
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Construction of cost-effective bimetallic nanoparticles on titanium carbides as a superb catalyst for promoting hydrolysis of ammonia borane†
Zhangwei Guo‡
a,
Tong Liu‡ *b,
Qingtao Wangb and
Guanhui Gao*c
*b,
Qingtao Wangb and
Guanhui Gao*c
aCollege of Ocean Science and Engineering, Shanghai Maritime University, Haigang Ave 1550, 201306 Shanghai, China
bCollege of Materials Science and Engineering, Qingdao University of Science and Technology, Zhengzhou Rd 53, 266000 Qingdao, China. E-mail: liutong@qust.edu.cn
cPaul-Drude-Institut für Festkörperelektronik, Hausvogteiplatz 5–7, 10117 Berlin, Germany. E-mail: gao@pdi-berlin.de
First published on 3rd January 2018
Abstract
Bimetallic cost-effective CoNi nanoparticles (NPs) are conveniently supported on titanium carbides (MXene) by a simple one-step wet-chemical method. The synthesized CoNi/MXene catalysts are characterized by XPS, TEM, STEM-HAADF and ICP-AES. The as-prepared CoNi NPs with a size of 2.8 nm are well dispersed on the MXene surface. It is found that among the CoNi bimetallic system, Co0.7Ni0.3 shows the best performance toward catalyzing ammonia borane (AB) decomposition with a turnover frequency value of 87.6 molH2 molcat−1 min−1 at 50 °C. The remarkable catalytic performance is attributed to the mild affiliation of MXene to NPs, which not only stabilizes NPs to maintain a good dispersion but also leaves sufficient surface active sites to facilitate the catalytic reaction.
Introduction
In order to solve the global problems caused by heavy use of fossil fuels in the world, it is highly desirable to develop clean and sustainable energy sources and in turn decrease the consumption of traditional fossil fuels.1–5 Hydrogen, as an environmentally clean energy carrier, has been regarded as one of the most promising candidates to meet the increasing demands for an efficient and clean energy supply.6–9 Among various chemical hydrogen storage materials, ammonia-borane (AB) with a high hydrogen content (19.6 wt%), high stability under ambient conditions, high solubility and nontoxicity, has been considered as a leading contender in promising chemical hydrogen-storage materials for various applications.10–12 Up to now, intensive efforts have been made to achieve efficient AB decomposition, generating 3 mol of H2 per mole of AB, through an efficient, economical and durable catalyst. Although, noble metal-based catalysts, such as Pt, Rh, and Ru,13 provided the highest activities to dehydrogenation of AB, the high cost and scarcity of these catalysts hinders their industrial application. Alternatively, cost-effective metals have been recently developed. Especially, it is found that bimetallic systems usually show higher catalytic kinetics than their monometallic counterparts, due to the synergistic geometric and electronic effects of bimetallic systems, such as Cu–Ni, Cu–Co, Co–Ni.14–16 However, the aggregation of these catalysts always resulted in the tremendous loss of their active sites. To address this problem, different carriers such as graphene, metal–organic frameworks and carbon nanotubes were used to disperse metal nanoparticles (NPs).17–20 The results show that these carriers can effectively restrain the agglomeration of metal NPs and improve their catalytic activities. Recently, graphene-like transition metal carbide (MXene, Ti3C2(OHxF1−x)2) has been widely investigated as a promising carrier for new nanocatalyst.21–26 The abundant functional groups on MXene surface, such as Ti–OH and Ti–F bonds, not only stabilize the nanoparticles during reduction but also improve the hydrophilicity of the resultant catalysts.27,28 However, there is rare report on catalytic properties of Ti3C2X2 supported NPs. In this work, for the first time, we synthesize a composite material of CoNi bimetallic nanoparticles supported on MXene via a simple one-step wet-chemical method. Unexpectedly, the resultant CoNi/MXene nanocatalysts with 100% of H2 selectivity show extremely high catalytic activity and excellent durability toward AB decomposition.Experiment
Synthesis of CoNi/MXene
The synthesis route of the CoNi/MXene nanocatalyst is shown in Scheme 1. In a typical experiment, 100 mg MXene was dissolved in 2 mL water in a two-neck round-bottom flask (30 mL). Ultrasonication was required to get a uniform dispersion for 30 min. Then, 200 μL cobalt chloride solution (0.07 mmol mL−1) and 200 μL nickel chloride (0.03 mmol mL−1) were added into the MXene solution. The resulted mixture was stirred for 20 min with a rotating shaker (220 rpm). A 0.5 mL aqueous solution of sodium borohydride (NaBH4) (1.3 mol L−1) was quickly added into the resulted mixture under vigorous stirring for 3 h at 0 °C using an ice bath to maintain the temperature. When the situ synthesis reaction was completed, 0.5 mL of aqueous solution containing 1 mmol AB was injected into the mixture using a syringe, a gas burette filled with water was connected to the reaction flask to measure the volume of released gas. For comparison, CoNi/vulcanxc-72 carbon (CoNi/XC-72), CoNi/graphene oxide (CoNi/GO) and CoNi/Al2O3 reduced by NaBH4 were synthesized respectively by the similar method.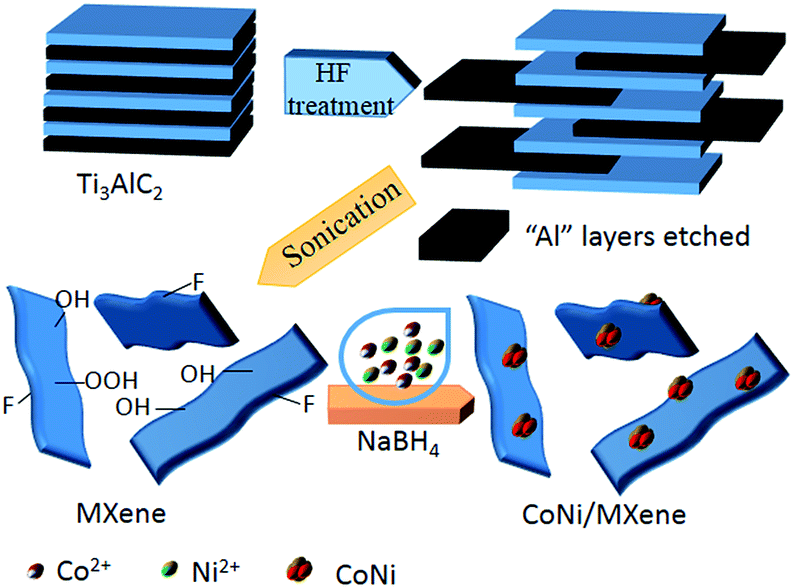 | ||
| Scheme 1 Schematic representation of synthesis of the CoNi/MXene catalyst. | ||
Morphological, structural and compositional characterizations
Transmission electron microscopy (TEM) images and high-resolution STEM measurements were obtained using Tecnai G2 F30 S-Twin instrument with a field emission gun operating at 200 kV. X-ray photoelectron spectroscopy (XPS) measurement was performed with ESCALAB 250Xi spectrophotometer. The metal contents of the catalyst were analyzed using inductively coupled plasma atomic emission spectroscopy (ICP-AES) on Leeman PROFILE SPEC. Mass spectrometry (MS) analysis of the generated gas was performed using an OmniStar GSD320 mass spectrometer, wherein Ar was chosen as the carrying gas.Results and discussions
Fig. 1 shows XPS spectra of CoNi/MXene in the survey, and C 1s, Co 2p and Ni 2p regions. As shown in Fig. 1a, the C 1s spectrum can be divided into several peaks, which corresponds to different kinds of bonds including C–Ti, C–C, C–OH, and HO–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. Additionally, the presence of F and O indicate the introduction of functional groups such as OH and F on Ti3C2X2 surface (Fig. S1 and S2†), which not only enhances the hydrophilicity of the support but also facilitates to the stabilization of the synthesized CoNi NPs, leading a fine dispersion and good stability of the synthesized nanocatalyst in this heterogeneous system. The two peaks located at 787.4 eV and 797.6 eV can be assigned to elemental Co0 2p3/2 and Co0 2p1/2, respectively. While the Ni peaks corresponding to Ni0 2p3/2 and Ni0 2p1/2 are observed at 856 eV and 874 eV, respectively.27,28 From the above results, it can be clearly demonstrated that the CoNi particles have been successfully anchored on MXene surface.
O, respectively. Additionally, the presence of F and O indicate the introduction of functional groups such as OH and F on Ti3C2X2 surface (Fig. S1 and S2†), which not only enhances the hydrophilicity of the support but also facilitates to the stabilization of the synthesized CoNi NPs, leading a fine dispersion and good stability of the synthesized nanocatalyst in this heterogeneous system. The two peaks located at 787.4 eV and 797.6 eV can be assigned to elemental Co0 2p3/2 and Co0 2p1/2, respectively. While the Ni peaks corresponding to Ni0 2p3/2 and Ni0 2p1/2 are observed at 856 eV and 874 eV, respectively.27,28 From the above results, it can be clearly demonstrated that the CoNi particles have been successfully anchored on MXene surface.
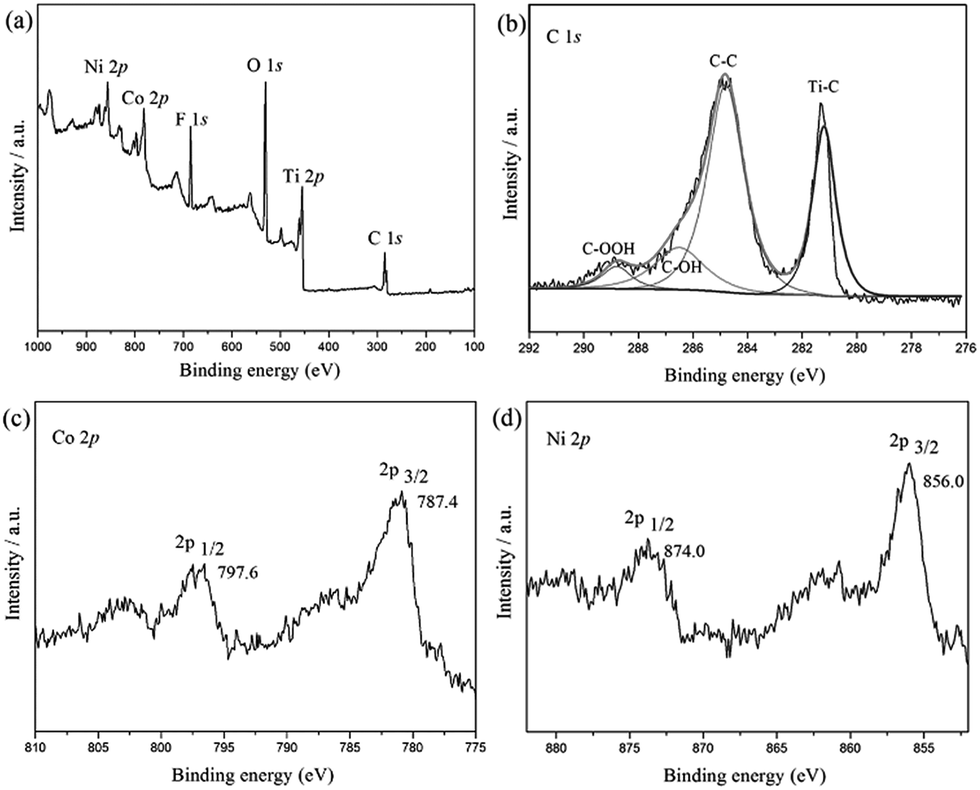 | ||
| Fig. 1 Spectroscopic characteristics of Co0.7Ni0.3/MXene. Survey spectrum of Co0.7Ni0.3/MXene (a), and XPS spectra of C 1s, Co 2p and Ni 2p (b–d). | ||
To further investigate the morphologies of resultant CoNi NPs, transmission electron microscopy (TEM) and high resolution TEM (HRTEM) are used. As seen in Fig. 2a and S3,† the Co0.7Ni0.3 NPs have the good dispersion and no obvious aggregation has been happened. The average diameter of the NPs is about 2.8 nm (Fig. 2c inset). The narrow size and shape distributions benefit from the ‘confine effect’ from the functional OH and F groups on Ti3C2X2 surface, which has been confirmed by XPS results. A high resolution TEM (HRTEM) analysis of a representative nanoparticle of Co0.7Ni0.3 suggests that the as-synthesized specimen has a clearly identified lattice fringe space of 0.2039 nm, which is less than that of the (111) plane of pure face-centered cubic (fcc) Co (0.2046 nm), and larger than that of the (111) plane of pure face-centered cubic (fcc) Ni (0.2033 nm). These results confirmed the as-synthesized Co0.7Ni0.3 NPs with an alloy structure. In addition, an energy dispersive X-ray (EDX) spectrum proves the presence of Co and Ni elements with an atomic ratio of 2.35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S4†), which is in good agreement with the ICP-AES results (Table S1†). The elemental mappings of Ni, Co, and C (Fig. 3) further reveal that Co and Ni elements were homogeneously dispersed throughout the whole MXene. From the above results, it can be seen that the as-synthesized CoNi nanocatalysts have been well dispersed on MXene surface, which benefits to enhancing catalytic property for AB decomposition in this bimetallic heterogeneous system.
:1 (Fig. S4†), which is in good agreement with the ICP-AES results (Table S1†). The elemental mappings of Ni, Co, and C (Fig. 3) further reveal that Co and Ni elements were homogeneously dispersed throughout the whole MXene. From the above results, it can be seen that the as-synthesized CoNi nanocatalysts have been well dispersed on MXene surface, which benefits to enhancing catalytic property for AB decomposition in this bimetallic heterogeneous system.
 | ||
| Fig. 2 TEM images (a–c), and HRTEM images (d) of the Co0.7Ni0.3/MXene. | ||
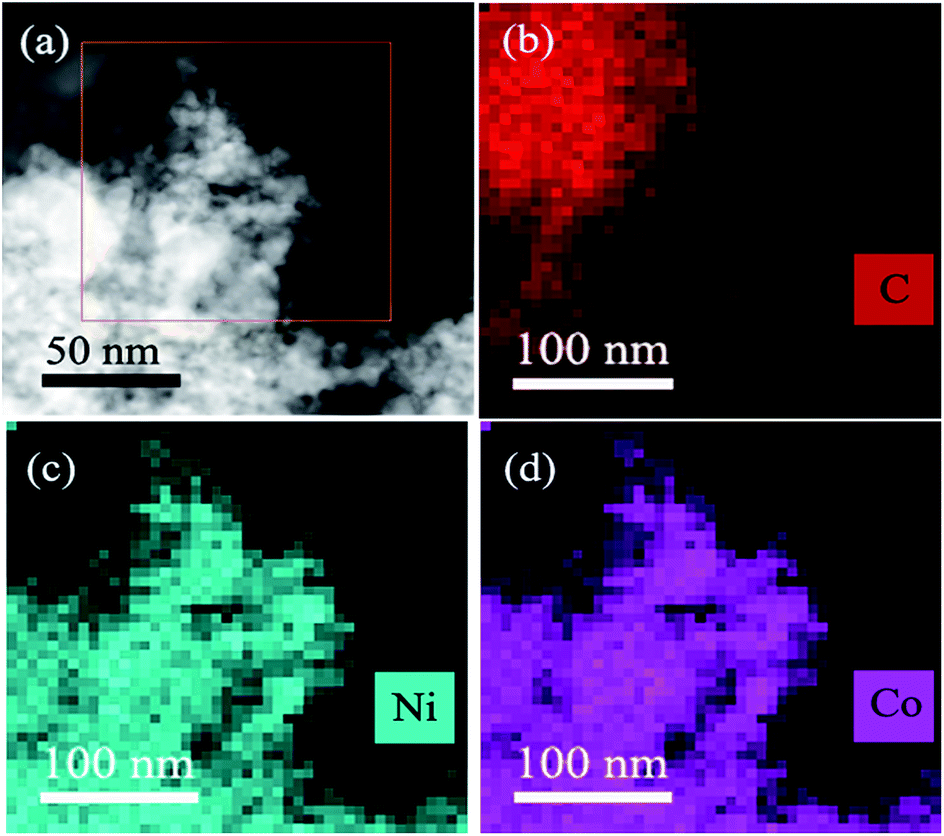 | ||
| Fig. 3 The STEM-HAADF images of the Co0.7Ni0.3/MXene (a) and the corresponding elemental mapping images for C (b), Ni (c) and Co (d). | ||
The as-synthesized CoxNi1−x/MXene (0 ≤ x ≤ 1) NPs have been applied as catalysts for the AB decomposition at 50 °C with a constant molar ratio of catalyst/AB = 0.02 (Fig. 4a). A fast hydrogen generation starts immediately without any induction period when the catalyst is mixed with AB solution under magnetic stirring at 50 °C. Namely, Co0.7Ni0.3 NPs exhibit the highest catalytic activity for AB decomposition, and the corresponding TOF value can achieve 87.6 molH2 molcat−1 min−1 at 50 °C. This value is one of the highest values among all the heterogeneous catalytic dehydrogenation of AB (Table 1). Mass spectrometry (MS) results further confirm the generated H2 and the absence of NH3 in the released gas (Fig. S5†). Notably, the composition of Co and Ni has the obvious effect on the catalytic performance of CoxNi1−x for AB decomposition. If absence of Co additives, reaction catalyzed by pure Ni shows the lowest catalytic activity and hydrogen generation quantity. By alloying Co to Ni, the catalytic activity and selectivity is significantly improved. When the molar ratio of Co (x value) increases from 0.1 to 0.7, the reaction time decreases from 6.6 min to 1.7 min and the corresponding TOF value increases from 22.7 molH2 molcat−1 min−1 to 87.6 molH2 molcat−1 min−1, respectively. However, when the molar ratio of Co reaches 1.0, namely, no addition of Ni, the reaction is completed in 3.75 min. Obviously, the catalytic activity of the CoxNi1−x alloy is better than those of pure Ni and Co. The reason for the enhanced performance of CoxNi1−x alloy may be mainly resulted from the synergetic effect between Co and Ni, which can efficiently tune surface electronic states of bimetallic nanoparticles, particularly related to local strain and effective atomic coordination number at the surface, leading an apparent improvement for AB decomposition.
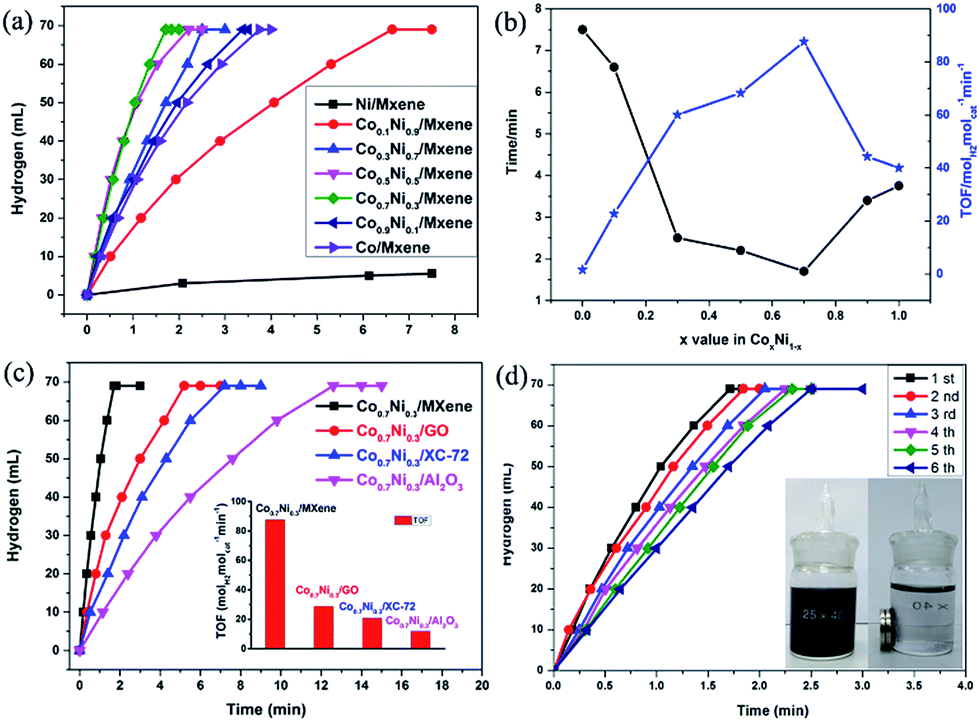 | ||
| Fig. 4 Time course plots for H2 generation from AB decomposition with different Co/Ni molar ratios (0 ≤ x ≤ 1) (a); the corresponding reaction time and TOF (b); over Co0.7Ni0.3/MXene, Co0.7Ni0.3/GO, Co0.7Ni0.3/XC-72 and Co0.7Ni0.3/Al2O3 at 50 °C (c); catalyzed by Co0.7Ni0.3/MXene from 1st to 6th cycles (d). The molar ratio of metal/AB = 0.02. | ||
| Catalyst | Temp. (K) | TOF (molH2 molcat−1 min−1) | Ea (kJ mol−1) | Ref. |
|---|---|---|---|---|
| CoNi/MXene | 323 | 87.6 | 36.9 | This work |
| 303 | 39.1 | 36.9 | This work | |
| CuCo@MIL-101 | 298 | 19.6 | — | 15 |
| Cu0.8Co0.2O@graphene oxide | 298 | 70 | 43.5 | 29 |
| Cu@Co/Co | 303 | 8.36 | 51.3 | 30 |
| NiCu@C nanofiber | 298 | 3.6 | 285.9 | 31 |
| CuCo2O4/Ti | 298 | 44 | 23.6 | 32 |
| Cu0.49Co0.51/C | 298 | 45 | 51.9 | 33 |
| Ni/CNTs | 298 | 26.2 | 32.3 | 16 |
| Co/PSMA | 298 | 25.7 | 34 | 34 |
| Co/graphene | 298 | 18.5 | 27.41 | 35 |
| Co/rGO | 298 | 13.8 | 32.7 | 36 |
| Cu0.1@Co0.45Ni0.45/graphene | 298 | 15.5 | 58 | 37 |
| Au–Co@CN | 298 | 28.4 | — | 38 |
| Ag@Co/graphene | 298 | 10.23 | 20.03 | 8 |
| Au@AuCo/CNT | 298 | 13.24 | 41.9 | 39 |
| Pd@Co/graphene | 298 | 408 | — | 40 |
| Cu@FeNi | 303 | 114 | 44 | 41 |
| Cu@FeCo | 303 | 10.5 | 38.75 | 42 |
Furthermore, the carrier materials also play an important role on determining the catalytic performance for AB decomposition. In order to evaluate the effect of the carrier materials on the catalytic performances toward the AB decomposition, Co0.7Ni0.3 NPs are loaded on different carriers such as GO (Co0.7Ni0.3/GO), XC-72 (Co0.7Ni0.3/XC-72), and Al2O3 (Co0.7Ni0.3/Al2O3) and their catalytic activities toward AB decomposition at 50 °C with a constant molar ratio of catalyst/AB = 0.02 are shown in Fig. 4c. The AB decomposition catalyzed by Co0.7Ni0.3/GO, Co0.7Ni0.3/XC-72 and Co0.7Ni0.3/Al2O3 are completed in 5.2, 7.2 and 12.6 min, respectively, and the corresponding TOF values are 28.8, 20.6 and 11.9 molH2 molcat−1 min−1 in the same reaction condition. Compared to Co0.7Ni0.3/GO, Co0.7Ni0.3/XC-72 and Co0.7Ni0.3/Al2O3, the prepared Co0.7Ni0.3/MXene show higher catalytic kinetics. The superior catalytic performance is mainly attributed that strong interface interaction between metal and carrier, especially for the defect-rich or oxygen group-deficient surfaces on MXene. Such interface interaction is considered to favour the formation of a tunable electronic state of metal NPs, which enhances the AB decomposition.
In order to obtain the activation energy (Ea) of the AB decomposition catalyzed by Co0.7Ni0.3/MXene catalysts, the reactions at different temperatures (30–60 °C) were carried out. Fig. S6a† shows that the plots of the generated H2 versus reaction time for AB decomposition in the presence of Co0.7Ni0.3 catalyst at different temperatures. The Arrhenius plot of lnTOF vs. 1/T for the catalyst is plotted in Fig. S6b,† and Ea is calculated to be 36.9 kJ mol−1.
For the practical application of catalysts, the durability/stability of catalysts is the key point. Therefore, the durability of the Co0.7Ni0.3/MXene nanocatalyst up to fifth run for AB decomposition was characterized by adding additional aliquots (1 mmol) of AB to the catalyst after the reaction completion for the last run. It is evident from Fig. 4d, it is found that a little decrease after a six-time recycle test was seen and the reaction time is prolonged from 1.7 to 2.5 min, indicating the as-prepared Co0.7Ni0.3/MXene possess a moderated durability in AB decomposition. It is due to that the functional OH and F groups on Ti3C2X2 surface, as an anchor, can stabilize the CoNi NPs, avoid the aggregation of CoNi NPs during the reaction process, which is confirmed by TEM images. As clearly seen from the TEM images (Fig. S7†), the CoNi NPs can well disperse on MXene surface and there is no obvious aggregation of the CoNi NPs on MXene. Furthermore, the in situ synthesized NPs are magnetic and thus can be separated from the reaction solution by an external magnet (Fig. 4d, inside), which makes the practical recycling application of the NPs more convenient.
Conclusions
In summary, cost-effective bimetallic CoNi NPs are well dispersed on MXene surfaces by a simple one-step wet-chemical method. Wherein the MXene plays an important role in stabilizing the CoNi NPs, leading a fine dispersion and good stability of the synthesized nanocatalyst in this heterogeneous system. By optimizing the fraction of Co component in Co–Ni system, the synthesized Co0.7Ni0.3/MXene have been proven to the most reactive nanocatalyst in this family reward to AB decomposition with 100% of H2 selectivity and excellent catalytic performance of 87.6 molH2 molcat−1 min−1 at 50 °C. Additionally, the synergetic effect between CoNi NPs and MXene also improve the kinetic toward AB decomposition. This simple synthetic method can be easily extended to facile preparation other MXene supported metal NPs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Project No. 51306166 and 51206101).Notes and references
- S. Young, Nature, 2001, 414, 487–488 CrossRef.
- P. P. Edwards, V. L. Kuznetsov and N. P. Brandon, Energy Policy, 2008, 36, 4356–4362 CrossRef.
- X. Yang and Q. Xu, Chin. J. Catal., 2016, 37, 1594–1599 CrossRef CAS.
- H. L. Wang, Q. L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS.
- J. Kotowicz, L. Bartela, D. Wecel and K. Dubiel, Energy, 2017, 118, 156–171 CrossRef CAS.
- H. M. Chen, C. K. Chen, R. S. Liu, L. Zhang, J. Zhang and D. P. Wilkinson, Chem. Soc. Rev., 2012, 41, 5654–5671 RSC.
- J. Graetz, Chem. Soc. Rev., 2009, 38, 73–82 RSC.
- L. Yang, W. Luo and G. Cheng, ACS Appl. Mater. Interfaces, 2013, 5, 8231–8240 CAS.
- J. Cheng, X. Gu, P. Liu, H. Zhang, L. Ma and H. Su, Appl. Catal., B, 2017, 218, 460–469 CrossRef CAS.
- J. M. Yan, Z. L. Wang, H. L. Wang and Q. Jiang, J. Mater. Chem., 2012, 22, 10990–10993 RSC.
- O. Metin, S. Duman, M. Dinc and S. Ozkar, J. Phys. Chem. C, 2011, 115, 10736–10743 CAS.
- M. Chandra and Q. Xu, J. Power Sources, 2006, 156, 190–194 CrossRef CAS.
- W. W. Zhan, Q. L. Zhu and Q. Xu, ACS Catal., 2016, 6, 6892–6905 CrossRef CAS.
- C. Yu, J. Fu, M. Muzzio, T. Shen, D. Su, J. Zhu and S. Sun, Chem. Mater., 2017, 29, 1413–1418 CrossRef CAS.
- J. Li, Q. L. Zhu and Q. Xu, Catal. Sci. Technol., 2015, 5, 525–530 CAS.
- W. Feng, L. Yang, N. Cao, C. Du, H. M. Dai, W. Luo and G. Z. Cheng, Int. J. Hydrogen Energy, 2014, 39, 3371–3380 CrossRef CAS.
- Q. L. Zhu, J. Li and Q. Xu, J. Am. Chem. Soc., 2013, 135, 10210–10213 CrossRef CAS PubMed.
- F. Z. Song, Q. L. Zhu, X. C. Yang and Q. Xu, ChemNanoMat, 2016, 2, 1003 CrossRef CAS.
- W. Y. Chen, J. Ji, X. Z. Duan, G. Qian, P. Li, X. G. Zhou, D. Chen and W. K. Yuan, Chem. Commun., 2014, 50, 2142–2144 RSC.
- S. Akbayrak and S. Ozkar, ACS Appl. Mater. Interfaces, 2012, 4, 6302–6310 CAS.
- X. Xie, S. Chen, W. Ding, Y. Nie and Z. Wei, Chem. Commun., 2013, 49, 10112–10114 RSC.
- M. Naguib, J. Come, B. Dyatkin, V. Presser, P.-L. Taberna and P. Simon, Electrochem. Commun., 2012, 16, 61–64 CrossRef CAS.
- X. Xie, Y. Xue, L. Li, S. Chen, Y. Nie, W. Ding and Z. D. Wei, Nanoscale, 2014, 6, 11035–11040 RSC.
- M. Ghidiu, M. R. Lukatskaya, M. Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- Q. Peng, J. Guo, Q. Zhang, J. Xiang, B. Liu, A. Zhou, R. Liu and Y. Tian, J. Am. Chem. Soc., 2014, 136, 4113–4116 CrossRef CAS PubMed.
- X. Li, G. Fan and C. Zeng, Int. J. Hydrogen Energy, 2014, 39, 14927–14934 CrossRef CAS.
- X. Li, C. Zeng and G. Fan, Int. J. Hydrogen Energy, 2015, 40, 3883–3891 CrossRef CAS.
- K. Feng, J. Zhong, B. Zhao, H. Zhang, L. Xu, X. Sun and S.-T. Lee, Angew. Chem., Int. Ed., 2016, 55, 11950–11954 CrossRef CAS PubMed.
- Y. S. Du, N. Cao, L. Yang, W. Luo and G. Z. Cheng, New J. Chem., 2013, 37, 3035–3042 RSC.
- A. Yousef, N. A. M. Barakat, M. El-Newehy and H. Y. Kim, Int. J. Hydrogen Energy, 2012, 37, 17715–17723 CrossRef CAS.
- Q. B. Liu, S. J. Zhang, J. Y. Liao, K. J. Feng, Y. Y. Zheng, B. G. Pollet and H. Li, J. Power Sources, 2017, 355, 191–198 CrossRef CAS.
- A. Bulut, M. Yurderi, I. E. Ertas, M. Celebi, M. Kaya and M. Zahmakiran, Appl. Catal., B, 2016, 180, 121–129 CrossRef CAS.
- Ö. Metin and S. Özkar, Int. J. Hydrogen Energy, 2011, 36, 1424–1432 CrossRef.
- M. Li, J. Hu and H. Lu, Catal. Sci. Technol., 2016, 6, 7186–7192 CAS.
- L. Yang, N. Cao, C. Du, H. Dai, K. Hu, W. Luo and G. Cheng, Mater. Lett., 2014, 115, 113–116 CrossRef CAS.
- X. Meng, L. Yang, N. Cao, C. Du, K. Hu, J. Su, W. Luo and G. Cheng, ChemPlusChem, 2014, 79, 325–332 CrossRef CAS.
- L. T. Guo, Y. Y. Cai, J. M. Ge, Y. N. Zhang, L. H. Gong and X. H. Li, ACS Catal., 2014, 5, 388–392 CrossRef.
- L. Yang, W. Luo and G. Cheng, ACS Appl. Mater. Interfaces, 2013, 5, 8231–8240 CAS.
- J. Wang, Y. L. Qin, X. Liu and X. B. Zhang, J. Mater. Chem., 2012, 22, 12468–12470 RSC.
- H. L. Wang, J. M. Yang, Z. L. Wang and Q. Jiang, Int. J. Hydrogen Energy, 2012, 37, 10229–10235 CrossRef CAS.
- F. Qiu, Y. Dai, L. Li, C. Xu, Y. Huang and C. Chen, Int. J. Hydrogen Energy, 2014, 39, 436–441 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10568a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |