
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA DFT study on the mechanism of the sulfonic acid + alcohol esterification reaction†
Luis Salvatella
Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, Pedro Cerbuna 12, E-50009 Zaragoza, Spain. E-mail: lsalvate@unizar.es
First published on 22nd January 2018
Abstract
Four alternative mechanisms for the benzenesulfonic acid + methanol esterification reaction have been studied at the B3LYP/aug-cc-pVTZ level. The participation of a pentacoordinate sulfur intermediate (in either neutral or protonated form) can be disregarded according to energy considerations. Instead, results show a low activation barrier for the SN1 pathway (through a sulfonylium cation intermediate) and a moderate barrier for the SN2 path (involving protonated methanol as an alkylating reagent).
Introduction
Sulfonate esters constitute a family of powerful alkylating reagents.1 Although such reactants have been typically prepared from sulfonyl chlorides and alcohols, increasing interest has been paid to the reaction of sulfonic acids with alcohol precursors (orthoesters,2 diethyl carbonate,3 and dialkyl acylfosfonates)4 in anhydrous conditions. In fact, sulfonate esters are a matter of concern in the pharmaceutical industry because of their undesired formation from sulfonate salts when alcohols are used as crystallization solvents.5 Recently, our research group turned its attention to this process because of the unexpected esterification of sulfonated hydrothermal carbon by methanol.6 As a result of our interest in such a process, the first theoretical study on the sulfonic acid + alcohol esterification reaction is presented here.Several alternative mechanisms can be envisaged for such an esterification process (shown in Scheme 1 for the benzenesulfonic acid + methanol reaction).7,8 Thus, two addition–elimination (Ad–E) pathways taking place through pentacoordinate sulfur intermediates7 in either neutral or protonated forms can be firstly regarded. As an alternative, an SN1 reaction through a sulfonylium cation intermediate (identical to those formed from sulfonyl chlorides in the presence of Lewis acids)9 can take place. Finally, an SN2 reaction involving methyl transfer from protonated methanol to a sulfonate anion (as proposed for the methyl methanesulfonate + methanol reaction)5 is considered here.
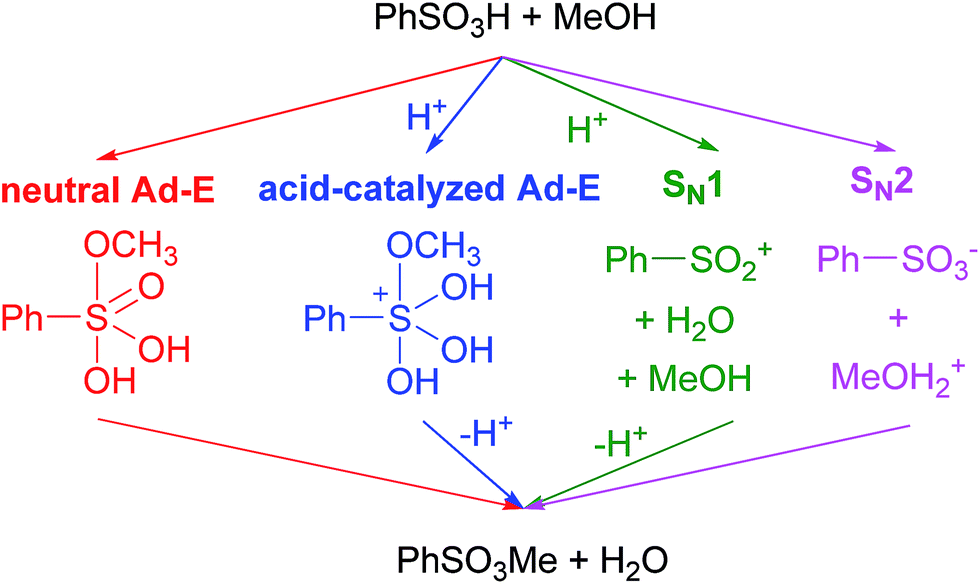 | ||
| Scheme 1 Alternative mechanisms. | ||
The mechanism of the benzenesulfonic + methanol reaction was studied as a model for the sulfonic acid + alcohol esterification. All aforementioned pathways have been regarded, though benzenesulfonate anion has been replaced by water in the SN2 pathway. The possible assistance of methanol molecules has been discussed in the ESI.†
Computational methods
B3LYP density functional was chosen because of its good performance on related systems (sulfonic esters,10 benzenesulfonic acid,11 protonated phosphoric acid,12 phosphonium ions).13 The aug-cc-pVTZ basis set was used because of its good description on the hydration reaction of metaphosphoric acid14 and the sulfuric acid–methanol–water system.15 The resulting B3LYP/aug-cc-pVTZ method has provided good results on geometries and energetics of the hydrogen-bonded HCl–HNO3–H2SO4 cluster.16Calculations were carried out by means of the Gaussian 09 software package.17 All stationary points were characterized by the correct number of imaginary frequencies (0 for minima, 1 for transition states, TS's). Basis set superposition error (BSSE) corrections were not considered in this work since those effects are typically negligible in studies using large basis sets (such as aug-cc-pVTZ).16 Free energies at 25 °C in gas phase and methanol solution (by means of the IEF-PCM continuum model) for the most stable conformation of every structure were used for the discussion of the results throughout the paper. Relative Gibbs free energies for the calculated structures in gas phase (in parenthesis) and methanol solution (in square brackets), shown in figures, have been calculated by comparison with stabilities of the non-interacting starting reactants for every mechanistic pathway. Figures were generated by using CYLView.18
Results and discussion
Neutral Ad–E pathway
Results on the neutral Ad–E path are shown in Fig. 1. Benzenesulfonic acid (1) shows a nearly orthogonal S–O bond/phenyl group arrangement as well as an O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S–O–H sp conformation (as computed for some substituted benzenesulfonic acids).19 The possible hydrogen bond donation from methanol (2) was considered, though the resulting complex (3) is disfavored in Gibbs free energy terms.
S–O–H sp conformation (as computed for some substituted benzenesulfonic acids).19 The possible hydrogen bond donation from methanol (2) was considered, though the resulting complex (3) is disfavored in Gibbs free energy terms.
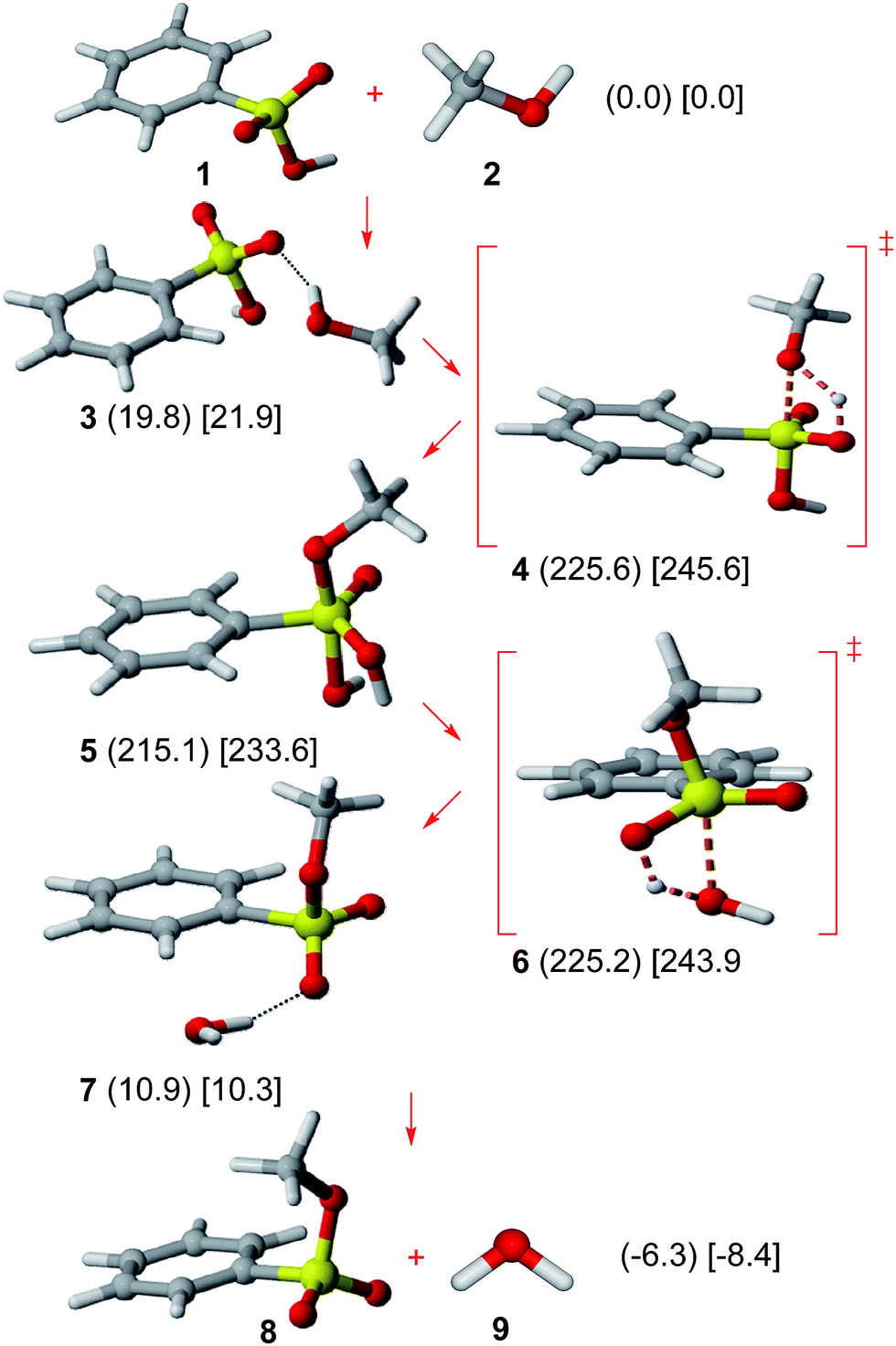 | ||
| Fig. 1 Neutral Ad–E mechanism. Relative Gibbs free energies (kJ mol−1) in gas phase (in parenthesis) and solution (in square brackets) of all involved structures are shown. | ||
A very high energy barrier (gas phase: 225.6 kJ mol−1; solution: 245.6 kJ mol−1) has been calculated in this work for the methanol addition (TS 4), similarly to the very high activation energy for the methanesulfonyl chloride + methanol addition previously reported (263.6 kJ mol−1 at HF/6-31G* level in gas phase).20
Methyl dihydrogen benzeneorthosulfonate (5), bearing a pentacoordinate sulfur atom, is thus formed. Such a species can be regarded as an analogue to tris(p-tolyl) tolueneorthosulfonate, which has been detected by 1H-NMR.21 Although all possible pseudorotamers of 5 were studied, the structure bearing both phenyl and oxo groups in equatorial positions is clearly favored by at least 16.4 kJ mol−1 (see the ESI†). The equatorial preference of the oxo group in that sulfur compound is analogous to the well-known low apicophilicity of π-donor substituents in pentacoordinate phosphorus derivatives.22 Nevertheless, even for the most stable pseudorotamer, the pentacoordinate sulfur species 5 shows a very high energy (gas phase: 215.1 kJ mol−1; solution: 233.6 kJ mol−1).
The subsequent water elimination step can readily proceed through TS 6 (activation barrier: 10.1 kJ mol−1 in gas phase; 10.2 kJ mol−1 in solution) to yield a hydrogen-bonded methyl benzenesulfonate + methanol complex (7). Such a complex can dissociate to yield methyl benzenesulfonate (8) (in OS–O–C sp conformation, consistently with X-ray diffraction data)23 and water (9) as the final products.
The energy profile for the neutral Ad–E pathway shows the occurrence of a reaction intermediate (5) presenting a prohibitive energy, which precludes the feasibility of such a mechanism. This outcome contrasts with the major role of anionic pentacoordinate sulfur intermediates in alkaline conditions for some related reactions (intramolecular sulfuryl transfers,24 hydrolysis of aryl sulfonates).7,25 The unstability of 5 can be attributed to the protonation of an equatorial oxygen.
The overall process is slightly favored in both gas phase (−6.3 kJ mol−1) and solution (−8.5 kJ mol−1) in Gibbs free energy terms. However, thermodynamic data from equilibria involving methyl methanesulfonate reactions in solution (basic hydrolysis26 and pKa),27 allows inferring a +23.4 kJ mol−1 Gibbs free energy for the corresponding esterification reaction (hence, a disfavored process). No significant solvent effects are observed for the Ad–E mechanism according to the close values for relative Gibbs free energies for every structure in gas phase and solution.
Acid-catalyzed Ad–E pathway
Results on the acid-catalyzed Ad–E mechanism are shown in Fig. 2. The interaction between benzenesulfonic acid (1) and protonated methanol cation (10) leads to the formation of a hydrogen-bonded complex (11), which is favored in gas phase (−119.3 kJ mol−1) and solution (−5.2 kJ mol−1). A huge activation barrier in the methanol addition step (through TS 12) from 11 (gas phase, 256.9 kJ mol−1; solution, 260.1 kJ mol−1) is found. The resulting protonated methyl dihydrogen benzeneorthosulfonate cation (13) is rather unstable (100.6 kJ mol−1 in gas phase; 216.5 kJ mol−1 in solution).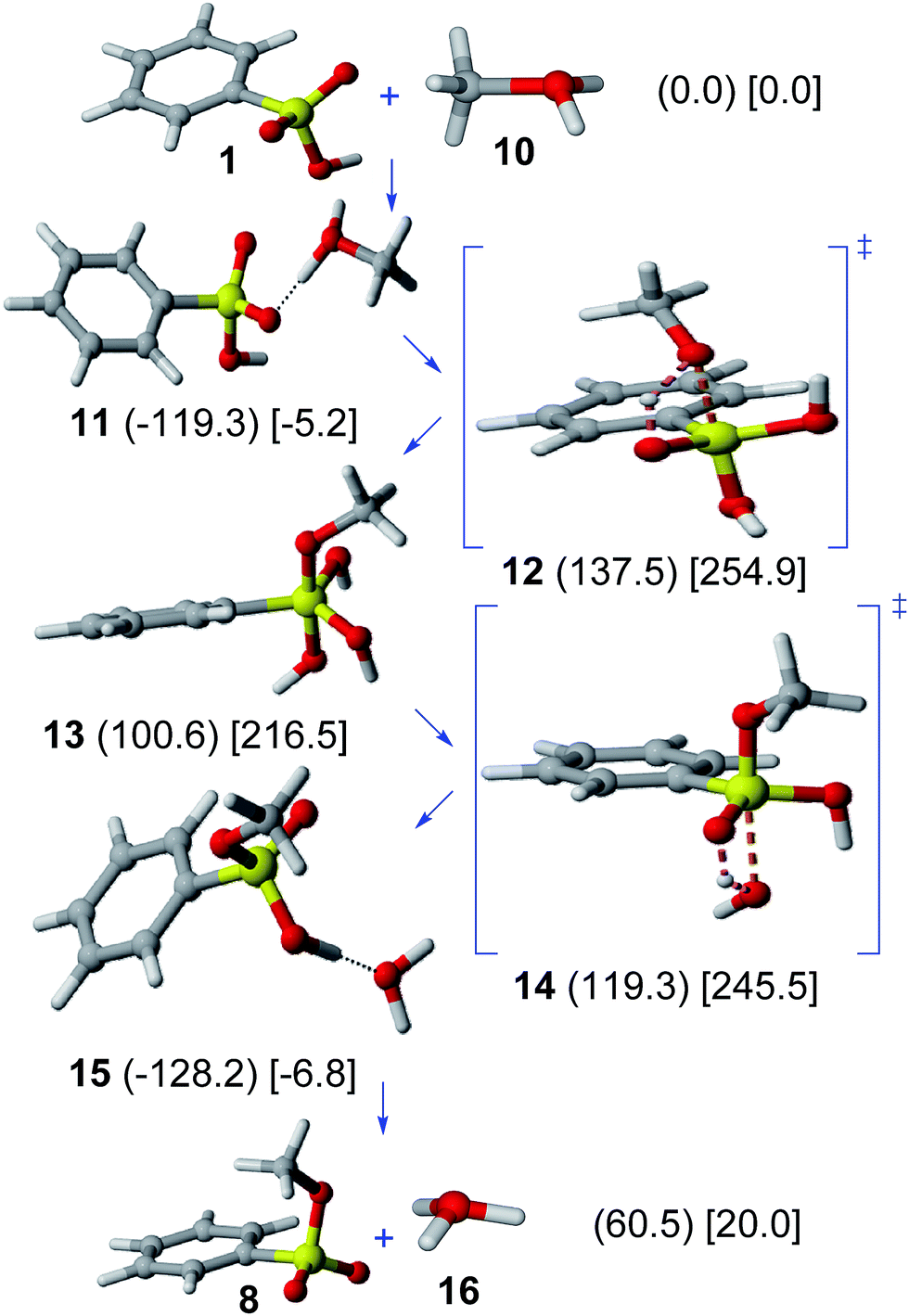 | ||
| Fig. 2 Acid-catalyzed Ad–E mechanism. Relative Gibbs free energies (kJ mol−1) in gas phase (in parenthesis) and solution (in square brackets) of involved structures are shown. | ||
The subsequent elimination step (through TS 14) leads to the formation of the hydrogen-bonded complex 15, which can finally dissociate to yield methyl benzenesulfonate (8) and oxonium cation (16).
The acid-catalyzed Ad–E mechanism can thus be discarded because of the prohibitive activation barrier involved in the addition step, hence refuting the previously assumed path for the esterification of sulfonated hydrothermal carbon.6 The lack of a protonated pentacoordinate sulfur intermediate in this reaction is consistent with the lack of sulfonyl oxygen exchange for phenyl benzenesulfonate in 10 M HCl.28
The possible methanol assistance in both neutral and acid-catalyzed Ad–E mechanisms (detailed in the ESI†) cannot avoid the high energy of the pentacoordinate sulfur intermediate (in either neutral or protonated forms), precluding thus the feasibility of such pathways. Such a conclusion contrasts with experimental studies on acid-catalyzed reactions of sulfinic acid derivatives (transesterification of sulfinates,29 hydrolysis of sulfinamides)30 showing the participation of hypervalent sulfurane intermediates.
SN1 pathway
Results on the SN1 mechanism are shown in Fig. 3. Thus, protonation of 1 yields oxo-protonated benzenesulfonic acid cation 17, consistently with NMR31 and X-ray diffraction32 data of other protonated sulfonic acid cations.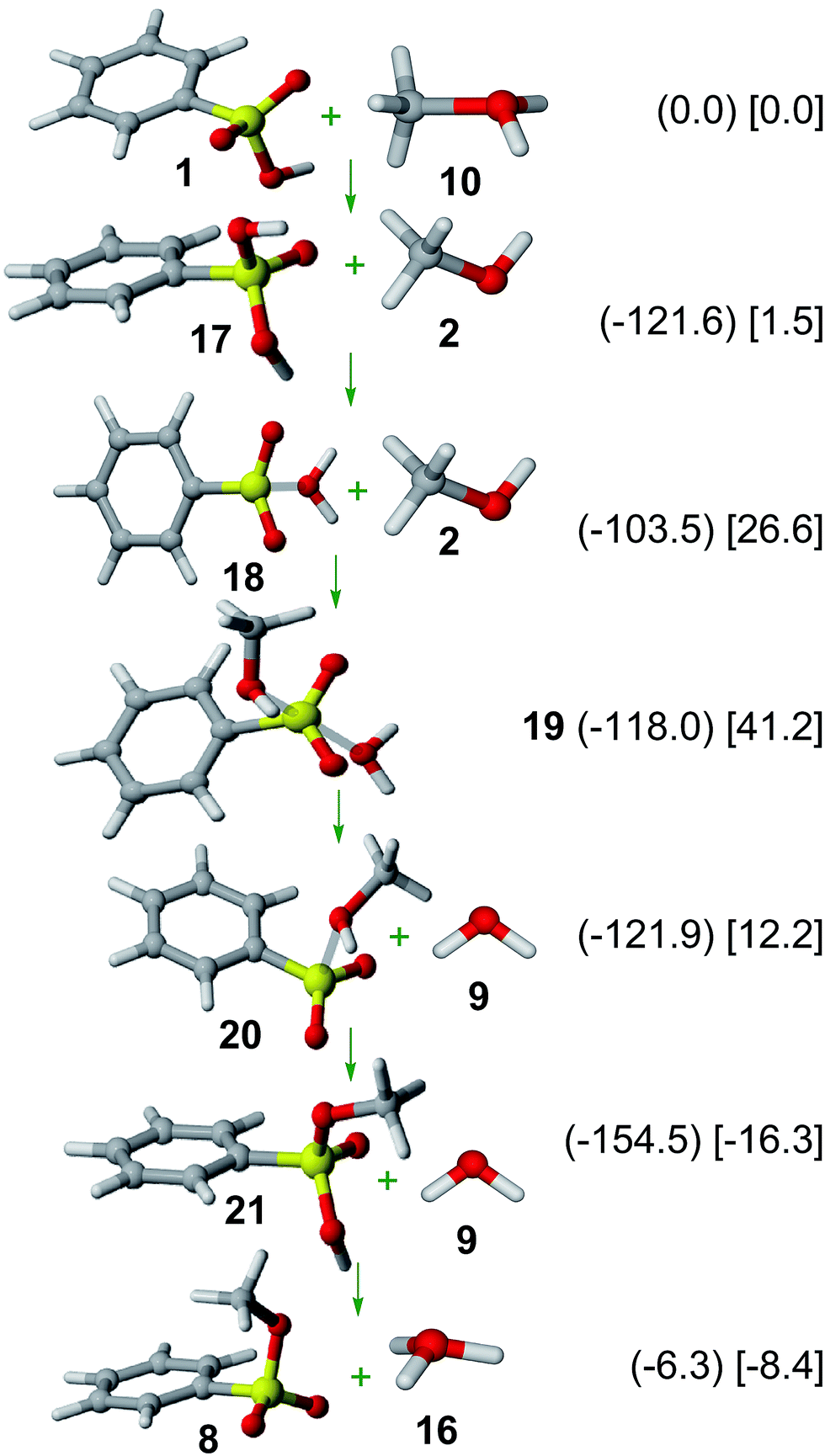 | ||
| Fig. 3 SN1 mechanism. Relative Gibbs free energies (kJ mol−1) in gas phase (in parenthesis) and solution (in square brackets) of involved structures are shown. | ||
Tautomerization of 17 yields OH-protonated benzenesulfonic acid cation (18), which is less stable (by 18.1 kJ mol−1 in gas phase; by 25.1 kJ mol−1 in solution). Interestingly, the almost planar sulfur arrangement and the long S⋯O distance (2.256 Å in gas phase; 2.082 Å in solution) indicates that 18 should be rather described as a weakly-bound water complex of benzenesulfonylium ion (interaction: −23.7 kJ mol−1 in gas phase; +3.0 kJ mol−1 in solution). The formation of such a sulfonylium cation in the sulfonic acid esterification is thus analogous to the mechanism of Friedel–Crafts sulfonylation reactions.8,33
Exchange of the water moiety in 18 by a methanol molecule through the energy minimum 19 yields methanol-coordinated benzenesulfonylium cation (20). Subsequent tautomerization yields oxo-protonated methyl benzenesulfonate cation (21), which can deprotonate to yield methyl benzenesulfonate (8) as the final product.
An inspection of the SN1 reaction mechanism shows a very low activation barrier by starting from 17 (18.1 kJ mol−1 in gas phase, 25.1 kJ mol−1 in solution), thus indicating that sulfonic esterification can easily proceed through a sulfonylium cation, analogously to acid-catalyzed hydrolysis of β-sultams.34
The formation of a sulfonylium cation as a key intermediate can thus explain the esterification of sulfonated hydrothermal carbon in the presence of methanol6 as well as the Friedel–Crafts sulfonylation reactions between sulfonic acids and arenes by using Nafion-H as a catalyst.35
SN2 pathway
An SN2 mechanism has been proposed for the esterification of methanesulfonic acid with methanol (involving methyl transfer from methyloxonium cation to the sulfonate anion).6 In order to avoid large solvation effects, benzenesulfonate anion was replaced by a water molecule. No significant variations on the energy profile should be expected36 for this replacement since both species show an identical attacking atom (oxygen) and close basicities (pKa values: benzenesulfonic acid, −2.8;27 H3O+, −1.74).37Results on the SN2 mechanism (Fig. 4) show that the pre-reactive methyloxonium cation–water complex38 (22) undergoes an SN2 reaction through TS 23 with a moderate gas phase activation barrier (39.1 kJ mol−1 in gas phase, 62.9 kJ mol−1 in solution). The affordable gas phase barrier is consistent with experimental data showing the fast oxygen exchange of methanol in acid solutions.39 Since the benzenesulfonate anion is a somewhat poorer nucleophile than water value, a moderate activation barrier can be estimated for the benzenesulfonate anion + methyloxonium cation reaction.
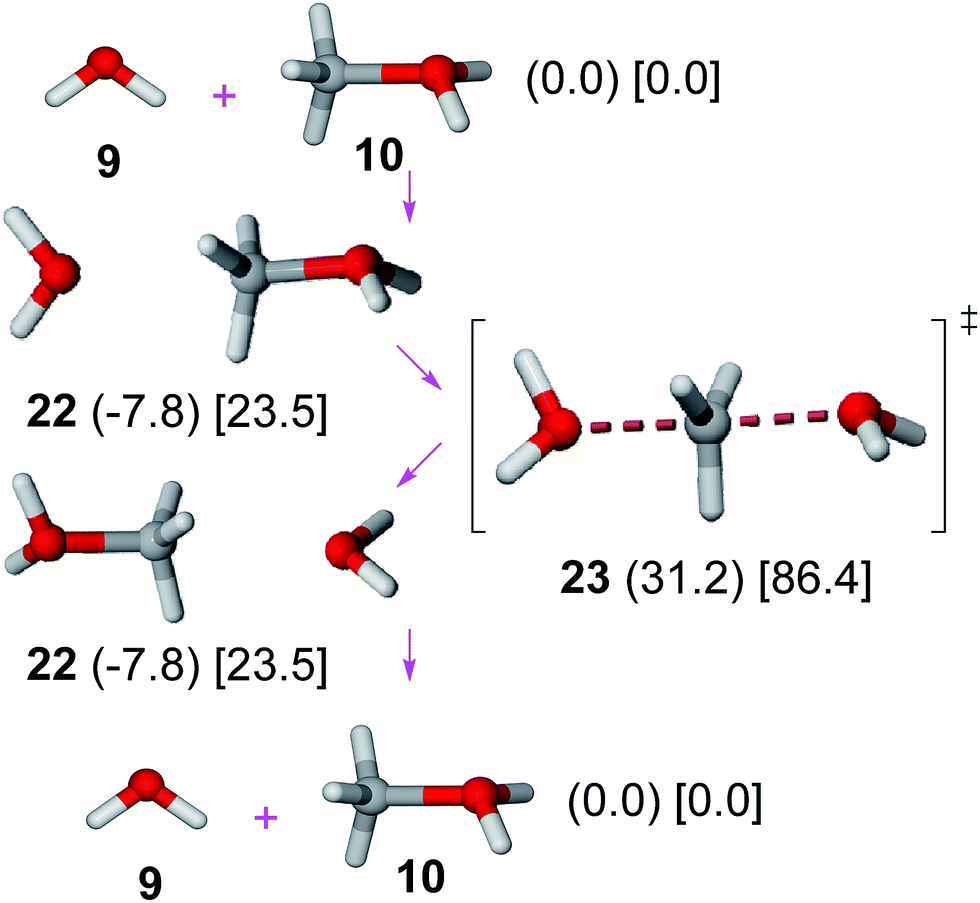 | ||
| Fig. 4 SN2 mechanism. Relative Gibbs free energies (kJ mol−1) in gas phase (in parenthesis) and solution (in square brackets) of involved structures are shown. | ||
Conclusions
Both neutral and acid-catalyzed Ad–E mechanisms can be ruled out on the basis of the high energy of the corresponding five-coordinate sulfur intermediates. As an alternative, a readily accessible path is found for the SN1 pathway (through a sulfonylium cation). The SN2 path (involving methyloxonium cation as an alkylating reagent) might also play a major role in the sulfonic acid esterification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I am indebted to the Instituto de Síntesis Química y Catálisis Homogénea (ISQCH) and the Instituto de Biocomputación y Física de Sistemas Complejos (BIFI) for allocation of computer time, as well the Ministerio de Economía y Competitividad (MINECO) of Spain (Project CTQ2014-52367-R), the Gobierno de Aragón (Consolidated Group E11) and the European Regional Development Fund (ERDF) for financial support.Notes and references
- J. Hoyle, in Sulphonic Acids, Esters and their Derivatives, ed. S. Patai and Z. Rappoport, Wiley, Chichester, UK, 1991, ch. 19, pp. 351–399 Search PubMed.
- (a) A. A. Padmapriya, G. Just and N. G. Lewis, Synth. Commun., 1985, 15, 1057–1062 CrossRef CAS; (b) J. I. Trujillo and A. S. Gopalan, Tetrahedron Lett., 1993, 34, 7355–7358 CrossRef CAS.
- Y. Nitta and Y. Arakawa, Chem. Pharm. Bull., 1985, 33, 1380–1386 CrossRef CAS.
- P. Golborn, Synth. Commun., 1973, 3, 273–275 CrossRef CAS.
- (a) A. Teasdale, S. C. Eyley, E. Delaney, K. Jacq, K. Taylor-Worth, A. Lipczynski, V. Reif, D. P. Elder, K. L. Facchine, S. Golec, R. S. Oestrich, P. Sandra and F. David, Org. Process Res. Dev., 2009, 13, 429–433 CrossRef CAS; (b) A. Teasdale, E. J. Delaney, S. C. Eyley, K. Jacq, K. Taylor-Worth, A. Lipczynski, W. Hoffmann, V. Reif, D. P. Elder, K. L. Facchine, S. Golec, R. S. Oestrich, P. Sandra and F. David, Org. Process Res. Dev., 2010, 14, 999–1007 CrossRef CAS.
- J. M. Fraile, E. García-Bordejé, E. Pires and L. Roldán, J. Catal., 2015, 324, 107–118 CrossRef CAS.
- J. P. Guthrie, in Reviews of Reactive Intermediate Chemistry, ed. M. S. Platz, M. Jones Jr and R. A. Moss, Wiley-Interscience, Hoboken NJ, 2007, ch. 1, pp. 3–46 Search PubMed.
- J. F. King, S. Skonieczny, K. C. Khemani and J. D. Lock, in Nucleophilicity, ed. J. M. Harris and S. P. McManus, American Chemical Society, Washington DC, 1987, ch. 9, pp. 137–153 Search PubMed.
- (a) G. A. Olah, S. Kobayashi and J. J. Nishimura, J. Am. Chem. Soc., 1973, 95, 564–569 CrossRef CAS; (b) E. Lindner and H. Weber, Chem. Ber., 1968, 101, 2832–2836 CrossRef CAS.
- O. Exner and S. Böhm, New J. Chem., 2008, 32, 638–642 RSC.
- S. N. Ivanov, N. I. Giricheva, T. V. Nurkevich and M. S. Fedorov, Russ. J. Phys. Chem. A, 2014, 88, 661–666 CrossRef CAS.
- S. Gevrey, A. Luna, V. Haldys, J. Tortajada and J.-P. Morizur, J. Chem. Phys., 1998, 108, 2458–2465 CrossRef CAS.
- S. Gevrey, A. Luna, M.-H. Taphanel, J. Tortajada and J.-P. Morizur, Int. J. Mass Spectrom., 2000, 195–196, 545–563 CrossRef.
- I. Alkorta, L. M. Azofra and J. Elguero, Theor. Chem. Acc., 2015, 134, 1–8 CrossRef CAS.
- M. Rozenberg, A. Loewenschuss and C. J. Nielsen, J. Phys. Chem. A, 2015, 119, 2271–2280 CrossRef CAS PubMed.
- M. Verdes and M. Paniagua, J. Mol. Model., 2014, 20, 2232 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, I. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- N. I. Giricheva, G. V. Girichev, M. S. Fedorov and S. N. Ivanov, Struct. Chem., 2013, 24, 807–818 CrossRef CAS.
- K. Yang, K. Kang, I. S. Koo and I. Lee, Bull. Korean Chem. Soc., 1997, 18, 1186–1191 CAS.
- G. R. Chalkley, D. J. Snodin, G. Stevens and M. C. Whiting, J. Chem. Soc., Perkin Trans. 1, 1978, 1580–1587 RSC.
- R. Hoffmann, J. M. Howell and E. L. Muetterties, J. Am. Chem. Soc., 1972, 94, 3047–3058 CrossRef CAS.
- R. Chênevert, R. Gagnon, D. Chamberland and M. Simard, Can. J. Chem., 1993, 71, 1225–1235 CrossRef.
- (a) D. R. Cameron and G. R. J. Thatcher, J. Org. Chem., 1996, 61, 5986–5997 CrossRef CAS; (b) J. F. King, M. S. Gill and D. F. Klassen, Pure Appl. Chem., 1996, 68, 825–830 CrossRef CAS; (c) S. G. Jarboe, M. S. Terrazas and P. Beak, J. Org. Chem., 2008, 73, 9627–9632 CrossRef CAS PubMed.
- C. A. Bunton and Y. F. Frei, J. Chem. Soc., 1951, 1872–1873 RSC.
- J. P. Guthrie and R. T. Gallant, Can. J. Chem., 2000, 78, 1295–1298 CrossRef CAS.
- J. P. Guthrie, Can. J. Chem., 1978, 56, 2342–2354 CrossRef CAS.
- S. Oae, T. Fukumoto and R. Kiritani, Bull. Chem. Soc. Jpn., 1963, 36, 346–348 CrossRef CAS.
- M. Mikołajczyk, J. Drabowicz and H. Ślebocka-Tilk, J. Am. Chem. Soc., 1979, 101, 1302–1303 CrossRef.
- (a) T. Okuyama, J. P. Lee and K. Ohnishi, J. Am. Chem. Soc., 1994, 116, 6480–6481 CrossRef CAS; (b) J.-P. Lee, H.-E. Yu, D.-I. Jung and I.-S. Koo, Bull. Korean Chem. Soc., 2010, 31, 1773–1775 CrossRef CAS.
- A. Koeberg-Telder and H. Cerfontain, J. Chem. Soc., Perkin Trans. 2, 1975, 226–229 RSC.
- T. Soltner, N. R. Goetz and A. Kornath, Eur. J. Inorg. Chem., 2011, 3076–3081 CrossRef CAS.
- (a) Y. Yoshii, A. Ito, T. Hirashima, S. Shinkai and O. Manabe, J. Chem. Soc., Perkin Trans. 2, 1988, 777–781 RSC; (b) F. R. Jensen and H. C. Brown, J. Am. Chem. Soc., 1958, 80, 4042–4045 CrossRef CAS.
- (a) N. J. Baxter, A. P. Laws, L. Rigoreau and M. I. Page, J. Chem. Soc., Perkin Trans. 2, 1996, 2245–2246 RSC; (b) N. J. Baxter, A. P. Laws, L. J. H. Rigoreau and M. I. Page, Chem. Commun., 1999, 2401–2402 RSC.
- G. A. Olah, T. Mathew and G. K. S. Prakash, Chem. Commun., 2001, 1696–1697 RSC.
- F. G. Bordwell, T. A. Cripe and D. L. Hughes, in Nucleophilicity, ed. J. M. Harris and S. P. McManus, American Chemical Society, Washington DC, 1987, ch. 9, pp. 137–153 Search PubMed.
- M. L. Campbell and B. A. Waite, J. Chem. Educ., 1990, 67, 386–388 CrossRef CAS.
- J. K. Laerdahl and E. Uggerud, Org. Biomol. Chem., 2003, 1, 2935–2942 CAS.
- R. A. McClelland, Can. J. Chem., 1980, 58, 393–398 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Discussions on the relative stability of 5 and related pseudorotamers, the role of methanol assistance on neutral and acid-catalyzed Ad–E pathways, and energies and Cartesian coordinates of all structures (PDF). See DOI: 10.1039/c7ra09833b |
| This journal is © The Royal Society of Chemistry 2018 |