisation of capture proteins on gold substrate, has become an alternative to optical techniques for biological-based studies.13 It is a powerful approach for determining mechanistic processes, and for providing a fingerprint for the monitored interaction via Δf–ΔD plots.14,15 However, this technique provides limited information about the conformational changes that take place during PPIs.
To map the spatial positions of interacting molecules in a system, it is necessary to use complementary techniques to gravimetric ones. Along this line, dual polarization interferometry (DPI) provides nanometric information on molecular interactions, which can be efficiently employed in bionanotechnology, surface science, crystallography or drug discovery.16 In all these cases, DPI measures distances as an optical average thickness value, and therefore the spatial position of a single interacting molecule has not yet been monitored to date.
TRIM21/Ro52/SS-A1, a 52–56 kDa member of the family of RING/B-box/coiled-coil tripartite motif protein's family, is an autoantigen recognized by antibodies in sera of control subjects and patients with Systemic Lupus Erythematosus (SLE) and also Sjögren's syndrome. Its activation results in the production of proinflammatory cytokines, modulation of natural killer stress ligands and induction of an antiviral state. Furthermore, the antibody-TRIM21α detection provides a potent and comprehensive activation of the intracellular immune response, neutralizing viral infection by virus degradation via ubiquitination.17,18 In this regard, anti-TRIM21α autoantibodies are one of the most common circulating antibodies found in patients with systemic autoimmune diseases, forming pathogenic immune complexes.19 However, the TRIM21-antibody recognition mechanism is still not well established, which could be of great importance in the treatment of these diseases.
Herein, we addressed conformational dynamics studies by developing a novel chip assay format, in which bait proteins are immobilized on a Self-Assembly Monolayer (SAM)-based biosensor. Upon incubation with the biological sample, interacting proteins are captured, which are then analyzed by combining the advantages of QCM-D and DPI. Next, we used this novel assay format to study the interaction of the TRIM21α protein with its circulating autoantibodies.
Experimental section
Ethics statement
Twenty SLE patients and eight healthy individuals provided full informed written consent and were enrolled in this observational study. Approval from the Biomedical Research Ethics Committee of the La Fe Hospital was obtained to conduct this study. All the patients agreed about taking part in the study, allowing their samples to be stored in La Fe Biobank, and giving their informed written consent to be included in the study. All the experiments were performed in accordance with relevant guidelines and regulations.Serum samples
Twenty-eight serum samples, twenty samples from anti-Ro+ patients with SLE and eight control samples (healthy subjects) were obtained from the Departamento de Reumatología, Hospital Universitario y Politécnico La Fe, Valencia (Spain). In this work and since no international standard for anti-TRIM21 autoantibodies was available, 20 anti-Ro+ patients with SLE with a high concentration of the anti-Ro antibodies (>200 U mL−1) were selected. Blood samples were collected by venipuncture from patient's forearm veins. All the samples were processed immediately following venipuncture and were handled by standard procedures and stored at −80 °C in La Fe Biobank. All the patients met the SLICC-ACR2012 classification criteria.20 The eight healthy subjects selected had an undetectable concentration of the anti-Ro antibodies, according to the reference assays. All the healthy subjects underwent a medical examination upon enrolment, which included full blood tests done to rule out any underlying diseases or conditions. The healthy controls were recruited with a mean age and gender ratio similar to that of the SLE patient group.IgG purification
As the antibody response is usually most heterogeneous, and in order to study only the dominant response, all the SLE patients selected for this work had an accentuated immune response to TRIM21. Furthermore, the study was performed using purified IgGs from a pool of 20 anti-Ro+ patients and a pool of eight healthy subject sera.IgGs were purified by affinity chromatography (GE Healthcare, HiTrap™ protein G HP) following the manufacturer's protocol. Columns were activated with a phosphate buffered saline solution composed of 0.0027 M KCl (Scharlau, reagent grade), 0.01 M Na2HPO4 (Scharlau, reagent grade), 0.0018 M KH2PO4 (Scharlau, extra pure) and 0.137 M NaCl (Scharlau, synthesis grade) at pH 7, called 1× PBS. Then, human sera were applied by pumping them thorough the column. Then, the columns were washed with 1× PBS until no material appeared in the effluent, which was quantified by spectrophotometry (Nanodrop 2000/2000c Spectrophotometer, Thermo Fischer Scientific). After the other sample components were washed away, IgGs were stripped from the support, which resulted in its purification from human sera. For that, IgGs were eluted with a 0.05 M glycine solution (Sigma-Aldrich, for electrophoresis ≥ 99%) at pH 2.5. When the appropriate aliquots were collected and quantified by spectrophotometry, the pH of the solution was immediately changed to 7.4 with a phosphate buffer composed of 0.023 M Na2HPO4 and 0.0018 M KH2PO4. The obtained IgGs were concentrated in a small volume by ultrafiltration filters with a membrane NMWL of 30 kDa (Pall Corporation, Macrosep® Advance Centrifugal Device, 30 K MWCO), the solution being reconstituted with 1× PBS. The concentration of the anti-TRIM21 autoantibodies was quantified by means of a human anti-SSA(Ro-52) ELISA kit (Signosis-Biosignal Capture). The optical density of each well was measured using a microplate reader (Wallac, Victor 1420 multilabel counter) at 450 nm. F(ab′)2 fragments were obtained with a Pierce™ F(ab′)2 preparation kit (ThermoFisher Scientific). The active human IgG Fc fragment proteins were obtained from the Abcam company.
QCM-D
QCM-D measurements were taken using gold Q-sense sensors (5 MHz, QSX 301, Biolin Scientific). Prior to the experiments, the sensor's surface was cleaned according to the manufacturer's protocol. The frequency and dissipation variations were monitored in a Q-Sense E1 (Biolin Scientific, Sweden) device equipped with a liquid flow cell setup. All the experiments were carried out in 1× PBS solution to dissolve protein and the resonance frequency was also stabilized in 1× PBS. The flow rate was 50 μL min−1 and the temperature was 25 °C.Self-assembly monolayer (SAM)
The unmodified piezoelectric quartz crystal chip was firstly cleaned for 10 minutes by irradiation with a UV-ozone cleaning system (UVOH 150 LAB, FHR, Ottendorf, Germany). Afterwards the chip was immersed in a 5:1:1 solution of Milli-Q water, 25% ammonia (Scharlau) and 30% hydrogen peroxide (Scharlau) at 75 °C. Then the chip was rinsed with Milli-Q water, and blown dry with high purity nitrogen. Finally, the chip was treated again with the UV-ozone cleaning system for 10 minutes. The self-assembled monolayer was formed by treating the quartz crystal with 10 mM of MPA (3-mercaptopropionic acid, Sigma-Aldrich) overnight, which was further activated with 46 mM of EDC –N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide, Sigma-Aldrich, pure grade-/NHS (98% N-hydroxysulphosuccinimide, Sigma-Aldrich) for 1 hour to produce the activated carboxyl chip.
Immobilisation of TRIM21 on a SAM chip
A hydrazine chip was prepared by converting the carboxyl group on SAM into a hydrazide group by treating with 5 mM 98% carbohydrazide (Sigma-Aldrich) before protein immobilisation, see Fig. 1. To immobilise the protein, 100 microliters of TRIM21 (recombinant human RO-52/SS-A expressed in E. coli, purity above 90 per cent, measured by SDS-PAGE, Sigma-Aldrich), was added at a concentration of 33 mg L−1 (around 49 ng cm−2, saturation probe), and incubated for 1 h to be cross-linked to the activated gold surface. The non-crosslinked residues were blocked with 1 M ethanolamine–HCl ≥98% (Sigma-Aldrich) and a blocking buffer that contained 1 mM ethylenediaminetetraacetic acid (Riser), 0.25% bovine serum albumin (BSA, Sigma-Aldrich, ≥98% agarose gel electrophoresis), and 0.05% Tween-20 (Scharlau, synthesis grade). The whole reaction time was 1 h and the sensor chip was cleaned with Milli-Q water after each step.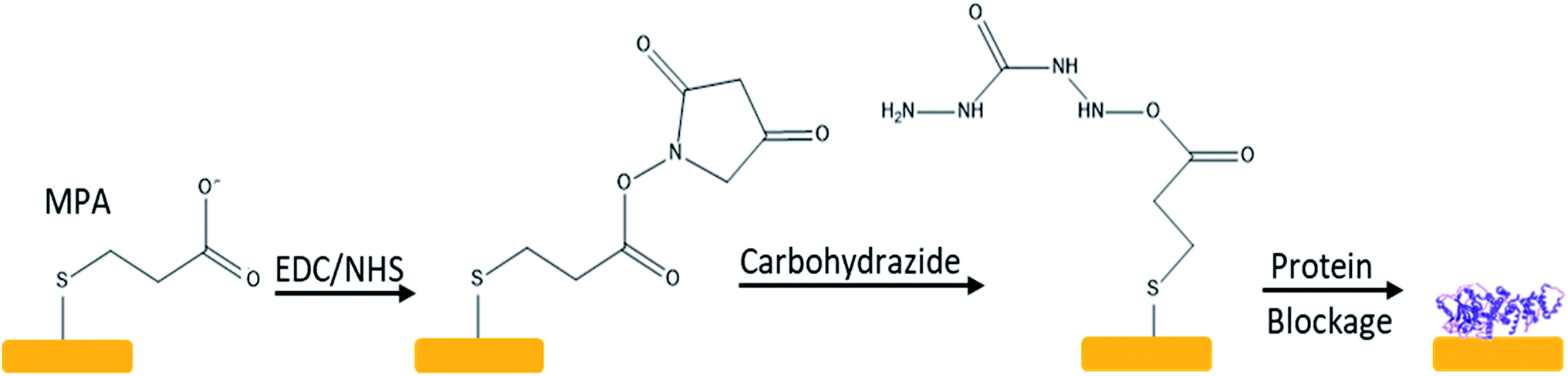 | ||
| Fig. 1 Scheme of the immobilization reaction mechanism. | ||
Immobilisation of polypeptides on a carboxyl chip
After activation, and as previously described, the chip was rinsed with Milli-Q water and the polypeptide sequences (peptides&elephants) were added at a concentration of 33.3 mg L−1. Each polypeptide was ended with a free amine group, and with an amide group at its C-terminus. These chips were blocked with 20 mg mL−1 of 95% D-glucamine (abcr) in 1× PBS, pH 9.75, and then they were rinsed with Milli-Q water.Characterization of immobilisation of polypeptide onto SAM chip
Static Water Contact Angle (CA) Measurements. The surface wettability was measured by automated static water contact angle measurements with a Krüss DSA 100 goniometer (volume of the drop of deionized water = 3.0 μL). The obtained values were the average of five droplets.Infrared Reflection Absorption Spectroscopy (IRRAS). IRRAS spectra were measured with a Bruker Tensor 27 FT-IR spectrometer (4000–800 cm−1), employing a variable-angle reflection unit (Auto Seagull, Harrick Scientific). A Harrick grid polarizer was mounted in front of the detector and used for registering spectra with p-polarized radiation. Single channel transmittance spectra were collected at 80° using 2048 scans. The raw data were subtracted by the data recorded on a freshly cleaned reference Au surface, after which a baseline correction was applied to give the reported spectra.
X-ray Photoelectron Spectroscopy (XPS). The XPS results was performed using a JPS-9200 photoelectron spectrometer (JEOL, Japan). All spectra were obtained under UHV conditions using monochromatic Al Kα X-ray radiation at 12 kV and 20 mA, and an analyser pass energy of 50 eV for wide scans and 10 eV for narrow scans. The emitted electrons were collected at 10° from the surface normal. Survey spectra were corrected with linear background before fitting, whereas high-resolution spectra were corrected with Shirley background. Atomic area ratios were determined after a baseline correction and normalizing the peak area ratios by the corresponding atomic sensitivity factors (1.00 for C 1s, 1.80 for N 1s, 2.93 for O 1s, 19.8 for Au 4d, and 1.68 for S 2p).
DPI
DPI measurements were taken with an Analight Bio200 Dual Polarisation Interferometer (Farfield Scientific Ltd., Crewe, UK), which consisted of a helium neon laser (λ = 632.8 nm and 20 mW), a 1024 × 1024 element-imaging device and a sensor chip clamped inside a thermally insulated chamber (temperature control within 1 mK). The state of polarisation of light was switched on a 2 ms cycle between the transversal electric and magnetic modes using a ferroelectric crystal before passing through the sensor. The interference pattern formed in the far-field was detected on the opposite side by the element-imaging device. Data acquisition and treatment were carried out by the Analight DAQ (Farfield) and the Analight Explorer (Farfield) software packages, respectively. Solutions were flowed by a double-channel precision syringe pump (Harvard Apparatus PHD 2000 Infusion, Kent, UK) and injections were carried out by using two high-performance chromatography valves connected in series. Throughout the experiment, the flow was maintained at a constant rate of 20 μL min−1, and all the solutions were injected into volumes of 250 μL.Chip functionalisation
Activation of DPI chips (FB100, Farfield, UK) was achieved by immersing them overnight in a chromic mixture (100 g L−1 K2Cr2O7 in 85% H2SO4). The chip was then rinsed with deionised water and dried with nitrogen before being submerged in 20% HNO3 for 2 h. After rinsing with deionised water and drying with nitrogen, the activated chip was immersed in 1% carboxyethylsilanetriol (abcr, sodium salt, 25% in water) for 1 h. After being cleaning with water, it was placed in an oven at 110 °C for 1 h. A phosphate buffered saline (PBS, 10 mM phosphate, 137 mM NaCl, 2.7 mM KCl, pH 7.4) solution, filtered through a 0.45 μm PVDF filter and degassed by sonication, was used as the carrier. This solution was also used to prepare the injected solutions, unless otherwise stated. When the treated chip was mounted on the setup, an injection of 0.2 M 3-(3-dimethylaminippropyl)-carbodiimide (EDC) and 0.05 M N-hydroxysuccinimide (NHS) in water, followed by an injection of 5 mM of carbohydrazide in water, was done. TRIM21α was then immobilised by covalent binding by injecting a 33 mg L−1 solution of this protein. An injection of 20 g L−1 of glucamine in PBS (pH 9.75) was next performed to block the surface against non-specific adsorption.TRIM21α structure
A preliminary model of the TRIM21α structure was predicted using homology-modelling by templating and threading methods using servers LOMETS and SWISS MODEL.21–24 The results given by SWISS MODEL were preferably used, and only the fragments not predicted by this server were taken from the results given by performing threading with LOMETS. The templates used for building the model were PDB ids 4CG4, 4AP4, 4TN3 and 2IWG. The different obtained pieces were manually modelled together on the graphics display and the final coordinates were subjected to mild energy minimisation with weak restraints using r−6 summed average over selected groups of atoms to ensure the pieces held together but without biasing the system to special orientations. In all instances of molecular mechanics (MM) and molecular dynamics simulations (MD) AMBER25 package was used. Once the system was relaxed in the AMBER force-field, short restrained molecular dynamics (r-MD) simulation at 300 K was performed. In all cases standard MD protocols were used, heating and equilibrating the system at 300 K and then using a thermal bath to keep temperature constant over the simulation. Once the system was stable at 300 K, another set of MD simulation was performed with no restraints at all to check if the system remain stable and how it behaved.Results and discussion
Characterisation by PM-IRRAS, XPS and SCA
The interaction between TRIM21α protein and autoantibodies (antigenic complex formation) was monitored by QCM-D. For this purpose, recombinant human TRIM21α protein was immobilized onto the gold chip using 3-mercaptopropionic acid as a monolayer precursor and subsequent covalent EDC coupling. The immobilization process was fully characterized by polarization modulation infrared reflection adsorption spectroscopy (PM-IRRAS), X-ray photoelectron spectroscopy (XPS) and static water contact angle (SCA) measurements (see Fig. 2 and 3 and Table 1). PM-IRRAS provided the qualitative analysis of the monolayer (see Fig. 2). The PM-IRRAS spectrum of the SAM shows the characteristic bands found in the infrared spectra of proteins, and polypeptides. Furthermore, the homogeneity and high-structural order in the gold surface was demonstrated by the high defined peaks, see Fig. 2.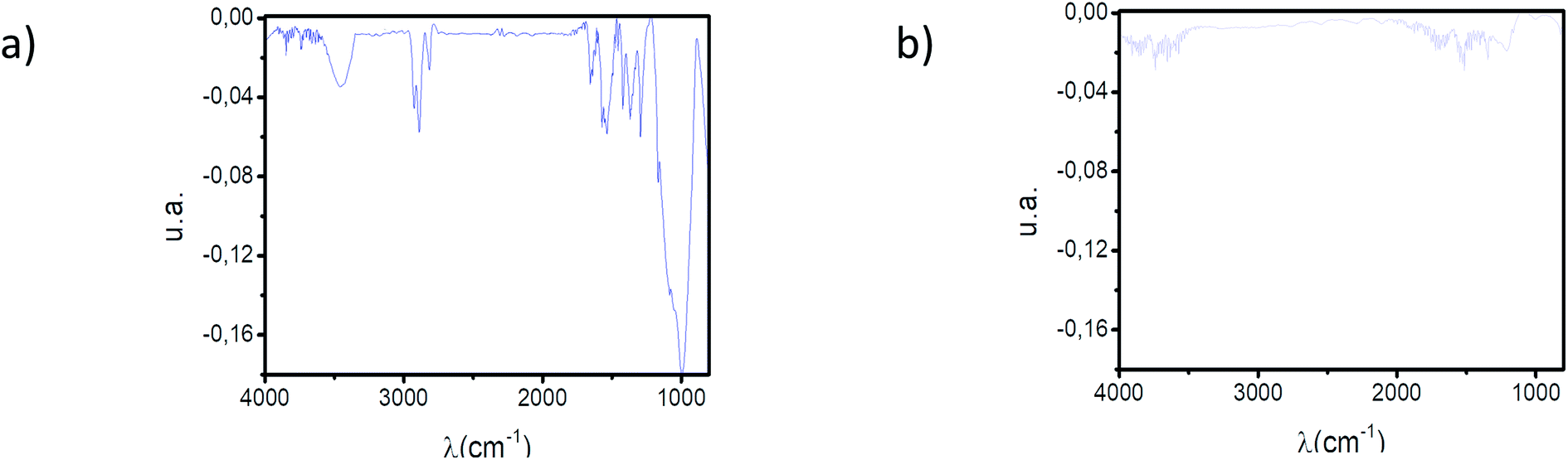 | ||
| Fig. 2 (a) PM-IRRAS spectrum of the TRIM21α SAM. (b) PM-IRRAS spectrum of the polypeptide SAM. | ||
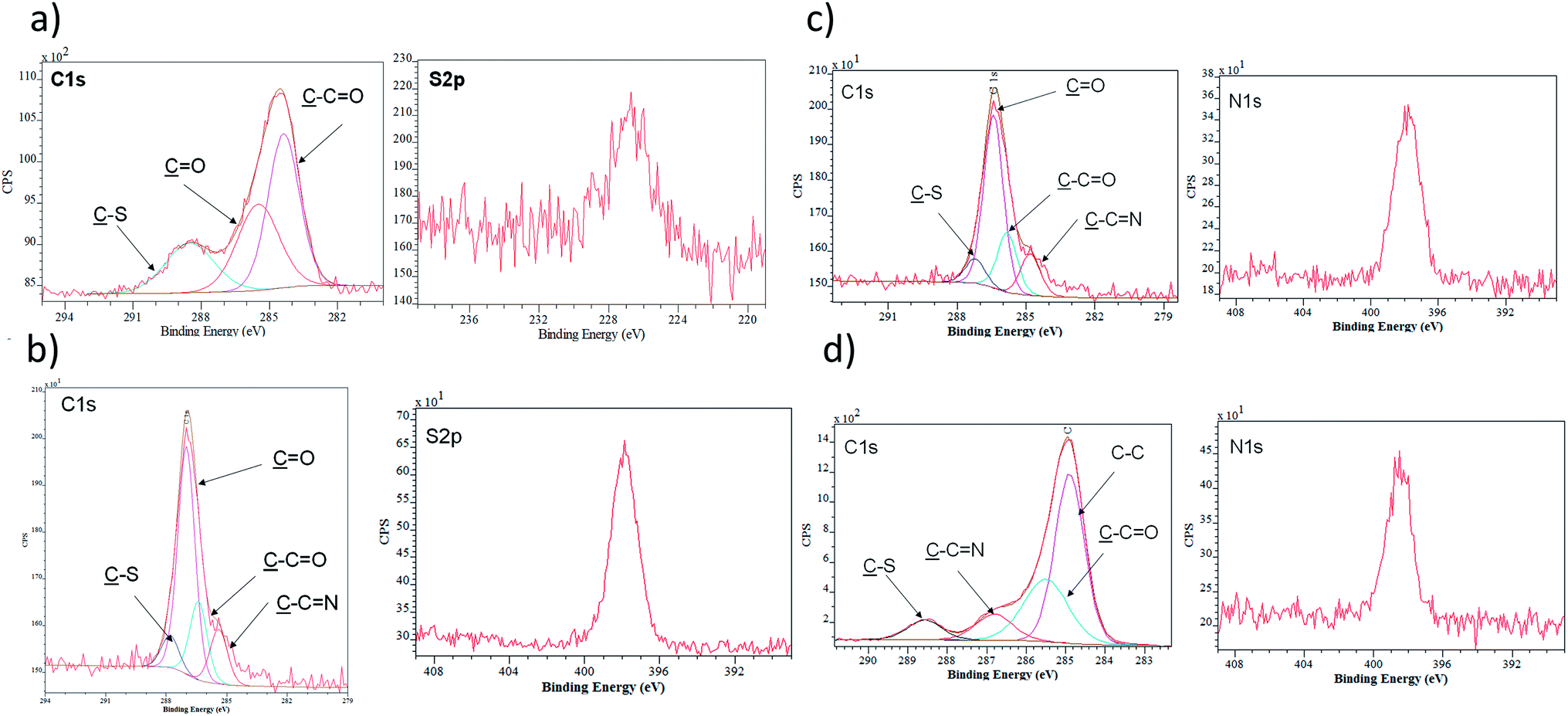 | ||
| Fig. 3 (a) XPS spectra of the C 1s region and S 2p of MPA monolayer. (b) XPS spectra of the C 1s region and N 1s of NHS-activated monolayer. (c) XPS spectra of the C 1s region and N 1s of the carbohydrazide-modified monolayer. (d) XPS spectra of the C 1s region and N 1s of the TRIM21α-modified monolayer. | ||
| Chip | % Au | % C | % O | % S | % N | SCA |
|---|---|---|---|---|---|---|
| Cleaned | 84.9 | 11.0 | 4.1 | — | — | 57.00° |
| MPA | 38.2 | 41.3 | 15.7 | 4.8 | — | 70.01° |
| EDC/NHS | 19.3 | 52.1 | 23.3 | 2.1 | 3.2 | 59.00° |
| Carbohydrazide | 26.1 | 55.7 | 12.0 | 2.6 | 3.6 | 55.60° |
| TRIM21α/blockage | 8.1 | 63.8 | 14.6 | 1.2 | 12.3 | 56.70° |
Following the characterization process, Fig. 3 and Table 1 present the data obtained by XPS analysis, starting from the cleaning of the surface until the immobilization of the protein and blockage of free active sites. After the addition of MPA acid, the sulphur percentage measured by XPS increased (0–4.8%). The EDC/NHS reaction and the addition of carbohydrazide induces an increase of the nitrogen percentage (0–3.6%). Finally, the immobilization of the protein and the blockage process rendered a clear increase carbon (55.7–63.8%) and nitrogen (3.6–12.3%) percentages, as expected after these immobilisation steps.
The static contact-angle measurement of each step of the construction of the self-assembly monolayer was also determined, see Table 1. The EDC/NHS reaction and the addition of carbohydrazide induced a decrease of the SCA (70.01–55.60°). Finally, the immobilisation of the TRIM21α protein and the blockage process rendered a SCA virtually constant (55.60–56.70°), decreasing when D-glucamine was added (55.60–26.60°). Consequently, this surface characterization corroborated the formation of hydrophilic SAMs. It is worth mentioning that the construction of the self-assembly monolayer with D-glucamine resulted in an almost super-hydrophilic surface, which reduces nonspecific interactions. Data corroborated the formation of a hydrophilic self-assembled monolayer of TRIM21α with high structural order and surface homogeneity.
Furthermore, BSA was used to probe whether all sensor chips were free of unspecific interactions. The results showed that there were no unspecific interactions within the range of concentrations tested. The control for immobilized BSA with the IgG pool was also zero at all concentrations tested. Consequently, it is possible to say that the employed sensor chips were free of unspecific interactions.
The function
In a QCM-D assay, the dissipation factor (D) and the acoustic resonant frequency (f) can be simultaneously measured in the PPIs that occurs on the sensing surface. In these assays, the raw laws module resonance frequency changes, whereas the dissipation factor is strongly affected by the structural changes caused by these interactions. Hence the ratio between both magnitudes shows how the adlayer structure changes per unit mass addition. The traditional way of presenting the effect of a reagent on a reactive surface is by using Δf–ΔD plots.14 Essentially, this plot suggests that a different process takes place where a change of slope occurs. Thus the Δf–ΔD plot can be used as a fingerprint for the monitored interaction. Processes with similar plots have comparable mechanisms of action. However, time is an implicit parameter, which is a handicap for highlighting mechanistic processes.To implement time into Δf–ΔD plots, the ratio between the derivatives of frequency and dissipation factors can be plotted against the reaction time. This real-time plot provides novel insights into the structural (viscoelasticity, μ1) properties of the adsorbed layers through reaction times (see eqn (1) and the S1 Note in the ESI† for details):
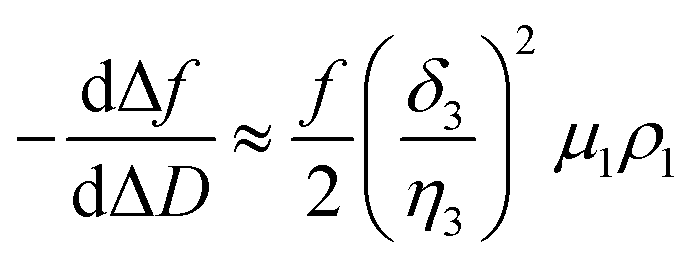 | (1) |
The plot of 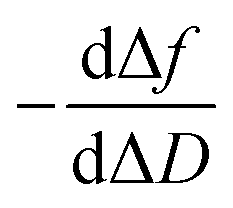 during a chip-based PPI essentially shows how the adlayer structure varies over time. It monitors how one type of biomolecule can form structures with different viscoelastic properties, depending on their interaction with each other, and how it is possible to distinguish between reagents (values at the start of the plot), products (values at the end of that plot), and reaction intermediates. Changes in this novel function enable us to trace and plot the reaction mechanism landscape. Where there is a change in the
during a chip-based PPI essentially shows how the adlayer structure varies over time. It monitors how one type of biomolecule can form structures with different viscoelastic properties, depending on their interaction with each other, and how it is possible to distinguish between reagents (values at the start of the plot), products (values at the end of that plot), and reaction intermediates. Changes in this novel function enable us to trace and plot the reaction mechanism landscape. Where there is a change in the 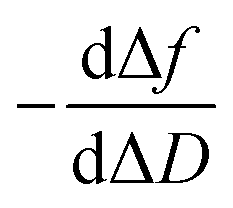 value this suggests that a process occurs in a given time. Hence, it can be used as a fingerprint for chip-based PPIs. This plot allows relations between proteins to be discovered. Proteins with similar trace plots have a comparable mechanism of action, which provides an understanding of which elementary steps should be responsible for that recognition mechanism. Thus this fingerprint can be used even to diagnose diseases.26
value this suggests that a process occurs in a given time. Hence, it can be used as a fingerprint for chip-based PPIs. This plot allows relations between proteins to be discovered. Proteins with similar trace plots have a comparable mechanism of action, which provides an understanding of which elementary steps should be responsible for that recognition mechanism. Thus this fingerprint can be used even to diagnose diseases.26
Next, guidelines are provided to ascertain what that two-dimensional points indicate. If the 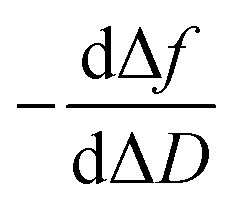 value increases, the recognition process makes the layer more rigid, but the layer becomes less rigid if it lowers. Thus where there is a critical (minimum or maximum or inflection point) point, two elementary steps converge with the same reaction rate. Consequently, a reaction intermediate accumulates. The combined ΔD and Δf information offers a unique tool to draw a reaction mechanism landscape. However, the catch is that the spatial positions of the interacting molecules that govern chip-based PPIs should also be monitored to draw the full reaction mechanism landscape. To map these positions, DPI monitors the adlayer dry thickness in real-time, which is related to the conformational dynamics of the studied interface processes.16
value increases, the recognition process makes the layer more rigid, but the layer becomes less rigid if it lowers. Thus where there is a critical (minimum or maximum or inflection point) point, two elementary steps converge with the same reaction rate. Consequently, a reaction intermediate accumulates. The combined ΔD and Δf information offers a unique tool to draw a reaction mechanism landscape. However, the catch is that the spatial positions of the interacting molecules that govern chip-based PPIs should also be monitored to draw the full reaction mechanism landscape. To map these positions, DPI monitors the adlayer dry thickness in real-time, which is related to the conformational dynamics of the studied interface processes.16
As in the QCM-D assays, the biophysical characterization of PPIs by DPI is based on immobilizing one reagent on the sensing surface, whereas the other is in solution. Thus any changes in adlayer thickness are caused only by the PPI between the immobilized protein and the ligand in solution. When this special case considered, it can be monitored (see eqn 2 and S2 Note in the ESI† for details):
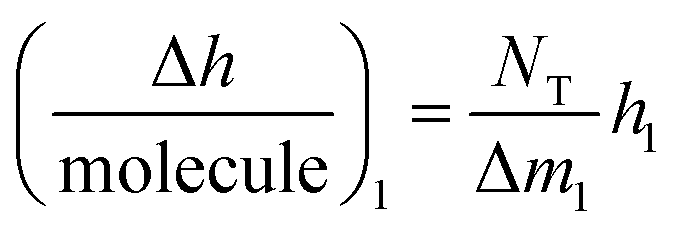 | (2) |
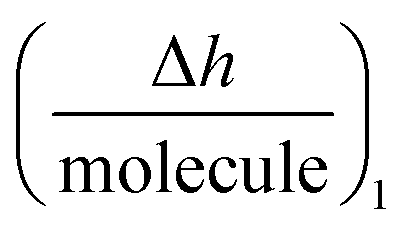 is the height change of the captured ligand (e.g., an antibody or another macromolecule) due to its binding with the receptor (e.g., an antigen or another ligand), NT is the maximum amount of species that can be captured, h1 is the change in adlayer thickness, and Δm1 is the amount of interacting complex.
is the height change of the captured ligand (e.g., an antibody or another macromolecule) due to its binding with the receptor (e.g., an antigen or another ligand), NT is the maximum amount of species that can be captured, h1 is the change in adlayer thickness, and Δm1 is the amount of interacting complex.
As NT, h1 and Δm1 can be easily measured in real-time by DPI, this novel technique allows the 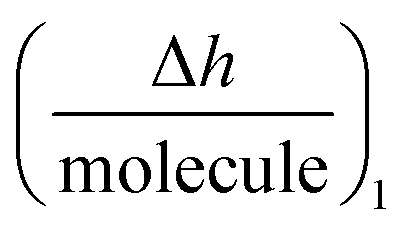 function to be also plotted. Thus its trace shows a structural analysis at the atomic level of the reactive species in chip-based PPIs (resolution < 0.1 Å, according to DPI's characteristics16). If we compare these values with the theoretical dimensions of the ligand, the spatial positions of the interacting molecules that govern PPIs can also be monitored in real-time. An interesting qualitative trend is identified as to how the
function to be also plotted. Thus its trace shows a structural analysis at the atomic level of the reactive species in chip-based PPIs (resolution < 0.1 Å, according to DPI's characteristics16). If we compare these values with the theoretical dimensions of the ligand, the spatial positions of the interacting molecules that govern PPIs can also be monitored in real-time. An interesting qualitative trend is identified as to how the 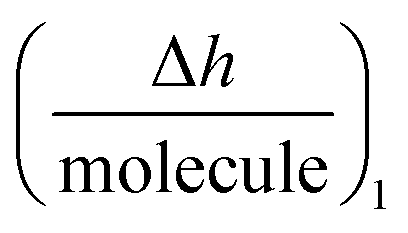 plot varies between different model systems. These trends can be understood qualitatively in terms of conformational dynamics, which facilitates the interpretation of the experimental results. A clear picture emerges as to how gravimetric assays (reaction pathway) can quantitatively correlate with the changes measured by DPI (structural dynamics). The use of both techniques allows molecular binding to be easily mapped by means of conformational dynamics measurements.
plot varies between different model systems. These trends can be understood qualitatively in terms of conformational dynamics, which facilitates the interpretation of the experimental results. A clear picture emerges as to how gravimetric assays (reaction pathway) can quantitatively correlate with the changes measured by DPI (structural dynamics). The use of both techniques allows molecular binding to be easily mapped by means of conformational dynamics measurements.
Monitoring the molecular recognition of TRIM21α by circulating autoantibodies in systemic lupus erythematosus (SLE) patients and healthy subjects demonstrates the advantage of simultaneously measuring chip-based PPIs by DPI and QCM-D. For this purpose, recombinant human TRIM21α protein was immobilized onto the gold chip.26 The formation of the TRIM21α-autoantibody antigenic complex was investigated by analysing the evolution of the piezoelectric signal for different concentrations of the anti-TRIM21α autoantibodies in the bulk.
As shown in Fig. 4a and b, the measured hypersurface was found to be different for the SLE patients and healthy subjects. The biorecognition mechanism is dependent on the selected autoantibody. To confirm this, linear epitope mapping for SLE patients and healthy subjects was performed (see S3 Note†). As shown in Fig. S2,† the anti-TRIM21α autoantibodies of both SLE patients and healthy subjects have different epitopes, which corroborate that they are not the same antibody, thus their reaction pathway may be different.
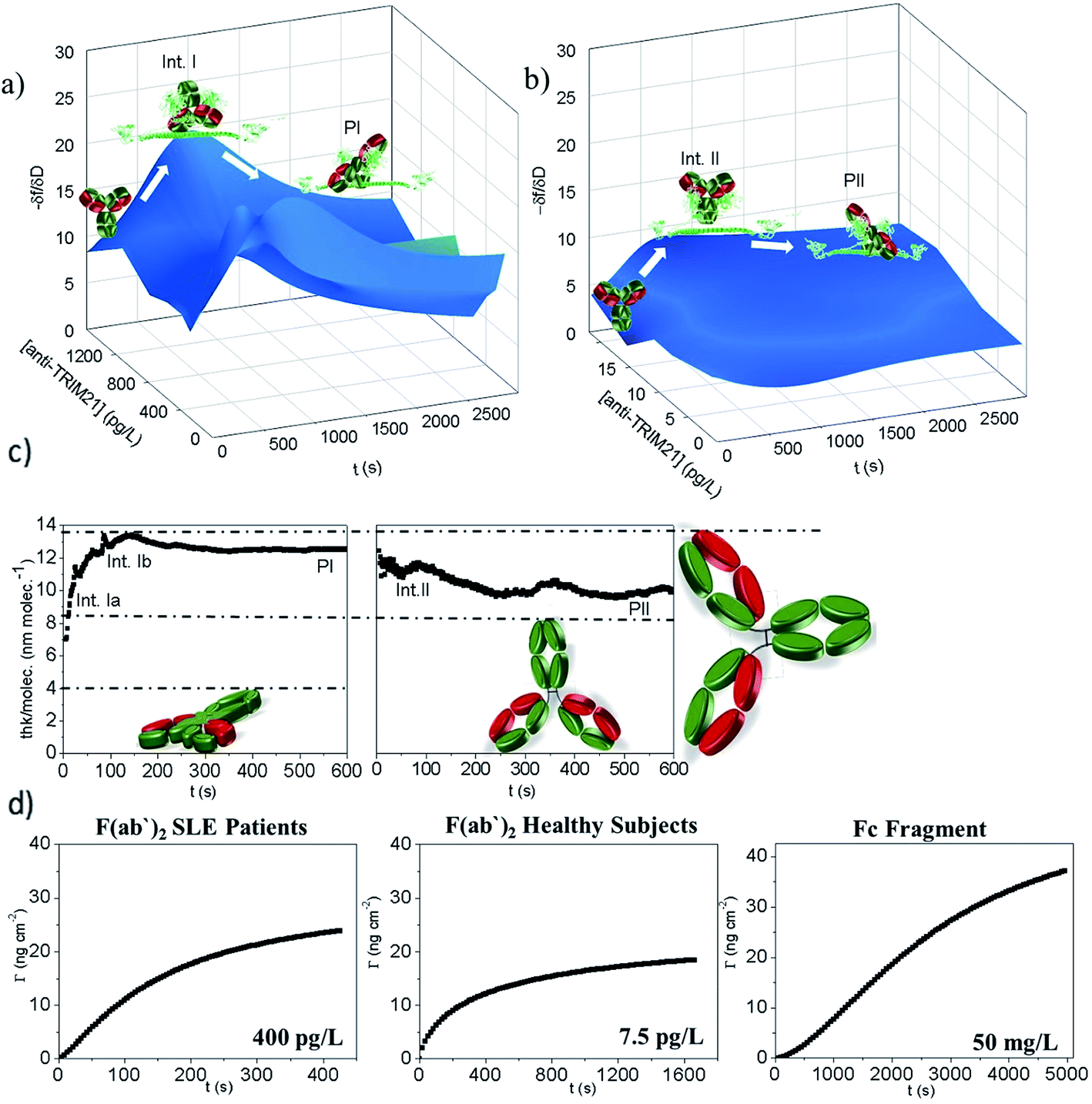 | ||
Fig. 4 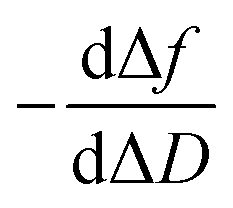 hypersurface representation according to the antibody concentration for: (a) SLE patients, (b) heatlhy subjects, (c) thickness per molecule (calculated from the DPI measurements) of 300 mg L−1 of purified IgGs from the SLE patients (left) and healthy subjects (right). Data are representative of two independent experiments. The interfacial mechanism can be established when considering the theoretical data (dash-dot lines). The IgG dimensions are about 14 × 8 × 4 (width × height × thick) nm, (d) monitoring the binding of the TRIM21α protein with the F(ab′)2 fragments from the SLE patients and healthy subjects and Fc fragments. hypersurface representation according to the antibody concentration for: (a) SLE patients, (b) heatlhy subjects, (c) thickness per molecule (calculated from the DPI measurements) of 300 mg L−1 of purified IgGs from the SLE patients (left) and healthy subjects (right). Data are representative of two independent experiments. The interfacial mechanism can be established when considering the theoretical data (dash-dot lines). The IgG dimensions are about 14 × 8 × 4 (width × height × thick) nm, (d) monitoring the binding of the TRIM21α protein with the F(ab′)2 fragments from the SLE patients and healthy subjects and Fc fragments. | ||
Moreover, the 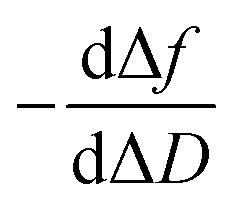 plots for both recognition events correspond to a peak-shape function, which is independent of the autoantibody concentration. As mentioned above, this characteristic shape comprises a similar collection of elementary steps for both populations. They resulted in two different molecular entities, namely the formation of a reaction intermediate (Int. I or II) and the final product (PI or PII), as shown in Fig. 4a and b.
plots for both recognition events correspond to a peak-shape function, which is independent of the autoantibody concentration. As mentioned above, this characteristic shape comprises a similar collection of elementary steps for both populations. They resulted in two different molecular entities, namely the formation of a reaction intermediate (Int. I or II) and the final product (PI or PII), as shown in Fig. 4a and b.
Using the 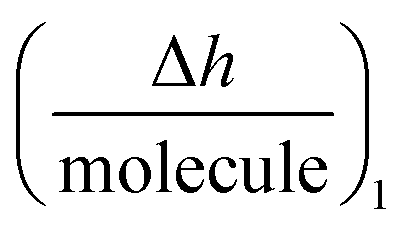 plot, it is provided information on the correlation in the molecular motions, which will be used later to identify the molecular regions that are highly coupled dynamically. Consequently, Fig. 4c shows how the autoantibody-TRIM21α interaction involves three and two different species for the SLE patients and the healthy subjects, respectively. The conformational dynamics of the interfacial process can be established if these values are compared with the theoretical dimensions of the antibody. In a first step, and for the SLE patients, the autoantibodies initially targeted the TRIM21α protein with both Fab fragments bound to the protein (∼8 nm per molecule, Int. Ia), which changed to an orientation with only one Fab fragment bound at intermediate times (∼14 nm per molecule, Int. Ib). For long times, the final product takes an intermediate orientation between both cases (∼13 nm per molecule, PI). In the same way for healthy subjects, the autoantibodies initially targeted the TRIM21α protein with only one Fab fragment (∼12 nm per molecule, Int. II), which changed to the above-mentioned intermediate orientation for long times (∼10 nm per molecule, PII or the final antigenic complex).
plot, it is provided information on the correlation in the molecular motions, which will be used later to identify the molecular regions that are highly coupled dynamically. Consequently, Fig. 4c shows how the autoantibody-TRIM21α interaction involves three and two different species for the SLE patients and the healthy subjects, respectively. The conformational dynamics of the interfacial process can be established if these values are compared with the theoretical dimensions of the antibody. In a first step, and for the SLE patients, the autoantibodies initially targeted the TRIM21α protein with both Fab fragments bound to the protein (∼8 nm per molecule, Int. Ia), which changed to an orientation with only one Fab fragment bound at intermediate times (∼14 nm per molecule, Int. Ib). For long times, the final product takes an intermediate orientation between both cases (∼13 nm per molecule, PI). In the same way for healthy subjects, the autoantibodies initially targeted the TRIM21α protein with only one Fab fragment (∼12 nm per molecule, Int. II), which changed to the above-mentioned intermediate orientation for long times (∼10 nm per molecule, PII or the final antigenic complex).
Although it has been hypothesised that this PPI involves two different reactive species in this case, it needs to be experimentally corroborated. Fig. 4d shows how the TRIM21α protein has multiple binding sites; autoantibodies may bind TRIM21α through their Fab and Fc arms. The half-life (t1/2) is 110 and 2474 s for the F(ab′)2 and the Fc fragments, respectively. The piezoelectric response obtained for 400 pg L−1 of the solution of the SLE F(ab′)2 fragments is similar to that measured for 50 mg L−1 of the Fc fragments (Fig. 4d). However, as Fig. 4d shows, the equilibrium conditions are reached at very different times. Indeed low concentrations of the Fc fragments (<1 mg L−1) were not detected. Consequently, it can be concluded from the experimental results that the F(ab′)2 fragments from the SLE patients' IgGs have more affinity for the TRIM21α protein than the Fc fragments. Similarly, the F(ab′)2 fragments from the healthy subjects' IgGs have a higher affinity for the TRIM21α protein than the Fc fragments (t1/2 = 218 s for the healthy F(ab′)2 fragments at 7.5 pg L−1). Hence the response at a higher affinity, the short times observed for the SLE patients and healthy subjects' IgGs may be related to the Fab region, whereas the response at a lower affinity (long times) may be due to the Fc region. The data and analysis presented herein clearly illustrate the value of the 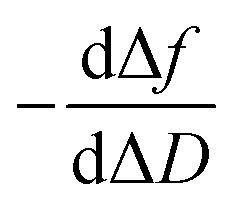 and
and 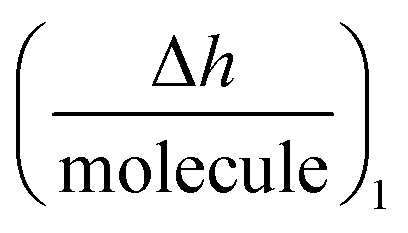 plots; i.e., there are two reaction steps in which the antibody binds an antigen via its Fab and Fc receptors (see the reaction kinetics in S4 Note†).
plots; i.e., there are two reaction steps in which the antibody binds an antigen via its Fab and Fc receptors (see the reaction kinetics in S4 Note†).
Next homology modelling and threading techniques were used to predict a preliminary model of the full TRIM21α structure for the first time (Fig. 4a). The obtained results predict a similar homodimeric structure to that indicated by Kuboshima et al.27 The TRIM21α protein homodimerizes by forming interdigitating antiparallel helical hairpins that position the N-terminal catalytic RING domain at opposite ends of the dimer and the C-terminal PRY-SPRY domain at the centre, according to the TRIM25 structure. The homodimer core also comprises an antiparallel coiled-coil with a distinctive symmetric pattern of a flanking heptad and central hendecad repeats, which appear to be conserved throughout the entire TRIM family.28
Fig. 5b shows a scheme of how the PPI takes place, which was deduced considering the preliminary structure of the TRIM21α homodimer and the experimental results obtained for this interaction. Hence, our results indicate that the Fab (fragments Fab extend, around 14 nm per molecule) and the Fc (at long times) fragments of the circulating autoantibodies bind sequentially. To this end, the PRY-SPRY domains of the homodimer are separated when the TRIM21 protein binds the Fc fragments of the antibody (see Fig. S1d†). This enables the flexible linker segments of the hitherto unknown structure to typically separate both the RING and B-box domains (L1, close to hot spot #1) and the coiled-coil and terminal effector domains (L2, hot spot #2 involves the hinge region of this linker). Consequently, the fold-back configuration of the TRIM21α subunits, and especially L1 and L2, may play an important role in the functional mechanism of this protein, according to the TRIM5α structure.29 Due to this mechanism, the modelled structure corroborates the 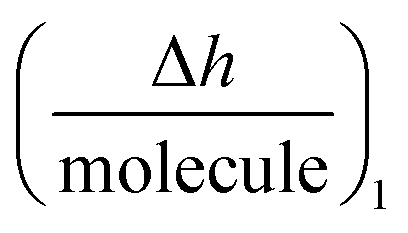 data, which thus confirms that this plot can help to better understand better the conformation dynamics of PPIs.
data, which thus confirms that this plot can help to better understand better the conformation dynamics of PPIs.
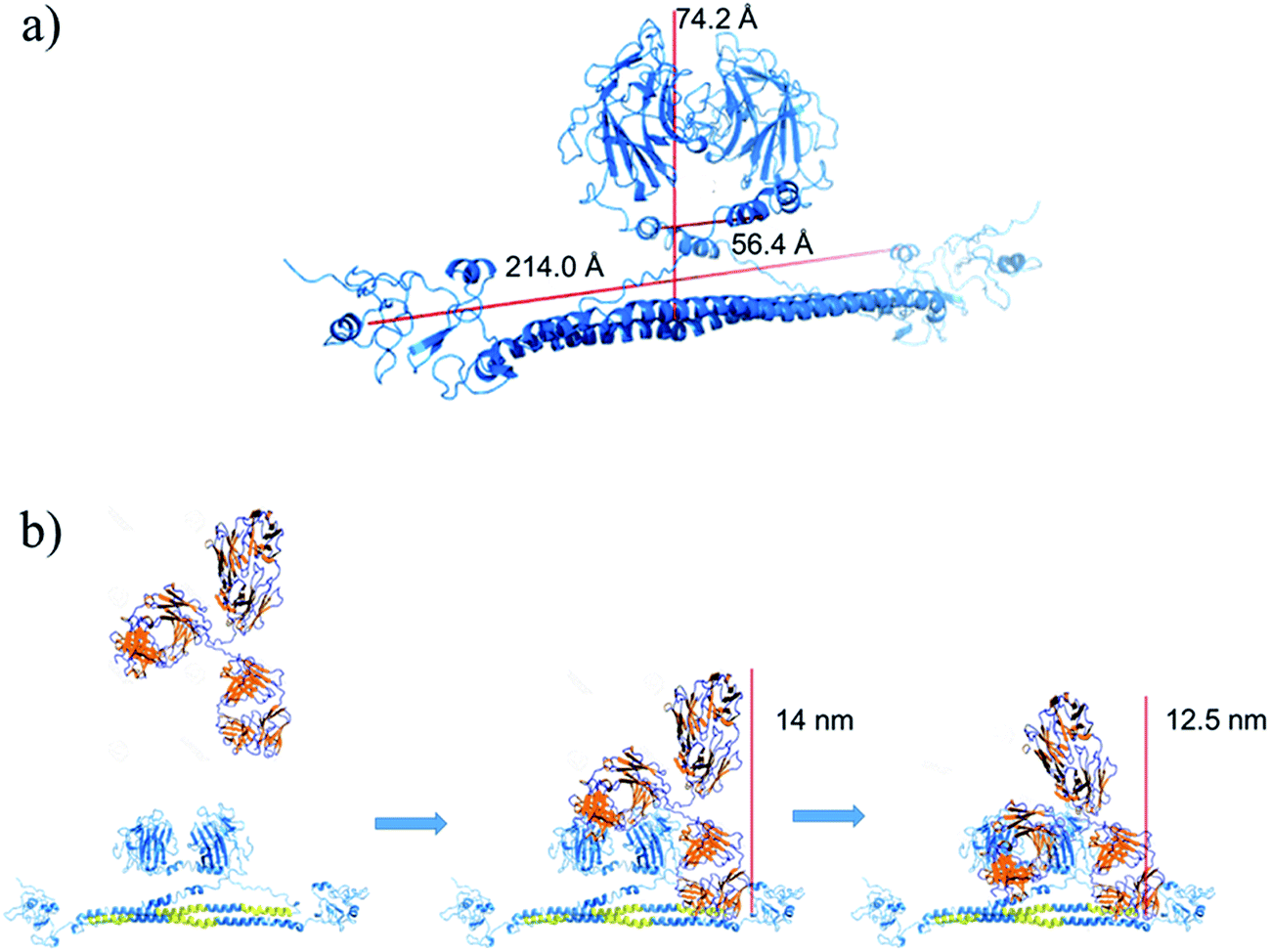 | ||
| Fig. 5 (a) Preliminary structure of the TRIM21α homodimer. (b) Scheme of the homodimer-IgG biorecognition. The PRY-SPRY domains open to interact with the Fc fragment of the antibody. | ||
Conclusions
A quantitative real-time determination of the reaction intermediates and protein orientation under dynamic conditions is an important challenge for a PPI analysis. Our results suggest that, by coupling of two surface analysis techniques (QCM-D and DPI), such large-scale interaction mapping endeavours can be adapted to a chip-based format. This provides a means to reconstruct at a functional level numerous dynamic molecular states, which is required to generate information on proteomes affected by disease or therapeutic treatments. Thus, the ability to monitor this molecular dynamics in a chip format may have great utility in drug discovery. The method could be applied to a wide variety of drug candidates with advances in bio-chip technology and a more comprehensive structural database of protein–protein interactions.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the Generalitat Valenciana (GVA-PROMETEO/2014/040), the Spanish Ministry of Economy and Competitiveness and the European Regional Development Fund (CTQ2013-45875-R and CTQ2013-42914-R) is acknowledged.References
- M. Zhou, Q. Li and R. Wang, Current Experimental Methods for Characterizing Protein–Protein Interactions, ChemMedChem, 2016, 11, 738–756 CrossRef CAS PubMed.
- M. Skwarczynska and C. Ottmann, Protein–protein interactions as drug targets, Future Med. Chem., 2015, 7, 2195–2219 CrossRef CAS PubMed.
- L.-G. Milroy, T. N. Grossmann, S. Hennig, L. Brunsveld and C. Ottmann, Modulators of protein–protein interactions, Chem. Rev., 2014, 114, 4695–4748 CrossRef CAS PubMed.
- M. R. Arkin, Y. Tang and J. A. Wells, Small-Molecule Inhibitors of Protein–Protein Interactions: Progressing toward the Reality, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS PubMed.
- A. H. Talasaz, M. Nemat-Gorgani, Y. Liu, P. Ståhl, R. W. Dutton, M. Ronaghi and R. W. Davis, Prediction of protein orientation upon immobilization on biological and nonbiological surfaces, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14773–14778 CrossRef CAS PubMed.
- H. Cui, E. T. Pashuck, Y. S. Velichko, S. J. Weigand, A. Heetham, C. Newcom and S. Stupp, Spontaneous and X-ray – Triggered Crystallization at Long Range in Self-Assembling Filament Networks, Science, 2010, 327, 555–559 CrossRef CAS PubMed.
- S. J. Ye, H. C. Li, F. Wei, J. Jasensky, A. P. Boughton, P. Yang and Z. Chen, Observing a Model Ion Channel Gating Action in Model Cell Membranes in Real Time in situ: Membrane Potential Change Induced Alamethicin Orientation Change, J. Am. Chem. Soc., 2012, 134, 6237–6243 CrossRef CAS PubMed.
- Y. S. Chen, M. Y. Hong and G. S. Huang, A protein transistor made of an antibody molecule and two gold nanoparticles, Nat. Nanotechnol., 2012, 7, 197–203 CrossRef CAS PubMed.
- G. Zhang, J. Li, P. Cui, T. Wang, J. Jiang and O. V. Prezhdo, Two-Dimensional Linear Dichroism Spectroscopy for Identifying Protein Orientation and Secondary Structure Composition, J. Phys. Chem. Lett., 2017, 8, 1031–1037 CrossRef CAS PubMed.
- B. Ding, A. Panahi, J. J. Ho, J. E. Laaser, C. L. Brooks, M. T. Zanni and Z. Chen, Probing Site-Specific Structural Information of Peptides at Model Membrane Interface In Situ, J. Am. Chem. Soc., 2015, 137, 10190–10198 CrossRef CAS PubMed.
- C. Consani, G. Aubock, F. van Mourik and M. Chergui, Ultrafast Tryptophan-to-Heme Electron Transfer in Myoglobins Revealed by UV 2D Spectroscopy, Science, 2013, 339, 1586–1589 CrossRef CAS PubMed.
- E. Callaway, The revolution will not be crystallized: a new method sweeps through structural biology, Nature, 2015, 525, 172–174 CrossRef CAS PubMed.
- P. Cleverton, R. de Freitas, R. Torresi, G. Picheth and M. Sierakowski, Piezoelectric immunochip coated with thin films of bacterial cellulose nanocrystals for dengue detection, Biosens. Bioelectron., 2017, 92, 47–53 CrossRef PubMed.
- G. A. McCubbin, S. Praporski, S. Piantavigna, D. Knappe, R. Hoffmann, J. H. Bowie, F. Separovic and L. L. Martin, QCM-D fingerprinting of membrane-active peptides, Eur. Biophys. J., 2011, 40, 437–446 CrossRef CAS PubMed.
- X. Li, S. Song, Q. Shuai, Y. Pei, Ç. T. Aastrup, Y. Pei and Z. Pei, Real-time and label-free analysis of binding thermodynamics of carbohydrate-protein interactions on unfixed cancer cell surfaces using a QCM biosensor, Sci. Rep., 2015, 5, 14066 CrossRef CAS PubMed.
- J. Escorihuela, M. A. Gonzalez-Martínes, J. L. López-Paz, R. Puchades, A. Maquieira and D. Gimenez-Romero, Dual-polarization interferometry: a novel technique to light up the nanomolecular world, Chem. Rev., 2015, 115, 265–294 CrossRef CAS PubMed.
- D. L. Mallery, W. A. McEwan, S. R. Bidgood, G. J. Towers, C. M. Johnson and L. C. James, Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21), Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19885–19890 CrossRef PubMed.
- W. A. McEwan, J. C. H. Tam, R. E. Watkinson, S. R. Bidgood, D. L. Mallery and L. C. James, Intracellular antibody-bound pathogens stimulate immune signaling via the Fc receptor TRIM21, Nat. Immunol., 2013, 14, 327–336 CrossRef CAS PubMed.
- A. H. Keeble, Z. Khan, A. Forster and A. L. C. James, TRIM21 is an IgG receptor that is structurally, thermodynamically, and kinetically conserved, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6045–6050 CrossRef CAS PubMed.
- M. Petri, A. M. Orbai, G. S. Alarcón, C. Gordon, J. T. Merrill, P. R. Fortin, I. N. Bruce, D. Isenberg, D. J. Wallace, O. Nived, G. Sturfelt, R. Ramsey-Goldman, S. C. Bae, J. G. Hanly, J. Sánchez-Guerrero, A. Clarke, C. Aranow, S. Manzi, M. Urowitz, D. Gladman, K. Kalunian, M. Costner, V. P. Werth, A. Zoma, S. Bernatsky, G. Ruiz-Irastorza, M. A. Khamashta, S. Jacobsen, J. P. Buyon, P. Maddison, M. A. Dooley, R. F. van Vollenhoven, E. Ginzler, T. Stoll, C. Peschken, J. L. Jorizzo, J. P. Callen, S. S. Lim, B. J. Fessler, M. Inanc, D. L. Kamen, A. Rahman, K. Steinsson, A. G. Franks Jr, L. Sigler, S. Hameed, H. Fang, N. Pham, R. Brey, M. H. Weisman, G. McGwin Jr and L. S. Magder, Derivation and validation of the systemic lupus international collaborating clinics classification criteria for systemic lupus erythematosus, Arthritis Rheum., 2012, 64, 2677–2686 CrossRef PubMed.
- S. Wu and Y. Zhang, LOMETS: a local metal-threading-server for protein structure prediction, Nucleic Acids Res., 2007, 35, 3375–3382 CrossRef CAS PubMed.
- M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T. Gallo Cassarino, M. Bertoni, L. Bordoli and T. Schwede, SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information, Nucleic Acids Res., 2014, 42, W252–W258 CrossRef CAS PubMed.
- L. Bordoli, F. Kiefer, K. Arnold, P. Benkert, J. Battey and T. Schwede, Protein structure homology modelling using SWISS-MODEL workspace, Nat. Protoc., 2008, 4, 1–13 Search PubMed.
- K. Arnold, L. Bordoli, J. Kopp and T. Schwede, The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling, Bioinformatics, 2006, 22, 195–201 CrossRef CAS PubMed.
- D. A. Case, R. M. Betz, W. Botello-Smith, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 11, University of California, San Francisco, 2010 Search PubMed.
- N. M. Do Nascimento, A. Juste-Dolz, E. Grau-García, J. A. Roman-Ivorra, R. Puchades, A. Maquieira, S. Morais and D. Gimenez-Romero, Label-free piezoelectric biosensor for prognosis and diagnosis of Systemic Lupus Erythematosus, Biosens. Bioelectron., 2017, 15, 166–173 CrossRef PubMed.
- M. Kuboshima, H. Shimada, T. L. Liu, F. Nomura, M. Takiguchi, T. Hiwasa and T. Ochiai, Presence of serum tripartite motif-containing 21 antibodies in patients with esophageal squamous cell carcinoma, Cancer Sci., 2006, 97, 380–386 CrossRef CAS PubMed.
- J. G. Sanchez, K. Okreglicka, V. Chandrasekaran, J. M. Welker, W. I. Sundquist and O. Pornillos, The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2494–2499 CrossRef CAS PubMed.
- N. Biris, Y. Yang, A. B. Taylor, A. Tomashevski, M. Guo, P. J. Hart, F. Diaz-Griffero and D. N. Ivanov, Structure of the rhesys monkey TRIM5α PRYSPRY domain, the HIV capsid recognition module, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13278–13283 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10617c |
| This journal is © The Royal Society of Chemistry 2018 |