
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsoastragaloside I suppresses LPS-induced tight junction disruption and monocyte adhesion on bEnd.3 cells via an activating Nrf2 antioxidant defense system
Hong-Li Li,
Jin-Mei Jin,
Chun Yang,
Ping Wang,
Fei Huang,
Hui Wu,
Bei-Bei Zhang,
Hai-Lian Shi* and
Xiao-Jun Wu *
*
Shanghai Key Laboratory of Compound Chinese Medicines, The Ministry of Education (MOE), Key Laboratory for Standardization of Chinese Medicines, Institute of Chinese Materia Medica, Shanghai University of Traditional Chinese Medicine, 1200 Cailun Road, Zhangjiang Hi-tech Park, Pudong District, Shanghai 201203, China. E-mail: shihailian2003@163.com; xiaojunwu320@126.com; Fax: +86 21-5132-2505; Tel: +86 21-5132-2577 Tel: +86 21-5132-2578
First published on 2nd January 2018
Abstract
Disruption of the blood-brain-barrier (BBB), a crucial event for the entry of inflammatory cells into brain, is a prerequisite for the pathogenesis of many cerebral diseases. Oxidative stress is one of the well-known factors that accounts for the leakage of the BBB, which can be alleviated by the activation of the nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway. Isoastragaloside I (ISOI) is a cycloartane glycoside isolated from Radix Astragali, a clinical widely used traditional Chinese medicine. In the present study, the effect of ISOI on LPS-induced bEnd.3 cells was assessed and the underlying molecular mechanisms were investigated. Our results showed that ISOI pre-treatment inhibited the reduction of TEER, prevented the increase of NaF extravasation and ROS production, and rescued the down-regulated tight-junction (TJ) protein level in bEnd.3 cells stimulated with LPS. Meanwhile, it significantly activated Nrf2 activity, increased its nuclear translocation, thereby, enhanced the expression of its downstream molecules such as NQO1 and HO-1. When Nrf2 was silenced by siRNA, ISOI could not rescue the decrease of TJ proteins induced by LPS any more. In addition, ISOI pre-treatment reduced the elevation of VCAM-1, IL-1β and TNF-α at either protein or mRNA levels, and abrogated the increased adhesion of the monocyte JAWS II cells onto bEnd.3 cells stimulated with LPS. Taken together, these results indicated that ISOI could protect the integrity of the BBB under inflammatory conditions, which was probably mediated through activating the Nrf2 antioxidant pathway.
Introduction
The blood-brain barrier (BBB) is a highly selective permeability barrier between the circulating blood and the brain extracellular fluid in the central nervous system (CNS). It maintains a stable environment for neurons to function properly by controlling the influx and efflux of substances in the brain. Disruption of the BBB is a crucial event occurring in the pathogenesis of many cerebral diseases that are accompanied with the infiltration of inflammatory cells,1 leading to the BBB permeability breakdown. The leakage of the BBB compromises synaptic and neuronal function, resulting in neurological dysfunction such as cognitive decline and dementia.2–5 Tight junction (TJ) proteins, such as occludin, claudin, and zonula occludens (ZO) sealing the space between adjacent brain endothelial cells, are important components of the BBB.3,6 TJ protein degeneration is concurrent with BBB leakage, which can be found in Parkinson's disease, Alzheimer's disease, intracerebral hemorrhage and cerebral ischemia.7–11 Therefore, drugs sustaining BBB permeability may facilitate the relief of cerebral diseases.Many factors exacerbate the leakage of the BBB, among which oxidative stress is a well-known factor for its deleterious effect on the permeability.12 Besides of regulating the magnitude of leukocyte extravasation into brain parenchyma, oxidative stress also can increase BBB permeability through modulating TJ protein expression.13 Nuclear factor erythroid 2-related factor 2 (Nrf2), a transcription factor, plays an important role in resistance to oxidative stress. It trans-activates the transcription of its downstream antioxidant defense enzymes, including hemeoxygenase 1 (HO-1) and NAD(P)H:quinone oxidoreductase I (NQO-1), and thus mitigates the damage of oxidative stress.14 Indeed, activation of Nrf2 signaling after brain injury has been shown to restore the loss of TJ and prevent BBB disruption,15,16 indicating its protective role in BBB integrity.
Isoastragaloside I (ISOI) is one of the active components of Radix Astragali, a famous traditional Chinese medicine used widely in clinic, with benefiting qi and nourishing blood activities.17 It has been found to alleviate hyperglycemia, glucose intolerance, and insulin resistance in both dietary and genetic obese mice by increasing the circulating adiponectin.17 So far, however, little is known about the effect of ISOI on the integrity of BBB under inflammatory condition. In present study, the effect of ISOI on LPS-induced bEnd.3 cells, an immortalized mouse brain endothelial cell line commonly used as in vitro BBB model, was evaluated and its possible underlying molecular mechanism was explored. Our results indicated that ISOI could maintain the integrity of BBB and prevent monocyte adhesion to the endothelial cells under inflammatory condition, the action of which might be exerted through activating Nrf2 mediated antioxidant defense system. The findings may pave the road for the clinical application of ISOI in the therapy of cerebral diseases.
Materials and methods
Cell culture
bEnd.3 cells obtained from ATCC were cultured in RPMI-1640 medium containing 25 mM HEPES (Wisent Inc., China), 10% fetal bovine serum (FBS, Gibco, Australia) and 1% penicillin/streptomycin. HEK293T cells obtained from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China) were cultured in DMEM medium containing 10% FBS (Gibco, Australia) and 1% penicillin/streptomycin. The murine dendritic cell line JAWS II was purchased from ATCC (CRL-11904™) and maintained in MEM medium supplemented with 5 ng mL−1 GM-CSF (Peprotech, USA) and 20% FBS. All cells were cultured at 37 °C in a humidified incubator with 5% CO2.Transendothelial electrical resistance (TEER) measurements
TEER is an indicator for the evaluation of the integrity of the BBB,18 which can be monitored with an electrical resistance system (Millicell ERS-2 epithelial volt-ohm-meter, Millipore, USA). bEnd.3 cells were seeded at a density of 2 × 105 cells per mL in 24-well transwell inserts (0.4 μm pore size, Corning, USA). Four days later, the cells were treated with ISOI (0, 25, 50 and 100 μM) for 2 h followed by LPS (1 μg mL−1) stimulation. Thereafter, the TEER was measured after LPS and ISOI co-treatment for 1, 3, 6, 12 and 24 h, respectively. The resistance measurements of cell-free filters were set as background resistance and were subtracted from that of the sample filters. The values of the treatment groups were presented as the percentage of that of the control group.Sodium fluorescein (NaF) transendothelial flux assay
NaF is a small molecule used as a permeability indicator. The flux of NaF across endothelial monolayers was determined as previously described.19,20 Briefly, the cells were cultured in the 24-well transwell inserts for 4 days as aforementioned. After pre-treated with ISOI (0, 25, 50 and 100 μM) for 2 h, the cells were challenged with LPS (1 μg mL−1) for 24 h. Consequently, NaF diluted in Hanks' Balanced Salt Solution (HBSS) to the final concentration of 100 μg mL−1 was loaded onto the luminal side of the filters (250 μL per well). Meanwhile, the lower compartments were added with fresh HBSS (1 mL per well). Sample solutions were collected from the lower compartments at 5, 10 and 20 min, respectively, and immediately replaced with the same volume of fresh HBSS. The concentration of NaF was determined using a fluorescence multiwell plate reader (Ex (λ) 485 nm; Em (λ) 530 nm). The values of the treatment groups were presented as the percentage of that of the control group.Reactive oxygen species (ROS) measurement
Intracellular ROS level in bEnd.3 cells was detected by 2′,7′-dichlorodihydrofluorescein (DCFH) as described previously.21 After ISOI pre-treatment for 2 h and co-treatment with LPS for another 24 h, the cells were incubated with 20 mM DCFH for 30 min. The dye was removed and washed twice with fresh HBSS. Fluorescence of the cells was measured immediately using a fluorescence microplate reader (Ex (λ) 485 nm; Em (λ) 535 nm). The values of the treatment groups were shown as the percentage of that of the control group.Western blotting analysis
The cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitor cocktail on ice for 30 min, then sonicated for 30 s. After centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min at 4 °C, the supernatants were collected and the protein concentration was quantified by BCA method. Total proteins (30 μg per sample) were separated by 10% SDS-PAGE and transferred onto PVDF membranes by wet transfer approach. The membranes were blocked with 5% (w/v) BSA solution and incubated with respective primary antibodies against ZO-1 (1:1000, #ab59720, Abcam, UK), occludin (1:2000, #GTX85016, GeneTex, USA), claudin-5 (1:500, #sc-28670, Santa Cruz, USA), Nrf2 (1:500, #sc-722, Santa Cruz, USA), HO-1 (1:500, #sc-10789, Santa Cruz, USA), NQO1 (1:1000, #sc-16464, Santa Cruz, USA), VCAM-1 (1:2000, #3540-1, Epitomics, USA) and β-actin (1:2000, #12413, CST, USA) overnight at 4 °C. After washed with 1× PBS containing 0.1% (v/v) Tween-20, the membranes were incubated with secondary antibodies conjugated with horseradish peroxidase. The protein bands were visualized with ECL-prime kit and quantified using Image J 1.46r software.
000 rpm for 15 min at 4 °C, the supernatants were collected and the protein concentration was quantified by BCA method. Total proteins (30 μg per sample) were separated by 10% SDS-PAGE and transferred onto PVDF membranes by wet transfer approach. The membranes were blocked with 5% (w/v) BSA solution and incubated with respective primary antibodies against ZO-1 (1:1000, #ab59720, Abcam, UK), occludin (1:2000, #GTX85016, GeneTex, USA), claudin-5 (1:500, #sc-28670, Santa Cruz, USA), Nrf2 (1:500, #sc-722, Santa Cruz, USA), HO-1 (1:500, #sc-10789, Santa Cruz, USA), NQO1 (1:1000, #sc-16464, Santa Cruz, USA), VCAM-1 (1:2000, #3540-1, Epitomics, USA) and β-actin (1:2000, #12413, CST, USA) overnight at 4 °C. After washed with 1× PBS containing 0.1% (v/v) Tween-20, the membranes were incubated with secondary antibodies conjugated with horseradish peroxidase. The protein bands were visualized with ECL-prime kit and quantified using Image J 1.46r software.
Immunocytochemistry
bEnd.3 cells seeded on coverslips in 24-well plates coated with rat tail collagen type I (#354236, Corning, USA) were pre-treated with ISOI for 2 h followed by LPS stimulation for 12 or 24 h. Thereafter, the cells were subjected to immunocytochemical staining as described previously.22 In brief, the cells were fixed with 4% PFA for 15 min followed by permeabilization with 0.3% Triton-X-100 in PBS for 10 min. After blocked with 3% normal donkey serum for 1 h at room temperature (RT), the cells were incubated with primary antibodies against ZO-1 (1:50) and Nrf2 (1:50) at 4 °C overnight followed by incubation with Alexa-488 conjugated second antibody (1:500) for 1 h at RT. The coverslips were mounted on glass slides with Vector mounting medium containing DAPI. Fluorescent pictures of the cells were taken under Olympus DX81 fluorescent microscope.
Real-time PCR analysis
Total RNA was extracted using TRIzol reagent (Life Technologies) and transcribed into cDNA with RevertAid First Strand cDNA Synthesis Kit (Thermo). The obtained cDNA was used as the template for the quantitative real-time PCR (qPCR) analysis, which was performed using SYBR Premix EX Taq (Roche) under the suggested conditions. The primers used for PCR amplification were listed in Table 1. Quantity of the target genes calculated by comparative Ct method was normalized to that of β-actin (endogenous reference) in the same sample.| Genes | Forward primer | Reverse primer |
|---|---|---|
| IL-1β | TTTGAAGTTGACGGACCCC | TGTGCTGCTGCGAGATTTG |
| TNF-α | GGAACACGTCGTGGGATAATG | GGCAGACTTTGGATGCTTGTT |
| VCAM-1 | GAACCCAAACAGAGGCAGAG | GGTATCCCATCACTTGAGCAG |
| ICAM-1 | CGCTGTGCTTTGAGAACTGT | AGGTCCTTGCCTACTTGCTG |
| β-Actin | CGGTTCCGATGCCCTGAGGCTCTT | CGTCACACTTCATGATGGAATTGA |
Monocyte adhesion assay
The monocyte adhesion assay was performed as mentioned previously.23 bEnd.3 cells were cultured in 96-well plates and co-treated with LPS and ISOI for 24 h as aforementioned. JAWS II cells were loaded with 1 μM calcein red™ dye (#22010, AAT Bioquest, USA) for 30 min after starvation for 6 h, and were diluted to a density of 2 × 106 cells per mL. Consequently, JAWS II cells were added onto the endothelial monolayers and co-cultured for 1 h at 37 °C in an incubator. After adhesion, the monolayers were washed three times with HBSS to remove the non-adherent cells. The fluorescence of the attached JAWS II cells was acquired with a fluorescent microplate reader (Ex (λ) 540 nm; Em (λ) 585 nm).Nrf2 luciferase reporter assay
To investigate the effect of ISOI on Nrf2 transactivation, HEK293T cells cultured in 96-well plate (1 × 104 cells per well) were transiently transfected with Nrf2/ARE luciferase reporter plasmid (Qiagen, Germany) and Renilla luciferase plasmid. Eight hours later, the cells were stimulated with ISOI (25, 50 and 100 μM) for 24 h. Thereafter, the cells were harvested, of which the firefly and Renilla luciferase activities were measured using dual-luciferase reporter assay system (Promega, USA). The final Nrf2 activity was expressed by normalizing firefly luciferase activity to the Renilla luciferase activity within the same sample.siRNA transfection
bEnd.3 cells were cultured in 6-well plates (4 × 105 cells per well). After 24 h, the cells were transfected with Nrf2 siRNA and control siRNA (GenePharma, China) using Lipofectamine® LTX & Plus Transfection Reagent (Life Technologies) according to the manufacturer's protocol. Twelve hours later, the cells were pre-treated with ISOI (100 μM) for 2 h followed by LPS stimulation for 12 or 24 h. Protein samples were extracted from the cells for western blot analysis.Statistical analysis
All data were presented as mean ± SEM. Differences among groups were analyzed by one-way analysis of variance with Dunnett's test using Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). The value of P < 0.05 was considered statistically significant.Results and discussion
ISOI prevented LPS-induced tight junction disruption in bEnd.3 cells
BBB dysfunction is an integral feature of many neurological disorders. Tight junctions are essential for the maintenance of BBB function, which not only create a paracellular diffusion constraint but also enable the vectorial transport across the endothelial monolayer.27 bEnd.3 cell is a convenient and useful model for the functional evaluation of drugs on BBB,24 since it highly expresses tight junction proteins, such as ZO-1, occludin and claudin-5. To assess the effect of ISOI on BBB integrity, the TEER and transendothelial NaF flux of bEnd.3 cells upon LPS stimulation were measured, respectively. As shown in Fig. 1A, LPS stimulation for 1, 3, 6, 12 and 24 h resulted in significantly reduced TEER of bEnd.3 cells (P < 0.001). ISOI pre-treatment at 25, 50 and 100 μΜ prevented the descent of the TEER (P < 0.05, P < 0.01 or P < 0.001). Accordingly, LPS stimulation increased the permeability of bEnd.3 cells as more NaF passed through the cell layer (Fig. 1B, P < 0.01 or P < 0.001). ISOI pre-treatment lessened the extravasion of NaF markedly (P < 0.05 or P < 0.001). In terms of tight junction proteins, compared to the control, LPS stimulation reduced the expression of ZO-1 in the cell membrane of bEnd.3 cells, which could be rescued by ISOI pre-treatment (Fig. 2A–C). Moreover, ISOI pre-treatment significantly prevented the decrease of occludin and claudin-5 (Fig. 2B and C, P < 0.01 and P < 0.05, respectively). Disrupted tight junctions are closely associated with BBB dysfunction characterized by increased permeability but reduced selectivity of BBB. ISOI treatment elevated the electrical resistance while reduced the permeability of LPS-stimulated bEnd.3 cells and enhanced the expression of tight junction proteins, including ZO-1, occludin and claudin-5, at either mRNA or protein level, indicating that ISOI could effectively mitigate the permeability breakdown and sustain the integrity of BBB under inflammatory condition.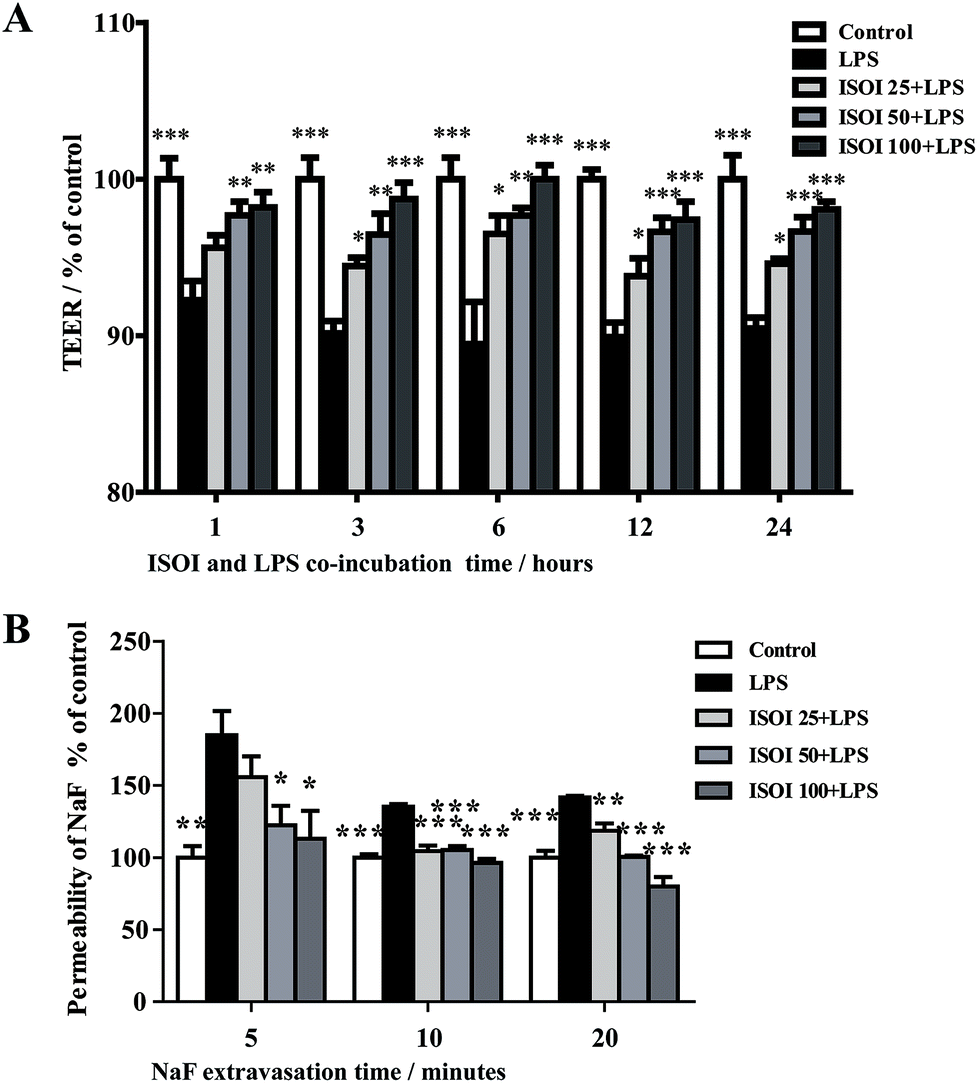 | ||
| Fig. 1 ISOI pretreatment improved the hyperpermeability induced by LPS in bEnd.3 cells. (A) The effect of ISOI on TEER. (B) The effect of ISOI on NaF extravasion. Values were expressed as mean ± SEM (n = 4). In the bar graph, ISOI 25 + LPS, ISOI 50 + LPS, and ISOI 100 + LPS denoted that the cells were co-treated with ISOI (25, 50, and 100 μM) and LPS, respectively. Data were analyzed by one-way analysis of variance with Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001 vs. LPS group. | ||
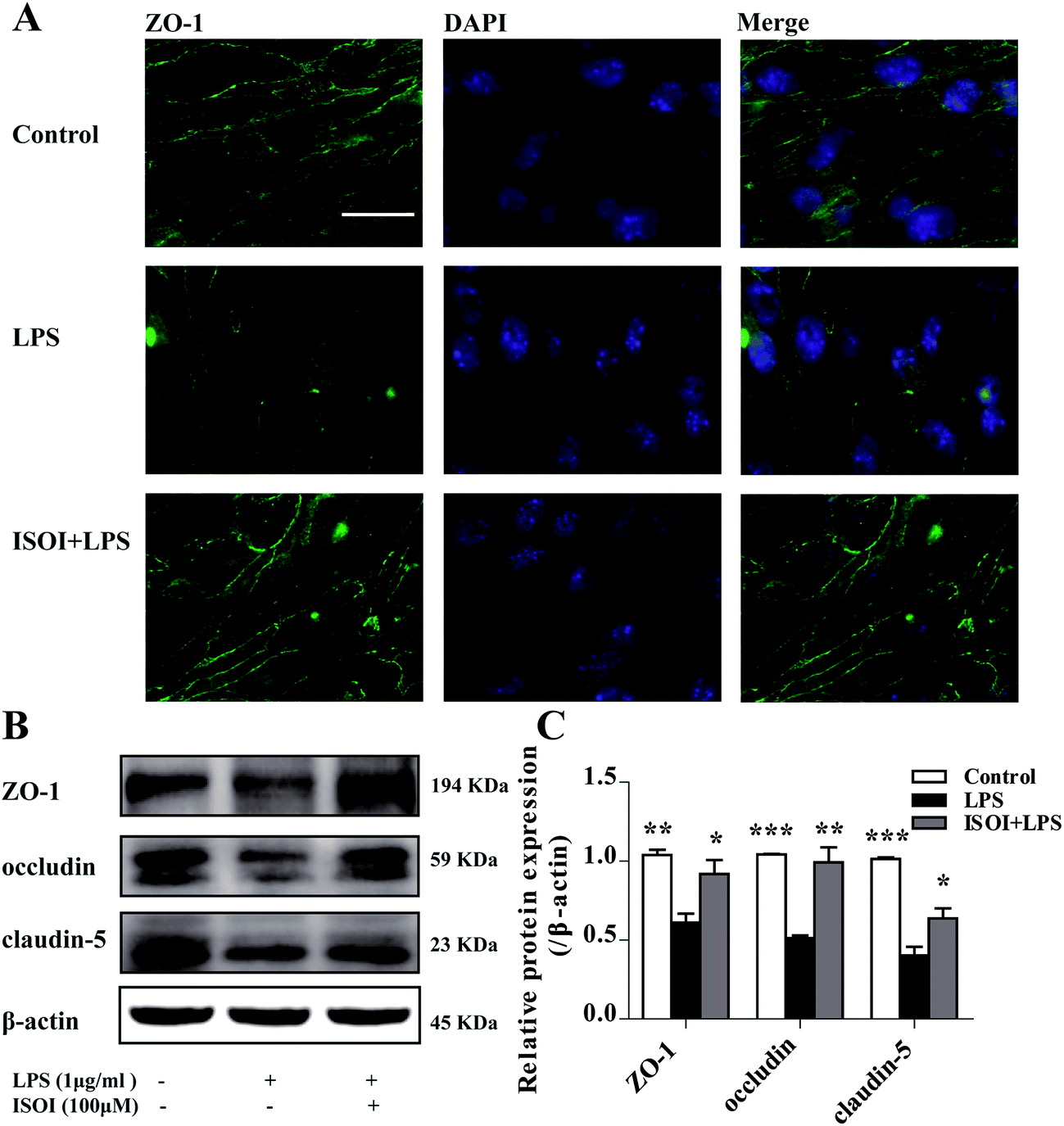 | ||
| Fig. 2 ISOI pretreatment prevented the decrease of TJ proteins after LPS stimulation for 24 h in bEnd.3 cells. (A) Immunocytochemistry of ZO-1. (B) and (C) Western blotting assay of ZO-1, occludin, claudin-5. ISOI + LPS denoted that the cells were co-treated with ISOI (100 μM) and LPS. Data were analyzed by one-way analysis of variance with Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001 vs. LPS group. | ||
ISOI ameliorated monocyte adhesion in bEnd.3 cells stimulated with LPS
Under inflammatory condition, activated brain endothelium attracts monocyte adhesion and mediates their transmigration across BBB. As shown in Fig. 3A, LPS stimulated bEnd.3 cells attracted more JAWS II adhesion compared with the un-stimulated ones (P < 0.001), which could be significantly attenuated by ISOI pre-treatment (P < 0.05 or P < 0.01). Furthermore, ISOI down-regulated pro-inflammatory cytokine's mRNA level, such as IL-1β and TNF-α, upon LPS stimulation (Fig. 3B, P < 0.001). Activation of the pro-inflammatory cytokines increases integrins such as VCAM-1 and ICAM-1 in brain microvessel endothelial cells, thus, enhances the interaction between monocytes and endothelial cells.25,26 Therefore, the effect of ISOI on the expression of VCAM-1 and ICAM-1 was next examined. As displayed in Fig. 3C, LPS stimulation enhanced the mRNA expression of both VCAM-1 and ICAM-1, which was suppressed by ISOI pre-treatment. Moreover, ISOI also restrained the increase of VCAM-1 at protein level upon LPS stimulation (Fig. 3D). These results implicated that ISOI could mitigate monocyte adhesion to endothelial cells and suppress their further transmigration across BBB.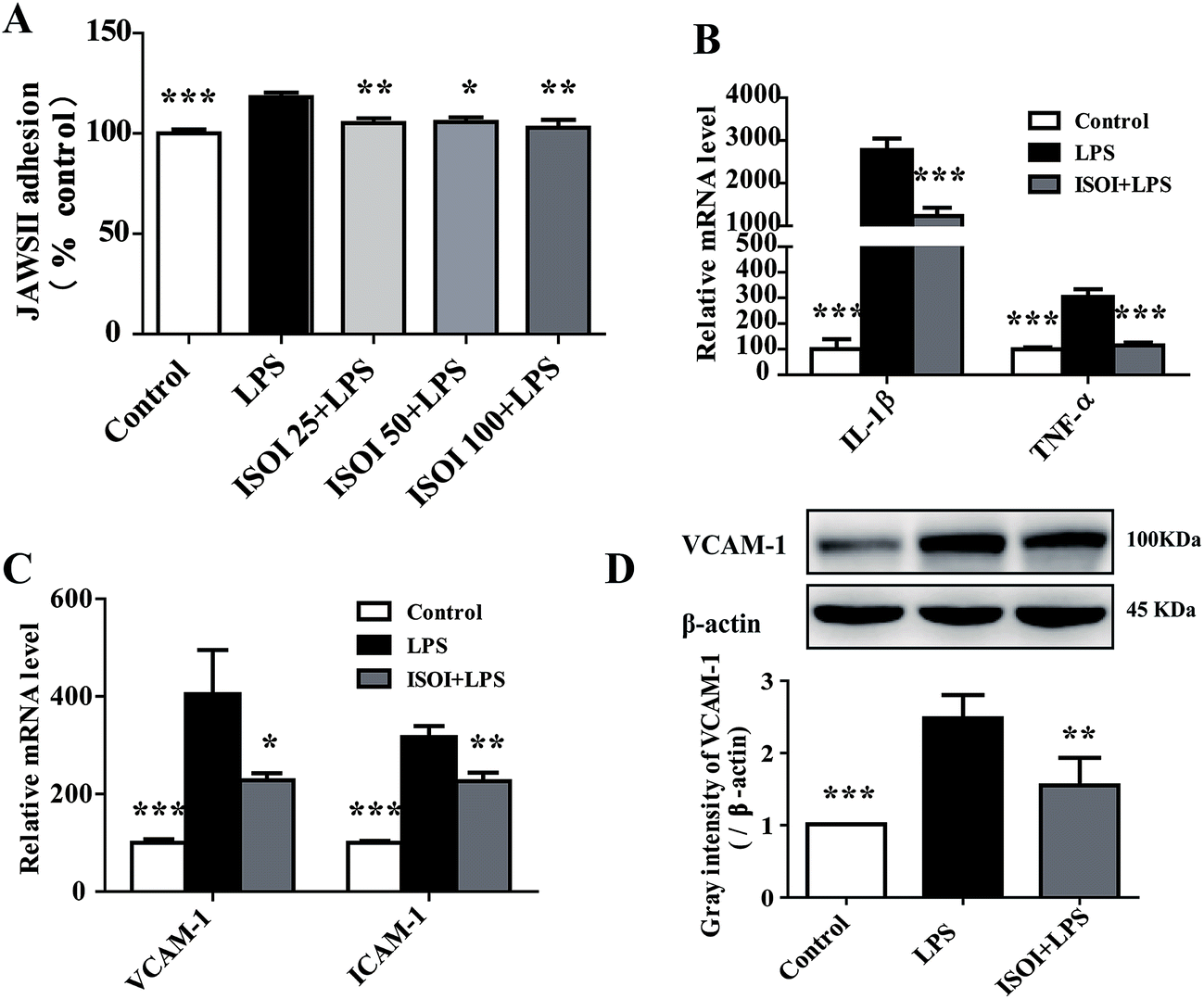 | ||
| Fig. 3 ISOI pretreatment relieved inflammatory response induced by LPS in bEnd.3 cells. (A) ISOI reduced JAWS II adhesion to bEnd.3 cells. ISOI 25 + LPS, ISOI 50 + LPS, and ISOI 100 + LPS denoted that the cells were co-treated with ISOI (25, 50, and 100 μM) and LPS, respectively. (B) and (C): ISOI down-regulated mRNA expression of IL-1β, TNF-α, VCAM-1 and ICAM-1. (D) ISOI depressed the elevated protein expression of VCAM-1. ISOI + LPS denoted that the cells were co-treated with ISOI (100 μM) and LPS. Data were analyzed by one-way analysis of variance with Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001 vs. LPS group. | ||
ISOI activated Nrf2 signaling pathway in bEnd.3 cells stimulated with LPS
ROS, such as superoxide, hydrogen peroxide, and hydroxyl radicals, are continuously produced in aerobic organism, the level of which is strictly modulated under physiological condition by the antioxidant defense system. However, oxidative stress and subsequent damage occur when the production of ROS overwhelms the cellular antioxidant capacity.28 It is well-known that DCFH is one of the fluorescent probes used for the assessment of cellular ROS level, which is oxidized into dichlorofluorescein (DCF) by ROS. As shown in Fig. 4A, LPS stimulation markedly increased the accumulation of DCF in cells (P < 0.01). ISOI treatment at 25, 50 and 100 μΜ could counteract the elevation of DCF (P < 0.001). Oxidative stress has been demonstrated to be one of the factors induce endothelial cells dysfunction, which can be alleviated by the activation of Nrf2 through induction of a variety of antioxidant proteins that exert cytoprotective effects.29 Nrf2 modulates the downstream gene expression by binding to antioxidant response element (ARE) in endothelial cells.30 As displayed in Fig. 4B, ISOI treatment at 25, 50 and 100 μΜ could significantly enhance Nrf2-luciferase activity in HEK293T cells (P < 0.05 or P < 0.01). Consistently, ISOI increased the nuclear fluorescence of Nrf2, suggesting that ISOI enhanced the nuclear translocation of Nrf2 (Fig. 4C and D). Furthermore, it rescued the down-regulated protein expression of Nrf2, HO-1 and NQO1 induced by LPS (Fig. 4E and F, P < 0.05 and P < 0.01). These results indicated that ISOI could activate Nrf2 signaling pathway and exert an antioxidant effect.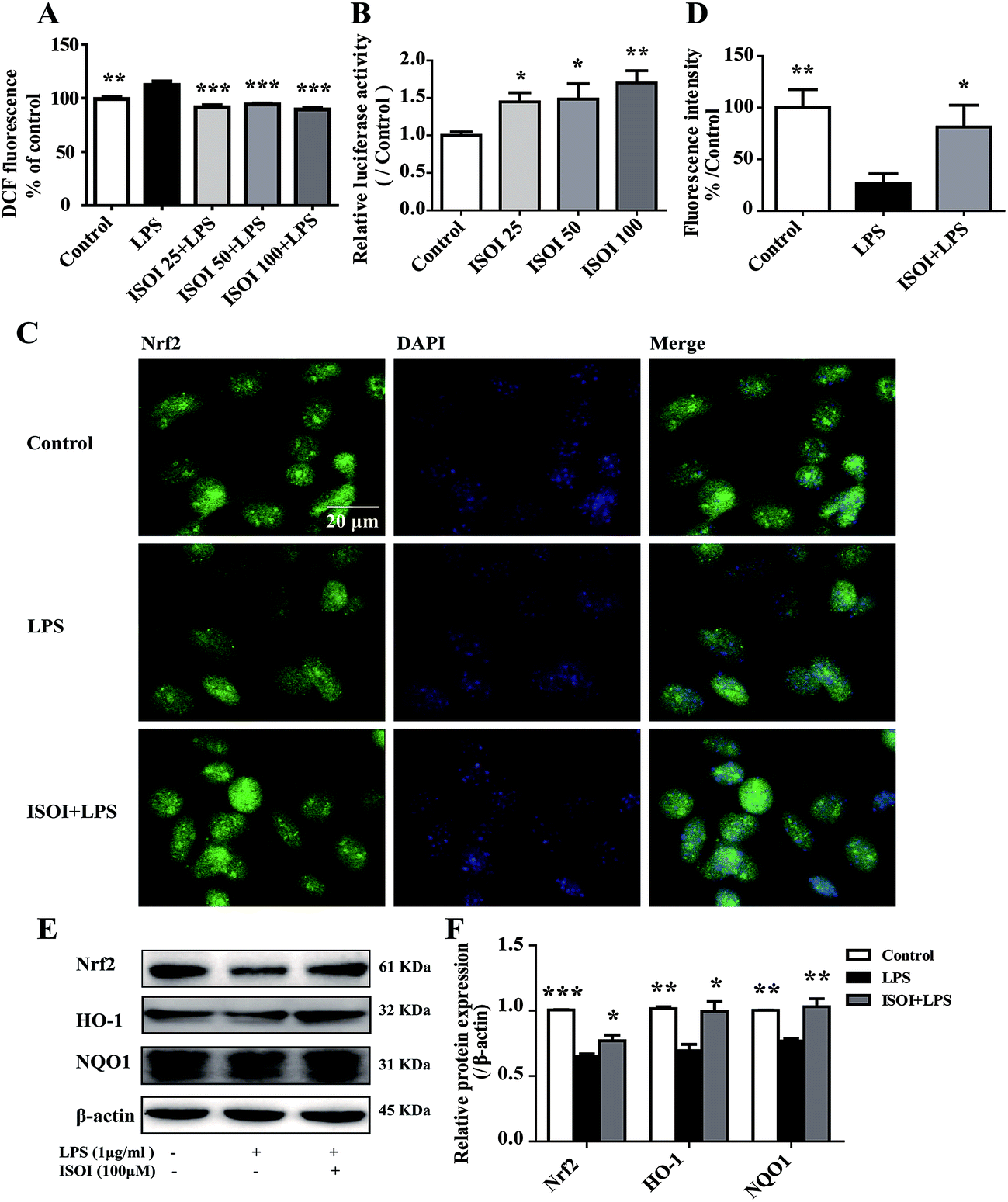 | ||
| Fig. 4 ISOI pretreatment activated the Nrf2 signaling pathway in bEnd.3 cells upon LPS stimulation. (A) The relative DCF fluorescence intensity with ISOI pretreatment for 2 h prior to LPS stimulation for 24 h. (B) Nrf2/ARE luciferase assay. ISOI 25 + LPS, ISOI 50 + LPS, and ISOI 100 + LPS denoted that the cells were co-treated with ISOI (25, 50, and 100 μM) and LPS, respectively. (C) The effect of ISOI on the nuclear translocation of Nrf2. (D) The fluorescence intensity analysis of nuclear Nrf2. (E) and (F) The effect of ISOI on protein expression of Nrf2, HO-1 and NQO1. ISOI + LPS denoted that the cells were co-treated with ISOI (100 μM) and LPS. Data were analyzed by one-way analysis of variance with Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001 vs. LPS group. | ||
Activation of Nrf2 has also been suggested to mediate the production or expression of pro-inflammatory mediators, including cytokines TNF-α and IL-1β, chemokines, integrins VCAM-1 and ICAM-1, matrix metalloproteinases, cyclooxygenase-2 and inducible nitric oxide synthase.31 Moreover, it suppresses TNF-α or IL-1β induced MCP-1 and VCAM-1 expression and monocyte adhesion in endothelial cells.30,32–36 To confirm the role of Nrf2 signaling pathway in the protective effect of ISOI on LPS-stimulated bEnd.3 cells, it was transiently knocked down with siRNA technique (Fig. 5A). As shown in Fig. 5B and C, when Nrf2 was silenced, LPS could not suppress the expression of tight junction proteins, including ZO-1, occludin and claudin-5. Meanwhile, ISOI did not show significant effect on these tight junction proteins. By contrast, LPS induced prominent expression of VCAM-1 in Nrf2 silenced bEnd.3 cells (Fig. 5D, P < 0.001). However, ISOI pretreatment did not reverse the elevation of VCAM-1 induced by LPS stimulation. These results suggested that ISOI could prevent further inflammatory injury to BBB through activating endothelial Nrf2 signaling pathway, thus, reduced monocytes transmigration across BBB into CNS.
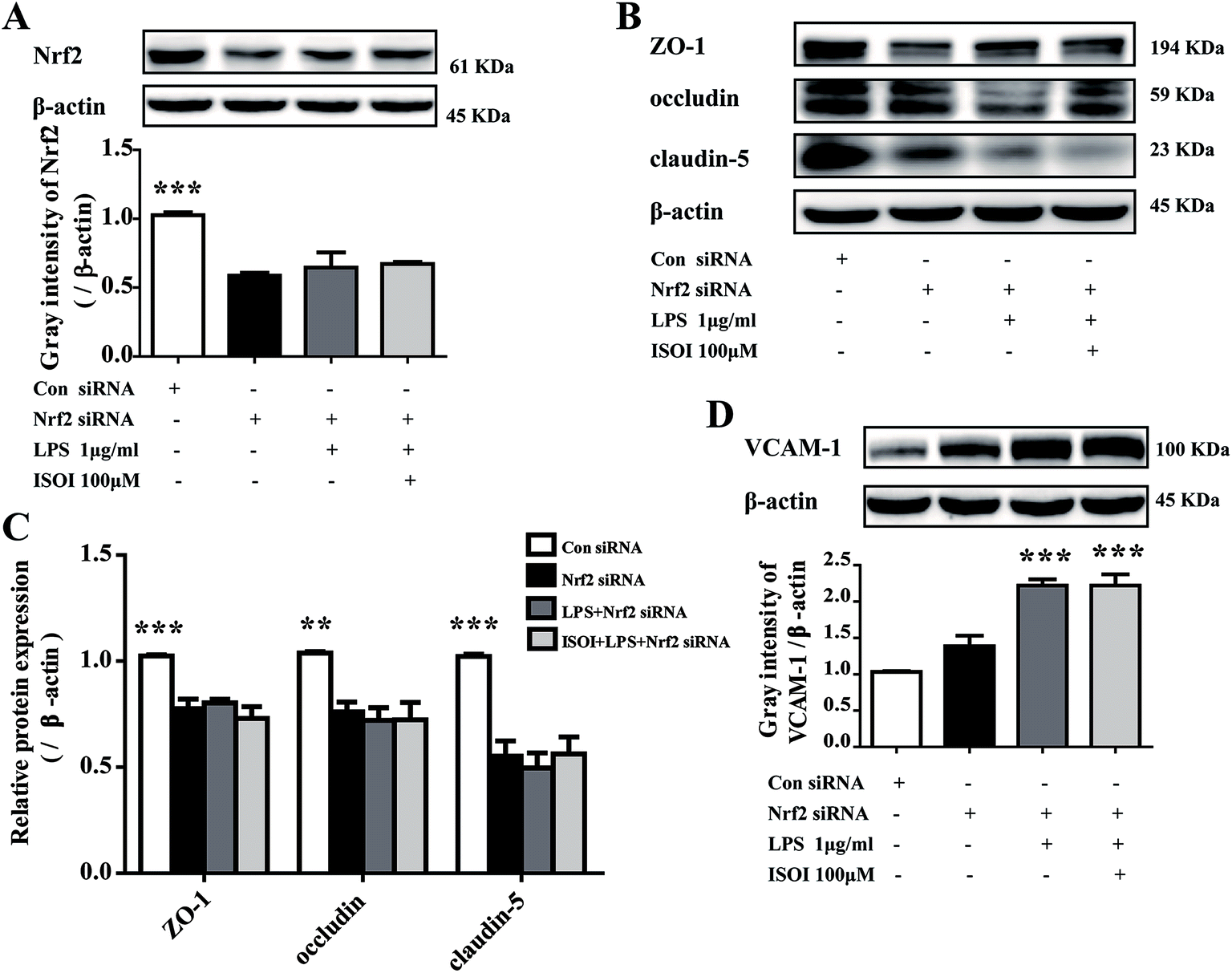 | ||
| Fig. 5 Silencing Nrf2 abolished the up-regulation of TJs and the down-regulation of VCAM-1 of ISOI in bEnd.3 cells stimulated with LPS. (A) Nrf2 protein expression was knocked down by transiently transfection of Nrf2 siRNA. (B) and (C): The effect of ISOI on TJ protein (ZO-1, occludin and claudin-5) expression after transiently transfection with Nrf2 siRNA. (D) The effect of ISOI on VCAM-1 protein expression after transiently transfection with Nrf2 siRNA. ISOI + LPS denoted that the cells were co-treated with ISOI (100 μM) and LPS. Data were analyzed by one-way analysis of variance with Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001 vs. LPS group. | ||
It is well-known that NF-κB is a ubiquitous pro-inflammatory transcription factor which mediates inflammatory response by regulating the expression of downstream inflammatory genes encoding cell adhesion molecules and cytokines, such as TNF-α, IL-1β and VCAM-1.37–39 Tremendous reports implicated the cross-talk between Nrf2 and NF-κB signaling pathway, in which Nrf2 activation was found to directly inhibit NF-κB signaling.38,40–43 However, in the present study, we do not know whether the inhibitory effect of ASI on the inflammatory responses in bEnd.3 cells via Nrf2 activation was dependent on NF-κB signaling pathway, which still need further investigation in the future.
Collectively, ISOI could maintain the BBB integrity and reduce monocyte adhesion to BBB endothelial cells stimulated by LPS, the effect of which was probably mediated by alleviation of oxidative stress through activating Nrf2 pathway. Our results indicated that ISOI could be a potential neuroprotective drug acting on BBB.
Conflicts of interest
All authors declare that they are no conflict of interest.Abbreviations
| ISOI | Isoastragaloside I |
| LPS | Lipopolysaccharide |
| Nrf2 | Nuclear factor-erythroid 2-related factor 2 |
| HO-1 | Hemeoxygenase 1 |
| NQO1 | NAD(P)H:quinone oxidoreductase I |
| IL | Interleukin |
| TNF-α | Tumor necrosis factor-alpha |
| TEER | Transendothelial electrical resistance |
| ROS | Reactive oxygen species |
| FBS | Fetal bovine serum |
| DCFH | 2′,7′-Dichlorodihydrofluorescein |
| NaF | Sodium fluorescein |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| ICAM-1 | Intercellular cell adhesion molecule-1 |
| BBB | Blood-brain barrier |
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81673626, 81603354), Shanghai Eastern Scholar Program (2013-59) and Shanghai E-research Institute of Bioactive Constituent in TCM plan.References
- S. F. Rodrigues and D. N. Granger, Tissue Barriers, 2015, 3, e978720 CrossRef PubMed.
- H. J. van de Haar, S. Burgmans, J. F. Jansen, M. J. van Osch, M. A. van Buchem, M. Muller, P. A. Hofman, F. R. Verhey and W. H. Backes, Radiology, 2016, 281, 527–535 CrossRef PubMed.
- B. V. Zlokovic, Neuron, 2008, 57, 178–201 CrossRef CAS PubMed.
- S. Burgmans, H. J. van de Haar, F. R. Verhey and W. H. Backes, Alzheimer's Dementia, 2013, 35, 859–873 Search PubMed.
- J. M. Wardlaw, F. N. Doubal, M. Valdes-Hernandez, X. Wang, F. M. Chappell, K. Shuler, P. A. Armitage, T. C. Carpenter and M. S. Dennis, Stroke, 2013, 44, 525–527 CrossRef PubMed.
- A. Prat, K. Biernacki, J. F. Lavoie, J. Poirier, P. Duquette and J. P. Antel, Arch. Neurol., 2002, 59, 391–397 CrossRef PubMed.
- Z. Liao, Z. Yang, A. Piontek, M. Eichner, G. Krause, L. Li, J. Piontek and J. Zhang, J. Neurosci., 2016, 327, 53–63 CrossRef CAS PubMed.
- X. Zhang, X. P. Chen, J. B. Lin, Y. Xiong, W. J. Liao and Q. Wan, Brain Res., 2017, 1655, 176–185 CrossRef CAS PubMed.
- M. Chen, L. Lai, X. Li, X. Zhang, X. He, W. Liu, R. Li, X. Ke, C. Fu, Z. Huang and C. Duan, Neurochem. Res., 2016, 41, 3095–3102 CrossRef CAS PubMed.
- A. C. Luissint, C. Artus, F. Glacial, K. Ganeshamoorthy and P. O. Couraud, Fluids Barriers CNS, 2012, 9, 23 CrossRef PubMed.
- J. Bednarczyk and K. Lukasiuk, Acta Neurobiol. Exp., 2011, 71, 393–408 Search PubMed.
- P. Lagrange, I. A. Romero, A. Minn and P. A. Revest, Free Radical Biol. Med., 1999, 27, 667–672 CrossRef CAS PubMed.
- X. Liu, B. Sui and J. Sun, Biomaterials, 2017, 121, 64–82 CrossRef CAS PubMed.
- C. Pang, Z. Zheng, L. Shi, Y. Sheng, H. Wei, Z. Wang and L. Ji, Free Radical Biol. Med., 2016, 91, 236–246 CrossRef CAS PubMed.
- A. Alfieri, S. Srivastava, R. C. Siow, D. Cash, M. Modo, M. R. Duchen, P. A. Fraser, S. C. Williams and G. E. Mann, Free Radical Biol. Med., 2013, 65, 1012–1022 CrossRef CAS PubMed.
- J. Zhao, A. N. Moore, J. B. Redell and P. K. Dash, J. Neurosci., 2007, 27, 10240–10248 CrossRef CAS PubMed.
- A. Xu, H. Wang, R. L. Hoo, G. Sweeney, P. M. Vanhoutte, Y. Wang, D. Wu, W. Chu, G. Qin and K. S. Lam, Endocrinology, 2009, 150, 625–633 CrossRef CAS PubMed.
- A. Patabendige, R. A. Skinner and N. J. Abbott, Brain Res., 2013, 1521, 1–15 CrossRef CAS PubMed.
- S. Nakagawa, M. A. Deli, S. Nakao, M. Honda, K. Hayashi, R. Nakaoke, Y. Kataoka and M. Niwa, Cell. Mol. Neurobiol., 2007, 27, 687–694 CrossRef CAS PubMed.
- S. Fukuda and S. Nakagawa, J. Mol. Neurosci., 2016, 59, 211–219 CrossRef CAS PubMed.
- Y. He, M. Du, Y. Gao, H. Liu, H. Wang, X. Wu and Z. Wang, PLoS One, 2013, 8, e76495 CAS.
- H. S. Liu, H. L. Shi, F. Huang, K. E. Peterson, H. Wu, Y. Y. Lan, B. B. Zhang, Y. X. He, T. Woods, M. Du, X. J. Wu and Z. T. Wang, Sci. Rep., 2016, 6, 19137 CrossRef CAS PubMed.
- J. Huppert, D. Closhen, A. Croxford, R. White, P. Kulig, E. Pietrowski, I. Bechmann, B. Becher, H. J. Luhmann, A. Waisman and C. R. Kuhlmann, FASEB J., 2010, 24, 1023–1034 CrossRef CAS PubMed.
- T. Watanabe, S. Dohgu, F. Takata, T. Nishioku, A. Nakashima, K. Futagami, A. Yamauchi and Y. Kataoka, Biol. Pharm. Bull., 2013, 36, 492–495 CAS.
- J. B. Dietrich, J. Neuroimmunol., 2002, 128, 58–68 CrossRef CAS PubMed.
- J. Quandt and K. Dorovini-Zis, J. Neuropathol. Exp. Neurol., 2004, 63, 350–362 CrossRef CAS PubMed.
- H. Bauer and A. Traweger, CNS Neurol. Disord.: Drug Targets, 2016, 15, 1016–1029 CAS.
- F. L. van Muiswinkel and H. B. Kuiperij, Curr. Drug Targets, 2005, 4, 267–281 CrossRef CAS.
- J. Mimura and K. Itoh, Free Radical Biol. Med., 2015, 88, 221–232 CrossRef CAS PubMed.
- X. L. Chen, G. Dodd, S. Thomas, X. Zhang, M. A. Wasserman, B. H. Rovin and C. Kunsch, Am. J. Physiol.: Heart Circ. Physiol., 2006, 290, H1862–H1870 CrossRef CAS PubMed.
- H. Chen, K. Xie, H. Han, Y. Li, L. Liu, T. Yang and Y. Yu, Int. Immunopharmacol., 2015, 28, 643–654 CrossRef CAS PubMed.
- J. Kim, Y. N. Cha and Y. J. Surh, Mutat. Res., 2010, 690, 12–23 CrossRef CAS PubMed.
- H. O. Pae, G. S. Oh, B. S. Lee, J. S. Rim, Y. M. Kim and H. T. Chung, Atherosclerosis, 2006, 187, 274–284 CrossRef CAS PubMed.
- X. L. Chen, G. Dodd and C. Kunsch, Inflammation Res., 2009, 58, 513–521 CrossRef CAS PubMed.
- H. P. Zhang, F. L. Zheng, J. H. Zhao, D. X. Guo and X. L. Chen, Arch. Med. Res., 2013, 44, 13–20 CrossRef CAS PubMed.
- X. L. Chen and C. Kunsch, Curr. Pharm. Des., 2004, 10, 879–891 CrossRef CAS PubMed.
- Z. H. Wang, D. Zhu, S. Xie, Y. Deng, Y. Pan, J. Ren and H. G. Liu, J. Cardiovasc. Pharmacol., 2017, 70, 102–109 CrossRef CAS PubMed.
- D. Fratantonio and A. Speciale, BioFactors, 2017, 43, 54–62 CrossRef CAS PubMed.
- A. Speciale, R. Canali, J. Chirafisi, A. Saija, F. Virgili and F. Cimino, J. Agric. Food Chem., 2010, 58, 12048–12054 CrossRef CAS PubMed.
- A. Speciale, S. Anwar, R. Canali, J. Chirafisi, A. Saija, F. Virgili and F. Cimino, Mol. Nutr. Food Res., 2013, 57, 1979–1987 CAS.
- A. Cuadrado, Z. Martin-Moldes, J. Ye and I. Lastres-Becker, J. Biol. Chem., 2014, 289, 15244–15258 CrossRef CAS PubMed.
- D. Fratantonio, A. Speciale, D. Ferrari, M. Cristani, A. Saija and F. Cimino, Toxicol. Lett., 2015, 239, 152–160 CrossRef CAS PubMed.
- C. Dai, B. Li, Y. Zhou, D. Li, S. Zhang, H. Li, X. Xiao and S. Tang, Food Chem. Toxicol., 2016, 95, 52–63 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |