
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activation of peroxymonosulfate by metal (Fe, Mn, Cu and Ni) doping ordered mesoporous Co3O4 for the degradation of enrofloxacin
Jing Deng,
Chen Ya,
Yongjian Ge,
Yongqing Cheng,
Yijing Chen,
Mengyuan Xu and
Hongyu Wang *
*
College of Civil Engineering and Architecture, Zhejiang University of Technology, 18 Chaowang Road, Xiacheng District, Hangzhou 310014, China. E-mail: hywangzjut@163.com; Fax: +86 571 88320180; Tel: +86 571 88320180
First published on 9th January 2018
Abstract
Various transition metals (Fe, Mn, Cu and Ni) were doped into ordered mesoporous Co3O4 to synthesize Co3O4-composite spinels. Their formation was evidenced by transmission electronic microscopy (TEM), X-ray diffraction (XRD) and Brunauer–Emmett–Teller (BET) analysis. It was found that Co3O4-composite spinels could efficiently activate peroxymonosulfate (PMS) to remove enrofloxacin (ENR) and the catalytic activity followed the order Co3O4–CuCo2O4 > Co3O4–CoMn2O4 > Co3O4–CoFe2O4 > Co3O4–NiCo2O4. Moreover, through the calculation of the specific apparent rate constant (ksapp), it can be proved that the Co and Cu ions had the best synergistic effect for PMS activation. The Co3O4-composite spinels presented a wide pH range for the activation of PMS, but strong acidic and alkaline conditions were detrimental to ENR removal. Higher reaction temperature could promote the PMS activation process. Sulfate radical was identified as the dominating reactive species in Co3O4-composite spinel/PMS systems through radical quenching experiments. Meanwhile, the probable mechanisms concerning Co3O4-composite spinel activated PMS were proposed.
1. Introduction
During the past years, sulfate radical (SO4−˙) based advanced oxidation processes (SR-AOPs) have attracted an increasing interest among researchers owing to their great potential in degradation or even mineralization of recalcitrant organic pollutants.1 Compared with the hydroxyl radical (˙OH), SO4−˙ possesses a longer lifespan, higher independence of pH and higher selectivity of oxidation.2,3 Peroxymonosulfate (PMS), a precursor of SO4−˙, is deemed as a cost-effective and environmental-friendly oxidant.4 PMS remains stable in aqueous solution and barely decomposes into SO4−˙ by itself, but it can be activated to produce SO4−˙ by the use of UV, transition metals, and some nonmetal catalysts.5–7 Among different activation technologies, transition metals have attracted much attention due to their lower energy consumption and higher activation efficiency. Actually, many transition metal ions such as Co2+, Mn2+, Ni2+, Fe2+, Ru3+, Ce3+ and so forth, have been proved as qualified catalysts for PMS activation.2 Of note, Co2+ has been found to possess the highest reactivity.8 Unfortunately, the Co2+/PMS process is unfavorable in practical application because of the toxicity of Co2+.In order to relieve the secondary pollution, heterogeneous cobalt-based catalysts have become a research hotspot. Anipsitakis et al. firstly employed Co3O4 to activate PMS and found Co3O4 presented an excellent catalytic behavior in the activation of PMS.9 Chen et al. successfully prepared nanoscale Co3O4 and tested its catalytic performance in PMS solution, results showed that 0.2 mM acid orange 7 (AO7) can be completely degraded within 30 min by 2 mM PMS in the presence of 0.5 g L−1 Co3O4.10 Pu et al. fabricated three types of Co3O4 using different metal organic frameworks, and found that all the Co3O4 exhibited outstanding catalytic activity and the difference in catalytic ability can be attributed to the difference in specific surface area.11 Consequently, Co3O4/PMS system is quite acceptable from the view of application due to the high activation efficiency and limitation of cobalt leaching. However, on the basis of the underlying threat of cobalt ions, it is essential to take measures to further limit the cobalt leakage during PMS activation.
It is reported that bimetallic oxides may be desirable catalysts to ease the conflict between catalytic performance and metal ions leaching, because intimate interactions between two metals can effectively suppress the leakage of metal ions, such as Fe–Co interactions in CoFe2O4.2 Moreover, bimetallic oxides are also prominent PMS activators. Su et al. synthesized a series of CoxFe3−xO4 nanoparticles and found that the higher cobalt content in CoxFe3−xO4 showed the higher catalytic activity towards PMS.12 The high catalytic behavior of CoFe2O4 was also illustrated in our previous study.13 Yao et al. reported that CoMn2O4 showed stronger catalytic activity than Co3O4, Mn2O3 and their physical mixture due to the synergistic effects of Co and Mn species.14 Similarly, CuCo2O4 also exhibited high catalytic performance and low metal leachability in PMS solution.15 In our previous research, order mesoporous Co3O4 (OM-Co3O4) was fabricated and showed superior catalytic ability toward PMS than its spinel counterpart, but the leakage of cobalt was up to 77.74 μg L−1 which was higher than conventional Co3O4 nanoparticles.16 Therefore, it can be reasonably speculated that cobalt leaching will reduce if some transition metals are doped to OM-Co3O4 to form mixed spinels with cobalt.
Herein, diverse transition metals (i.e., Fe, Mn, Cu and Ni) was introduced into OM-Co3O4 to synthesized a series of Co3O4-composite spinels which were characterized by transmission electronic microscope (TEM), X-ray diffraction (XRD), Brunauer–Emmett–Teller (BET) and Zeta potential analysis. Due to the ubiquitous detection in aquatic environment,17 enrofloxacin (ENR) was selected as target pollutant in this study. The catalytic activities of as-prepared Co3O4-composite spinels were systematically compared through apparent rate constant, PMS consumption, intensity of electron paramagnetic resonance (EPR) signal and specific apparent rate constant. Moreover, the effects of initial pH and reaction temperature during PMS activation were also investigated. Finally, a possible mechanism of PMS activation was proposed through quenching tests. To be best of our knowledge, it is the first time to apply order mesoporous Co3O4-composite spinels as effective PMS activators for the control of organic pollutants.
2. Materials and methods
2.1 Chemicals
ENR, acetonitrile (HPLC grade), Oxone (KHSO5·0.5KHSO4·0.5K2SO4, PMS, KHSO5 ≥ 47%) and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) were obtained from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). The silica templates KIT-6 were purchased from Nanjing XFNANO Materials Tech Co., Ltd. (Jiangsu, China). Other chemical reagents were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Deionized water (18 MΩ cm) was produced from an Ulupure water purification system (Shanghai, China).2.2 Catalysts preparation and characterization
OM-Co3O4 was synthesized using nanocasting route with KIT-6 as hard template, and the procedure was conducted as described before.16 For the preparation of Co3O4-composite spinels, 1.0 g OM-Co3O4 was dispersed in 2.5 mL ethanol of Fe(NO3)3·9H2O, Mn(NO3)2·4H2O, Cu(NO3)2·3H2O and Ni(NO3)2·6H2O, respectively, and Co/M molar ratio was controlled at 3. After magnetic stirring for 1 h, the mixture was dried overnight at 60 °C and then calcined at 450 °C for 5 h (the heating rate was set at 2 °C min−1). Finally, the obtained composite spinels were referenced as Co3O4–CoM2O4 (M = Fe, Mn) and Co3O4–MCo2O4 (M = Cu, Ni) which depended on the oxidation state of dopant.The crystal structures of catalysts were characterized by X'Pert PRO diffractometer (PANalytical, Holland) with Cu Kα radiation. The morphologies and structures of Co3O4-composite spinels were observed using transmission electron microscopy (TEM) (Philips, Holland). N2 adsorption and desorption isotherms were measured using ASAP 2010 analyzer (Micromeritics, USA) at liquid nitrogen temperature (−196 °C). The pH at point of zero charge (pHpzc) was determined by Zetasizer Nano analyzer (Malvern, UK).
2.3 Catalytic experimental procedure
The catalytic degradation experiments were performed with a 100 mL ENR solution at 10 mg L−1 in 250 mL brown glass bottles, which were installed in a controlled temperature water bath stirring apparatus. In a typical run, specific amount of catalysts was added to ENR solution to receive adsorption–desorption equilibrium, followed by pH adjustment with H2SO4 and NaOH solution (100 mM) to ensure a desirable pH value after PMS addition. Subsequently, an appropriate amount of PMS was charged into the reaction solution to initiate experiment. At defined time intervals, 1 mL samples were collected and quenched by 0.1 mL Na2SO3 (100 mM). The resulting mixtures were immediately filtered by a 0.22 μm syringe filter for further analysis. All experiments were carried out in duplicates and the mean values were reported (with error bar).2.4 Analytical methods
ENR concentrations were measured through a high-performance liquid chromatograph (HPLC, Agilent 1200, USA) with an Eclipse XDB-C18 column (5 μm particle, 150 × 4.5 mm), the concentrations were measured at λ = 278 nm using a mobile phase consisting of a mixture of acetonitrile and phosphoric acid (pH = 2.5) (v/v = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80) at a flow rate of 1.0 mL min−1. The PMS concentrations were measured by the method of Waclawek et al.18 EPR analysis were performed on a Bruker A300 spectrometer (Germany) with DMPO as a spin-trapping agent. The parameters of EPR spectrometer were center field was 3360.67 G, sweep width was 100 G, static field was 3310.66 G, microwave frequency was 9.42 GHz, microwave power was 2.03 mW, modulation amplitude was 1.0 G and sweep time was 30.72 s.
:80) at a flow rate of 1.0 mL min−1. The PMS concentrations were measured by the method of Waclawek et al.18 EPR analysis were performed on a Bruker A300 spectrometer (Germany) with DMPO as a spin-trapping agent. The parameters of EPR spectrometer were center field was 3360.67 G, sweep width was 100 G, static field was 3310.66 G, microwave frequency was 9.42 GHz, microwave power was 2.03 mW, modulation amplitude was 1.0 G and sweep time was 30.72 s.
3. Results and discussion
3.1 Characterization
The crystalline phases of OM-Co3O4 and Co3O4-composite spinels were displayed in Fig. 1. It was worth noting that the precursors and OM-Co3O4 would result in the formation of composite spinels at high temperature. The lattice of Co3O4 would host other cations through the replacement of cobalt cations.19 However, no significant difference can be observed between OM-Co3O4 and Co3O4-composite spinels in Fig. 1. The well-defined diffraction peaks of 2θ = 19.00°, 31.27°, 36.85°, 38.54°, 44.81°, 55.66°, 59.36° and 65.24° were corresponded to (111), (220), (311), (222), (400), (422), (511) and (440), respectively. This might be ascribed to that the unit cell parameters of CoFe2O4, CoMn2O4, CuCo2O4 and NiCo2O4 were very close to that of Co3O4,19 thus these phases cannot be distinguished through XRD analysis. But according to Debye–Scherrer equation, the calculated mean crystallite sizes of OM-Co3O4, Co3O4–CoFe2O4, Co3O4–CoMn2O4, Co3O4–CuCo2O4 and Co3O4–NiCo2O4 were 17.43, 25.32, 22.87, 27.35 and 24.86 nm, respectively. Compared with OM-Co3O4, the increase of mean crystallite sizes in Co3O4-composite spinels indicated that the introduction of metal dopants destroyed original structure of OM-Co3O4.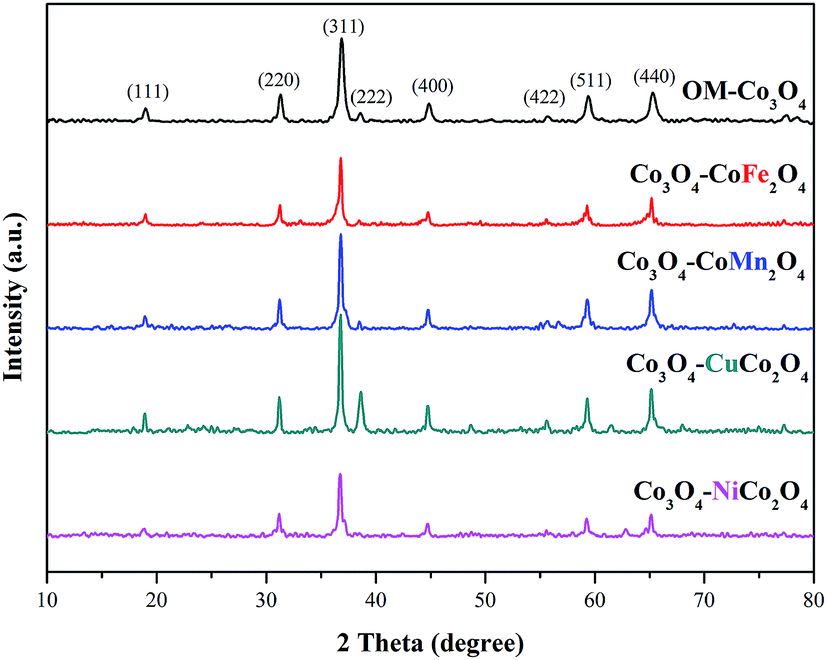 | ||
| Fig. 1 XRD patterns of OM-Co3O4 and Co3O4-composite spinels. | ||
The TEM and HR-TEM images of OM-Co3O4 and Co3O4-composite spinels were showed in Fig. 2. It can be clearly seen that OM-Co3O4 showed a highly ordered mesoporous structure, and the spacing distances between two fringes are 0.285 and 0.467 nm, which were in conformity with (220) and (111) planes, respectively. Obviously, after the introduction of metal dopants, ordered mesoporous structure was partly or completely destroyed, which may be attributed to the formation of Co3O4-composite spinels. The lattice fringes can be clearly observed in HR-TEM images, indicating that highly crystalline nature of Co3O4-composite spinels, which was corresponded to the strong and sharp diffraction peaks in XRD analysis. Similarly with OM-Co3O4, the spacing distances between two fringes in Co3O4–CoFe2O4 were 0.281 and 0.471 nm, corresponding to (220) and (111) planes, respectively. And that in Co3O4–CoMn2O4 were 0.279 and 0.469 nm, which were also assigned to (220) and (111) planes, respectively. However, as for Co3O4–CuCo2O4 and Co3O4–NiCo2O4, the (111) plane was not observed, and the lattice spacing of 0.275 and 0.286 nm was corresponded to (220) plane.
 | ||
| Fig. 2 TEM and HR-TEM images of OM-Co3O4 and Co3O4-composite spinels. (a)–(c) OM-Co3O4; (d)–(f) Co3O4–CoFe2O4; (g)–(i) Co3O4–CoMn2O4; (j)–(l) Co3O4–CuCo2O4; (m)–(o) Co3O4–NiCo2O4. | ||
The surface areas and pore size distributions of OM-Co3O4 and composite materials were investigated by N2 adsorption–desorption isotherms. As shown in Fig. 3(a), all materials showed type IV isotherms, and the specific surface areas of OM-Co3O4, Co3O4–CoFe2O4, Co3O4–CoMn2O4, Co3O4–CuCo2O4 and Co3O4–NiCo2O4 were 66.91, 52.34, 50.92, 30.39 and 29.53 m2 g−1, respectively. The reduction of specific surface areas in Co3O4-composite spinels was ascribed to the deterioration of ordered mesoporous structure after the impregnation of metal cations, which was in accordance with the observation of TEM images. Moreover, compared with Co3O4–CoFe2O4 and Co3O4–CoMn2O4, the more significant decrease of specific surface areas in Co3O4–CuCo2O4 and Co3O4–NiCo2O4 may be related to the oxidation state of the doped metal ions. The formation of Co3O4–MCo2O4 consumed more Co3O4 than Co3O4–CoM2O4,19 thus the deterioration of ordered mesoporous structure in Co3O4–MCo2O4 was more significant than that of Co3O4–CoM2O4, which can also be observed from TEM images. The changes of pore volume and pore diameter also demonstrated the conclusion, as seen in the Fig. 3(b), the pore volume and pore diameter all followed the order of OM-Co3O4 > Co3O4–CoM2O4 > Co3O4–MCo2O4. The textural parameters of OM-Co3O4 and Co3O4-composite spinels were summarized in Table 1.
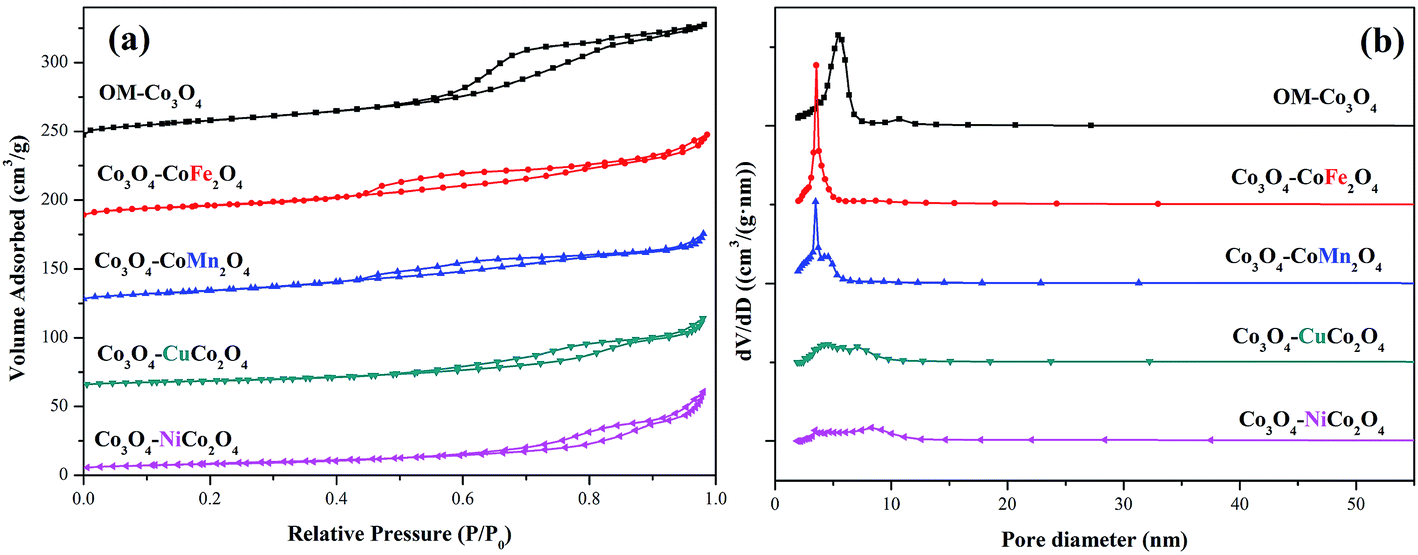 | ||
| Fig. 3 (a) Nitrogen adsorption–desorption isotherms of OM-Co3O4 and Co3O4-composite spinels and (b) their pore size distributions. | ||
| Samples | XRD | N2 adsorption–desorption | pHpzc | ||
|---|---|---|---|---|---|
| Crystallite size (nm) | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore diameter (nm) | ||
| OM-Co3O4 | 17.43 | 66.91 | 0.135 | 8.08 | 5.85 |
| Co3O4–CoFe2O4 | 25.32 | 52.34 | 0.105 | 7.19 | 4.21 |
| Co3O4–CoMn2O4 | 22.87 | 50.92 | 0.094 | 6.32 | 3.93 |
| Co3O4–CuCo2O4 | 27.35 | 30.39 | 0.083 | 5.91 | 4.76 |
| Co3O4–NiCo2O4 | 24.86 | 29.53 | 0.086 | 6.21 | 5.37 |
XPS analysis can be used to determine the surface composition and chemical oxidation states of OM-Co3O4 and Co3O4-composite spinels. In XPS spectra of OM-Co3O4 (Fig. 4(a)), the sharp peak emerged at 779.6 eV was assignable to Co 2p3/2, which could be deconvoluted into octahedral Co3+ at 779.4 eV and tetrahedral Co2+ at 780.7 eV.20 The proportions of Co2+ and Co3+ were determined to be 63.01% and 36.99%, respectively. The O 1s envelope (Fig. 4(b)) could be deconvoluted into two parts, namely the lattice oxygen (Olatt) at 529.2 eV and surface adsorbed oxygen (Oads) at 530.8 eV.16 Based on this deconvolution, the proportions of Olatt and Oads were found to be 50.86% and 49.14%, respectively. After the doping of iron into OM-Co3O4, the content of Co2+ increased from 63.01% to 65.34%, which could be ascribed to the substitution of Co3+ with Fe3+ in OM-Co3O4. It was worth noting that the content of Oads increased from 50.86% to 51.74%, which was conductive to the PMS activation.4 As seen in Fig. 4(c), the doped iron existed in the form of positive trivalent.
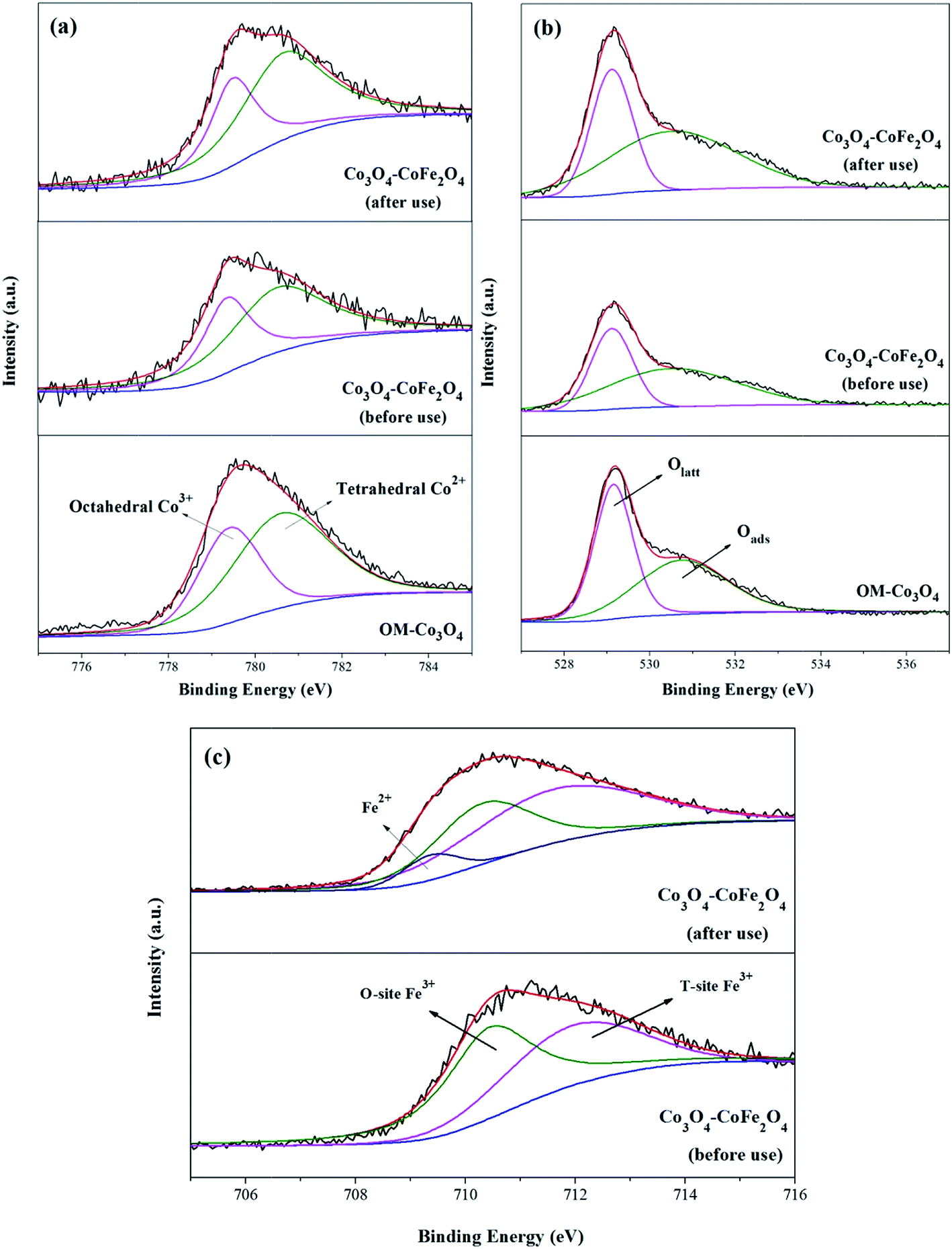 | ||
| Fig. 4 XPS survey spectrum of Co 2p3/2 (a), O 1s (b) and Fe 2p3/2 (c) for OM-Co3O4 and Co3O4–CoFe2O4. | ||
3.2 Catalytic activity of Co3O4-composite spinels
ENR removal in different systems was presented in Fig. 5(a). Adsorption tests showed that all the catalysts exerted a low efficiency in ENR adsorption, and the highest efficiency was received by Co3O4–CoFe2O4 with 3.22% of ENR adsorption within 30 min, which may be attributed to the largest specific surface area. Although PMS is a strong oxidizing agent with oxidation potential of 1.82 V,21 only 16.36% ENR could be removed by PMS in the absence of activator. However, the ENR degradation was greatly enhanced in the presence of both Co3O4-composite spinels and PMS. ENR can be completely removed in Co3O4–CuCo2O4/PMS and Co3O4–CoMn2O4/PMS systems, and the removal efficiencies were 96.37% and 94.56%, respectively. Furthermore, the ENR degradation well followed a pseudo-first-order kinetics pattern:
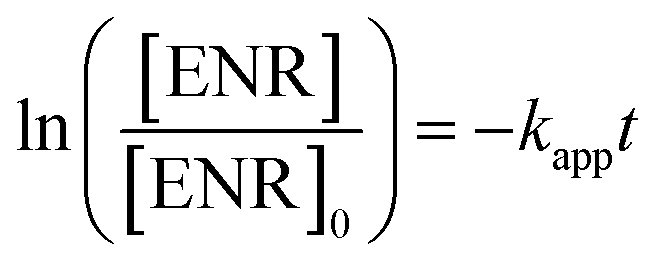 | (1) |
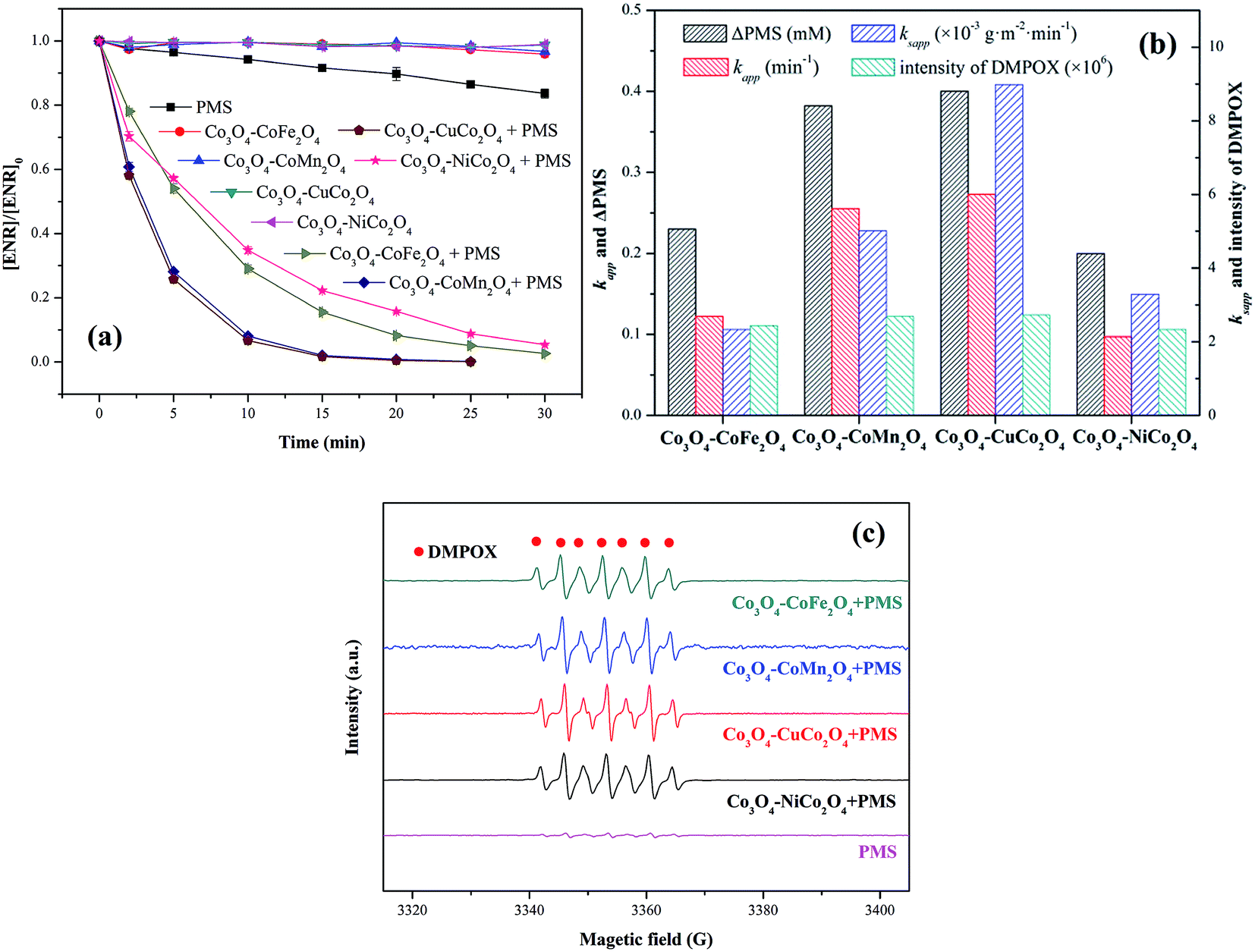 | ||
| Fig. 5 (a) ENR degradation in different systems; (b) values involved in different Co3O4-composite spinels/PMS systems; (c) EPR spectra in different composite spinels/PMS systems (DMPO = 25 mM). Experimental condition: [ENR] = 10 mg L−1, [catalyst] = 0.1 g L−1, [PMS] = 1 mM, pH0 = 6, T = 25 °C. | ||
EPR experiments were also performed for the comparison of the catalytic activity of Co3O4-composite spinels. As presented in EPR spectra (Fig. 5(c)), there was no distinctive EPR signal obtained by PMS alone. Nevertheless, simultaneous use of Co3O4-composite spinels and PMS could lead to obvious EPR signals. These EPR signals indicated the formation of 5,5-dimethyl-2-oxo-pyrroline-1-oxyl (DMPOX),22,23 which was ascribed to the fast activation of PMS and efficient oxidation of DMPO,24 also proofing the high catalytic performance of Co3O4-composite spinels. In addition, the signals of DMPOX caused by Co3O4–CuCo2O4/PMS and Co3O4–CoMn2O4/PMS systems were stronger than that caused by Co3O4–CoFe2O4 or Co3O4–NiCo2O4 activated PMS system (Fig. 5(b)), further authenticating the order of catalytic activity of Co3O4-composite spinels.
It was suggested that the catalytic performance of Co3O4-composite spinels not only depended on the specific surface area, but relied on dopant itself. In order to eliminate the difference in the specific surface area, the specific apparent rate constant ksapp which defined as the ratio of kapp to the BET surface area was introduced:
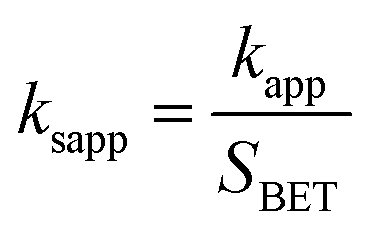 | (2) |
3.3 Effect of initial pH
The influence of initial pH ranging from 3 to 11 on ENR degradation was investigated in the PMS activation process, and the results were displayed in Fig. 6. From Fig. 6(a)–(e), it can be seen that four Co3O4-composite spinels all showed a wide pH range for PMS oxidation and higher ENR removals were obtained in pH range of 5 to 9 while lower removals occurred at strong acidic and alkaline conditions. Similar results were also reported by the previous investigations, such as degradation of orange II in MnFe2O4/PMS process and removal of acetaminophen in Fe3O4/PMS system.25,26 The ENR degradation was significantly inhibited at strong acidic condition might be originated from the attachment of H+ to the peroxide bond (O–O) of PMS (eqn (3)) and the change of catalyst surface charge (eqn (4)), so that the interfacial repulsion would result in a weaker catalytic performance.27 The retardation of ENR removal at strong alkaline condition can be ascribed to the following reasons: (1) the increase of catalyst surface negative charges. The pHpzc of Co3O4–CoFe2O4, Co3O4–CoMn2O4, Co3O4–CoCo2O4 and Co3O4–NiCo2O4 was 4.21, 3.93, 4.76 and 5.37, respectively (Table 1). The surface charges of catalysts were negative when solution pH was higher than pHpzc. Higher solution pH would cause higher amount of negative charges on catalyst surface, which could enhance the electrostatic repulsion between catalyst surface and PMS anions. Consequently, the catalytic performance decreased at strong alkaline condition. (2) The transform of dominant PMS species. Given that pKa1 of H2SO5 was less than 0 and pKa2 was 9.4, SO52− would replace HSO5− and become dominant PMS species when solution pH was higher than 9.4. Compared with HSO5− (E0(HSO5−/SO42−) = 1.75 V), SO52− (E0(SO52−/SO42−) = 1.22 V) was less oxidative and more difficult to react.28 Additionally, SO52− could also lead to a stronger electrostatic repulsion between catalyst surface and PMS anions.| SO2 − O − O − H + H+ → SO2 − O − O − H2+ | (3) |
| [Cat − OH] + H+ ↔ [Cat − OH2+] | (4) |
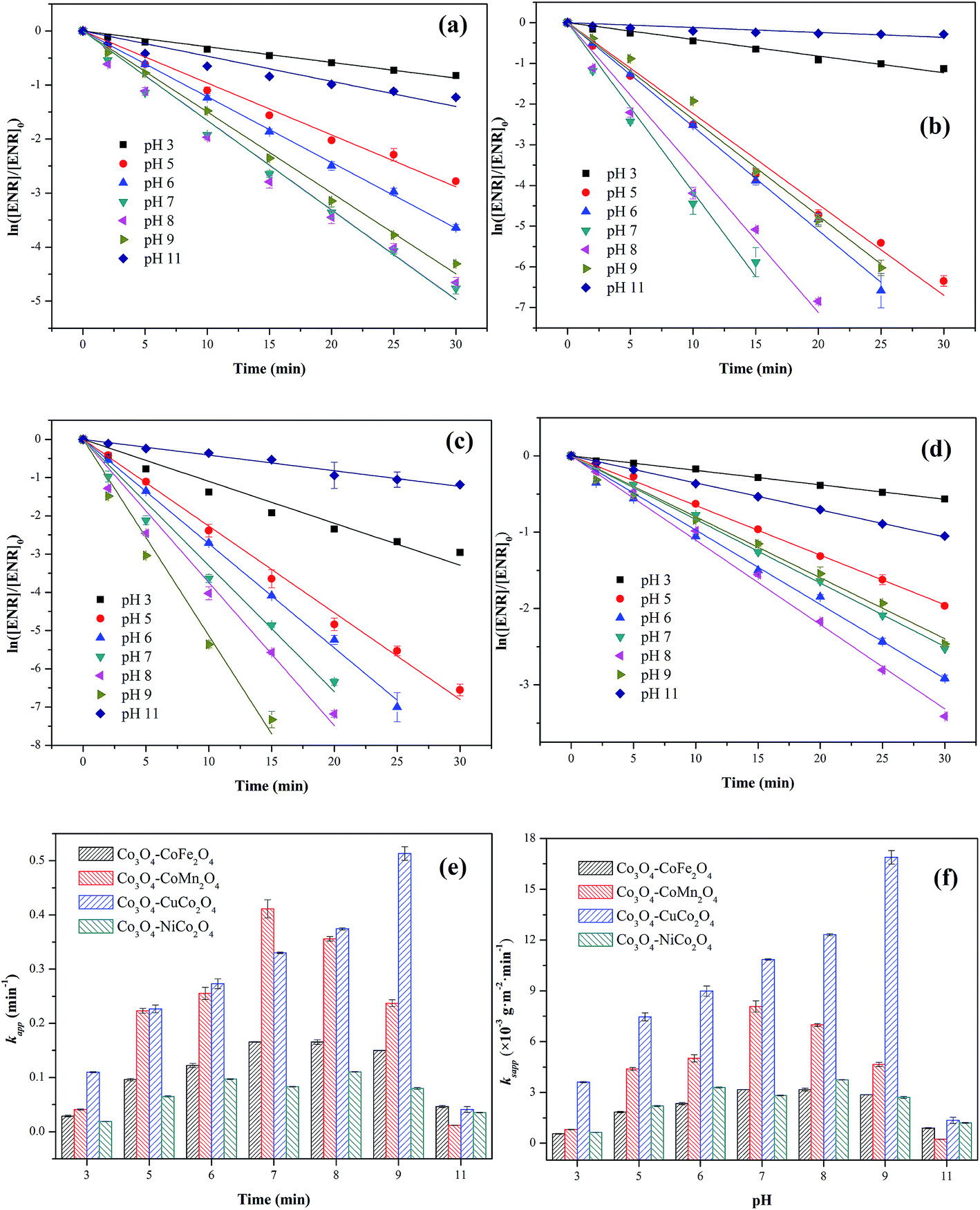 | ||
| Fig. 6 Effect of initial pH on ENR degradation in different Co3O4-composite spinels/PMS systems: (a) Co3O4–CoFe2O4; (b) Co3O4–CoMn2O4; (c) Co3O4–CuCo2O4; (d) Co3O4–NiCo2O4. Values involved in different pH and Co3O4-composite spinels/PMS systems: (e) kapp; (f) ksapp. Experimental condition: [ENR] = 10 mg L−1, [catalyst] = 0.1 g L−1, [PMS] = 1 mM, T = 25 °C. | ||
The values of ksapp were also calculated and the results were presented in Fig. 6(f). It could be more intuitionistic to compare the catalytic performances of four Co3O4-composite spinels in different conditions due to the elimination of difference in specific surface area. As shown in Fig. 5(f), Co3O4–CuCo2O4 maintained the highest ksapp values with pH varied from 3 to 11, suggesting that Co and Cu species were the best combination for PMS activation among these four spinels. However, it should be noted that the lowest ksapp value was showed by Co3O4–CoMn2O4 at pH 11 other than Co3O4–CoFe2O4 or Co3O4–NiCo2O4. This might be closely related to pHpzc of catalysts, the pHpzc of Co3O4–CoMn2O4 was 3.93 which was much lower than that of other three Co3O4-composite spinels. It manifested that Co3O4–CoMn2O4 could present lower performance at strong alkaline condition than others, thus Co3O4–CoMn2O4 possessed the highest ksapp value.
3.4 Effect of temperature
The effect of reaction temperature (25, 35, 45 and 55 °C) on ENR removal in the process of PMS activation was studied. As displayed in Fig. 7(a–d), the ENR degradation in four Co3O4-composite spinels/PMS systems presented the similar trend, catalytic performances of Co3O4-composite spinels significantly increased with the increase of reaction temperature. As reaction temperature increased from 25 to 55 °C, kapp values of Co3O4–CoFe2O4, Co3O4–CoMn2O4, Co3O4–CuCo2O4 and Co3O4–NiCo2O4 increased from 0.122, 0.255, 0.273, 0.097 min−1 to 0.496, 0.872, 0.898, 0.498 min−1, respectively. This result may be due to the fact that higher reaction temperature simplified the rupture of O–O bond and generation of SO4−˙.29,30 In addition, higher reaction temperature was beneficial for reactant molecules to overcome activation energy barrier.17 The activation energy (Ea) could be determined by plotting lnkapp against 1/T based on Arrhenius equation (Fig. 7(e)). The obtained Ea values were 40.73, 33.85, 33.07 and 45.51 kJ mol−1 in Co3O4–CoFe2O4, Co3O4–CoMn2O4, Co3O4–CuCo2O4 and Co3O4–NiCo2O4 activated PMS systems, respectively. The lower Ea value signified the higher catalytic reactivity, and the order of Ea was well corresponded to the sequence of catalytic activity. Moreover, all the Ea values were much higher than that of the diffusion-controlled reactions, which usually ranged from 10 to 13 kJ mol−1.31 This implied that the apparent reaction rate for ENR removal during Co3O4-composite spinels activated PMS processes was dominated by the rate of intrinsic chemical reactions on the catalyst surface. It was reported that out-sphere interactions were usually diffusion-controlled reactions, thus PMS activation by Co3O4-composite spinels was most likely an inner-sphere electron-transfer process.32 The consumption of PMS during ENR oxidation processes was monitored (Fig. 7(f)). Similar with the trend of kapp, PMS consumption also increased as the reaction temperature increased. The PMS consumption caused by Co3O4–CoFe2O4, Co3O4–CoMn2O4, Co3O4–CuCo2O4 and Co3O4–NiCo2O4 increased from 0.230, 0.372, 0.400 and 0.200 mM to 0.796, 0.954, 0.968 and 0.696 mM with the reaction temperature increased from 25 to 55 °C, suggesting that higher reaction temperature was conducive to PMS activation and ENR degradation, which was well correspond to the conclusions by the observations of kapp values. In addition, the higher PMS consumption also reflected higher catalytic reactivity. From Fig. 7(f), the PMS consumption caused by Co3O4-composite spinels always showed the sequence of Co3O4–CuCo2O4 > Co3O4–CoMn2O4 > Co3O4–CoFe2O4 > Co3O4–NiCuFe2O4 even in different reaction temperatures, indicating that Co3O4–CuCo2O4 possessed the highest catalytic activity among the four Co3O4-composite spinels.
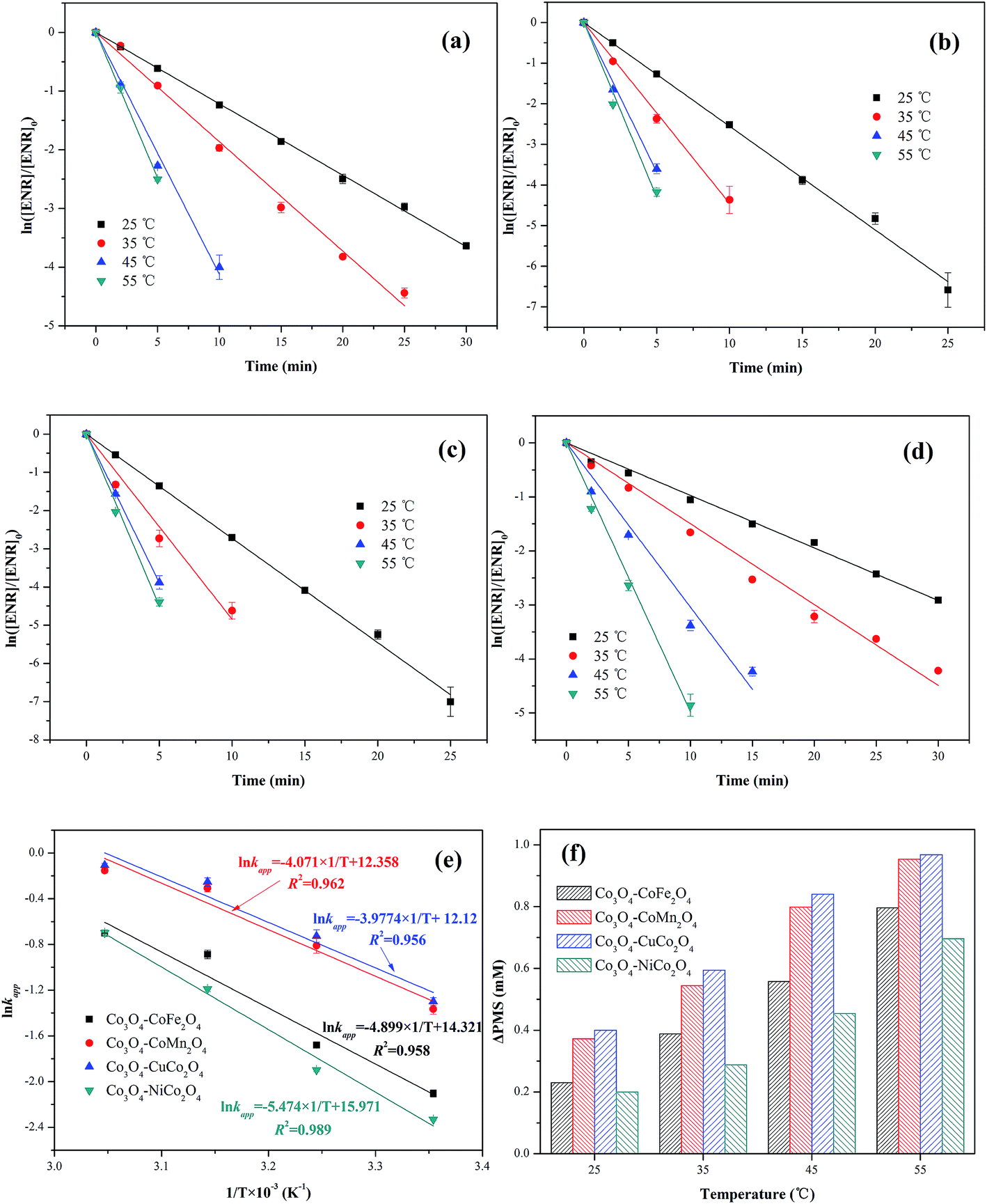 | ||
| Fig. 7 Effect of reaction temperature on ENR degradation in different Co3O4-composite spinels/PMS systems: (a) Co3O4–CoFe2O4; (b) Co3O4–CoMn2O4; (c) Co3O4–CuCo2O4; (d) Co3O4–NiCo2O4. Parameters involved in different reaction temperatures and Co3O4-composite spinels/PMS systems: (e) Arrhenius curves; (f) ΔPMS. Experimental condition: [ENR] = 10 mg L−1, [catalyst] = 0.1 g L−1, [PMS] = 1 mM, pH0 = 6. | ||
3.5 Radical identification and catalytic mechanism
Three different scavengers, tert-butyl alcohol (TBA), ethanol (EtOH) and phenol were employed to identify the dominant radical species in Co3O4-composite spinels/PMS systems. TBA can rapidly react with ˙OH (k˙OH = 3.8–7.6 × 108 M−1 s−1) but has a much lower reactivity with SO4−˙ (kSO4−˙ = 4–9.1 × 105 M−1 s−1),33 and EtOH is a well scavenger for ˙OH and SO4−˙ (k˙OH = 1.2–2.8 × 109 M−1 s−1, kSO4−˙ = 1.6–7.7 × 107 M−1 s−1).34 Phenol can also react with ˙OH and SO4−˙ at a high rate (k˙OH = 6.6 × 109 M−1 s−1, kSO4−˙ = 8.8 × 109 M−1 s−1).35 In view of the difference in reaction rates, using TBA, EtOH and phenol as scavengers was a feasible program for the identification of primary active species.As presented in Fig. 8(a–d), only a slight reduction of ENR removal could be obtained in the presence of 10 or 100 mM TBA, implying that ˙OH was involved in Co3O4-composite spinels activated PMS processes. With the addition of 10 mM EtOH, the ENR degradation was significantly inhibited and the removal efficiencies in Co3O4–CoFe2O4, Co3O4–CoMn2O4, Co3O4–CuCo2O4 and Co3O4–NiCo2O4 activated PMS processes were decreased from 97.37%, 100%, 100%, 94.56% to 66.84%, 69.92%, 82.42%, 57.81%, respectively. More addition of EtOH (100 mM) would cause further reduction of ENR removal efficiency with 47.90%, 47.70% 53.96% and 40.43%, respectively. In order to further confirm the contribution of SO4−˙, 10 mM phenol was introduced into solutions which resulted in a more significant inhibition to ENR removal, less than 18% of ENR could be decomposed in the four Co3O4-composite spinels/PMS processes. The quenching tests clearly suggested that SO4−˙ was the primary reactive species during PMS activation by Co3O4 composite spinels and ˙OH was also involved in these processes.
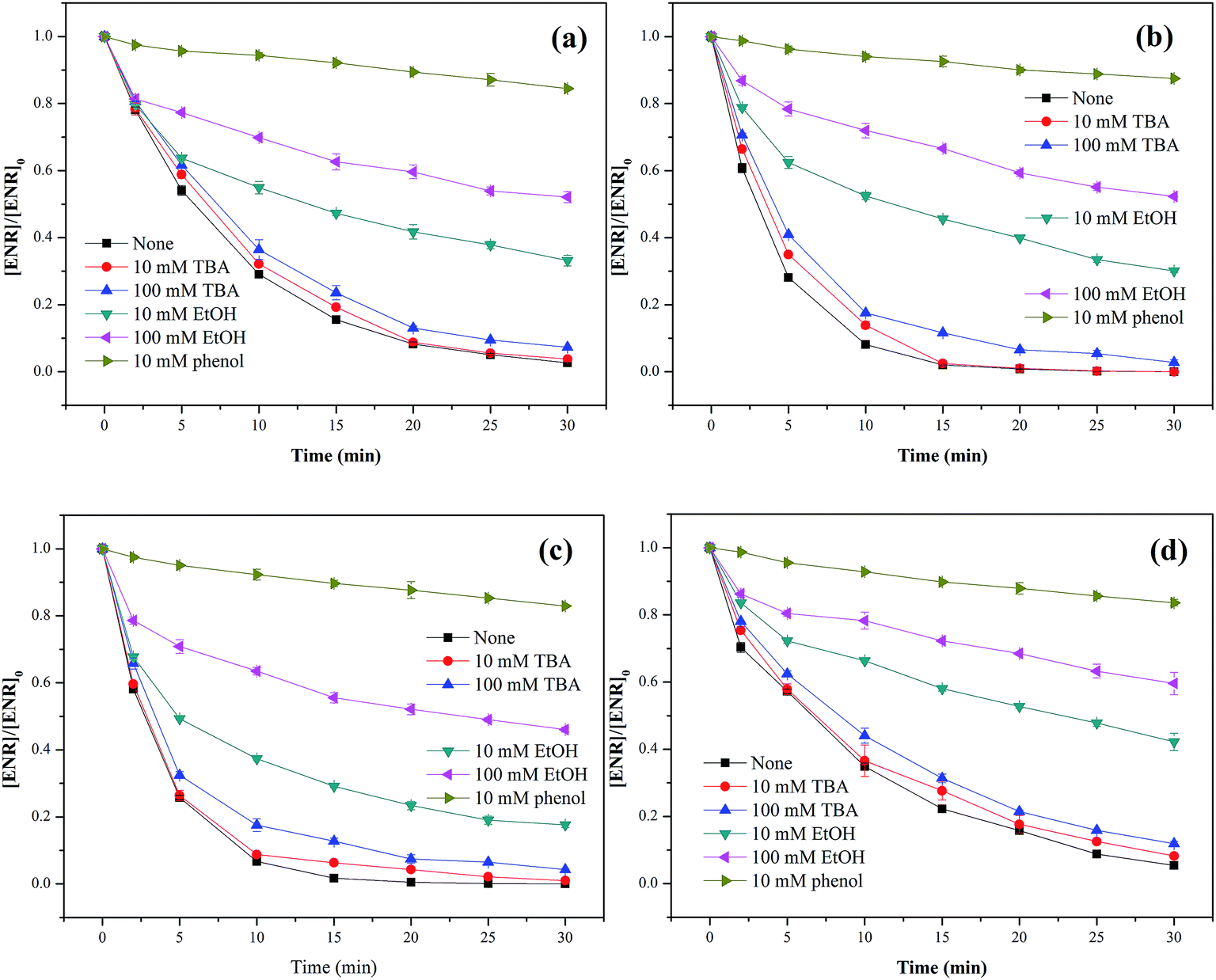 | ||
| Fig. 8 Effect of quenchers on ENR degradation in different Co3O4-composite spinels/PMS systems: (a) Co3O4–CoFe2O4; (b) Co3O4–CoMn2O4; (c) Co3O4–CuCo2O4; (d) Co3O4–NiCo2O4. Experimental condition: [ENR] = 10 mg L−1, [catalyst] = 0.1 g L−1, [PMS] = 1 mM, pH0 = 6, T = 25 °C. | ||
XPS analysis of Co3O4–CoFe2O4 before and after catalytic oxidation was also performed to illustrate the heterogeneous catalytic mechanism (Fig. 4). As shown in Fig. 4(a), before catalytic oxidation, the contents of Co2+ and Co3+ was determined to be 65.34% and 34.66%. After catalytic oxidation, the proportions of Co2+ and Co3+ were changed to 61.93% and 38.07%, respectively. The partial increase of Co3+ was ascribed to the electrons donating of Co2+ during the oxidation process. In the case of O 1s spectra (Fig. 4(b)), the content of Olatt decreased from 48.26% to 46.35%, and the proportion of Oads increased from 51.74% to 53.65%. The increment of Oads can be attributed to the generation of Co–OH or O2 adsorbed on the surface of Co3O4–CoFe2O4. It has been reported that ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Co2+ − −OH was the critical species for the generation of radicals during the process of PMS activation.36 Of note, the Fe 2p3/2 envelope could be deconvoluted into Fe2+ at 709.5 eV, which indicated that the redox reactions between Co and Fe were involved in the PMS activation.
Co2+ − −OH was the critical species for the generation of radicals during the process of PMS activation.36 Of note, the Fe 2p3/2 envelope could be deconvoluted into Fe2+ at 709.5 eV, which indicated that the redox reactions between Co and Fe were involved in the PMS activation.
Based on the results of quenching experiments and XPS analysis, the plausible mechanisms of Co3O4-composite spinels activated PMS were put forward. Taking Co3O4–CoFe2O4/PMS system as example, H2O molecules were firstly physically absorbed on the part of Co2+ sites to generate Co2+–−OH. Then, Co2+ − −OH would react with HSO5− to form SO4−˙ after introduction of PMS (eqn (5)), and could regenerate through the reaction between formed Co3+ − −OH species and HSO5− (eqn (6)). Similarly, Fe3+ could also combine with dissociative adsorption of H2O molecules to form Fe3+–−OH, which would transform to Fe2+ − −OH (eqn (7)) and generate SO4−˙ by reacting with HSO5− (eqn (8)). In addition, due to the standard redox potential of Co3+/Co2+ was 1.92 V,8 while the standard redox potential of Fe3+/Fe2+ was 0.77 V,37 the reduction of Co3+ by Fe2+ was thermodynamically feasible (eqn (9)). The efficient regeneration of surface Co2+ by this process may be able to remain the stability and high efficiency of Co3O4–CoFe2O4. Besides, Co2+ or Fe2+ on catalyst surface could also react with PMS to produce ˙OH (eqn (10) and (11)), which could also be generate by the transformation of SO4−˙ (eqn (12) and (13)). Co3O4–CoMn2O4, Co3O4–CuCo2O4 and Co3O4–NiCo2O4 activated PMS processes all presented the similar mechanism with Co3O4–CoFe2O4 activated PMS process, and the reactions which were involved in Co3O4-composite spinels/PMS systems were listed in Table 2.
| Systems | Reactions | |
|---|---|---|
| Co3O4–CoFe2O4/PMS | Co2+ − −OH + HSO5− → Co3+ − −OH + SO4−˙ + OH− |
(5) |
| Co3+ − −OH + HSO5− → Co2+ − −OH + SO5−˙ + H+ |
(6) | |
| Fe3+ − −OH + HSO5− → Fe2+ − −OH + SO5−˙ + H+ |
(7) | |
| Fe2+ − −OH + HSO5− → Fe3+ − −OH + SO4−˙ + OH− |
(8) | |
| Fe2+ + Co3+ → Fe3+ + Co2+ |
(9) | |
| Co2+ + HSO5− → Co3+ + SO42− + ˙OH |
(10) | |
| Fe2+ + HSO5− → Fe3+ + SO42− + ˙OH |
(11) | |
| SO4−˙ + H2O → HSO4− + ˙OH | (12) | |
| SO4−˙ + OH− → SO42− + ˙OH | (13) | |
| Co3O4–CoMn2O4/PMS | Co2+ − −OH + HSO5− → Co3+ − −OH + SO4−˙ + OH− |
|
| Co3+ − −OH + HSO5− → Co2+ − −OH + SO5−˙ + H+ |
||
| Mn3+ − −OH + HSO5− → Mn2+ − −OH + SO5−˙ + H+ |
||
| Mn3+ − −OH + HSO5− → Mn4+ − −OH + SO4−˙ |
||
| Mn2+ − −OH + HSO5− → Mn3+ − −OH + SO4−˙ |
||
| Mn4+ − −OH + HSO5− → Mn3+ − −OH + SO5−˙ + H+ |
||
| Mn2+ + Co3+ → Mn3+ + Co2+ |
||
| Mn3+ + Co3+ → Mn4+ + Co2+ |
||
| Co2+ + HSO5− → Co3+ + SO42− + ˙OH |
||
| Mn2+ + HSO5− → Mn3+ + SO42− + ˙OH |
||
| SO4−˙ + H2O → HSO4− + ˙OH | ||
| SO4−˙ + OH− → SO42− + ˙OH | ||
| Co3O4–CuCo2O4/PMS | Co2+ − −OH + HSO5− → Co3+ − −OH + SO4−˙ + OH− |
|
| Co3+ − −OH + HSO5− → Co2+ − −OH + SO5−˙ + H+ |
||
| Cu2+ − −OH + HSO5− → Cu+ − −OH + SO5−˙ + H+ |
||
| Cu2+ − −OH + HSO5− → Cu3+ − −OH + SO4−˙ + OH− |
||
| Cu+ − −OH + HSO5− → Cu2+ − −OH + SO4−˙ + OH− |
||
| Cu3+ − −OH + HSO5− → Cu2+ − −OH + SO5−˙ + H+ |
||
| Cu3+ + Co2+ → Cu2+ + Co3+ |
||
| Cu+ + Co3+ → Cu2+ + Co2+ |
||
| Co2+ + HSO5− → Co3+ + SO42− + ˙OH |
||
| Cu2+ + HSO5− → 3+ + SO42− + ˙OH |
||
| SO4−˙ + H2O → HSO4− + ˙OH | ||
| SO4−˙ + OH− → SO42− + ˙OH | ||
| Co3O4–NiCo2O4/PMS | Co2+ − −OH + HSO5− → Co3+ − −OH + SO4−˙ + OH− |
|
| Co3+ − −OH + HSO5− → Co2+ − −OH + SO5−˙ + H+ |
||
| Ni2+ − −OH + HSO5− → Ni3+ − −OH + SO4−˙ + OH− |
||
| Ni3+ − −OH + HSO5− → Ni2+ − −OH + SO5−˙ + H+ |
||
| Co2+ + HSO5− → Co3+ + SO42− + ˙OH |
||
| Ni2+ + HSO5− → Ni3+ + SO42− + ˙OH |
||
| SO4−˙ + H2O → HSO4− + ˙OH | ||
| SO4−˙ + OH− → SO42− + ˙OH |
3.6 Reusability and stability of different Co3O4-composite spinels
The reusability of heterogeneous catalysts is an important indicator to assess the industrial application potential of heterogeneous catalysts. In this study, the reusability of different Co3O4-composite spinels was evaluated and the results were presented in Fig. 9(a). As shown, after being reused for three times, all the Co3O4-composite spinels still remained high catalytic activity toward PMS. Taking Co3O4–CoFe2O4 as example, after three consecutive runs, just a slight reduction in ENR removal was observed and their values were 94.89%, 91.32% and 89.27%, respectively. The slight loss of catalytic activity was mainly ascribed to the leaching of metal ions during the consecutive runs. Therefore, the stability of different Co3O4-composite spinels was further investigated and the results were reported in Fig. 9(b). As exhibited, the leaching of cobalt was clearly observed after each run. With the increase of cycle times, the leaching concentration decreased. Of note, after doped different transition metals into OM-Co3O4, the cobalt leakage can be effectively controlled, which was attributed to the intimate interactions between two metals.2 Therefore, Co3O4-composite spinels are ideal PMS activator for environmental remediation.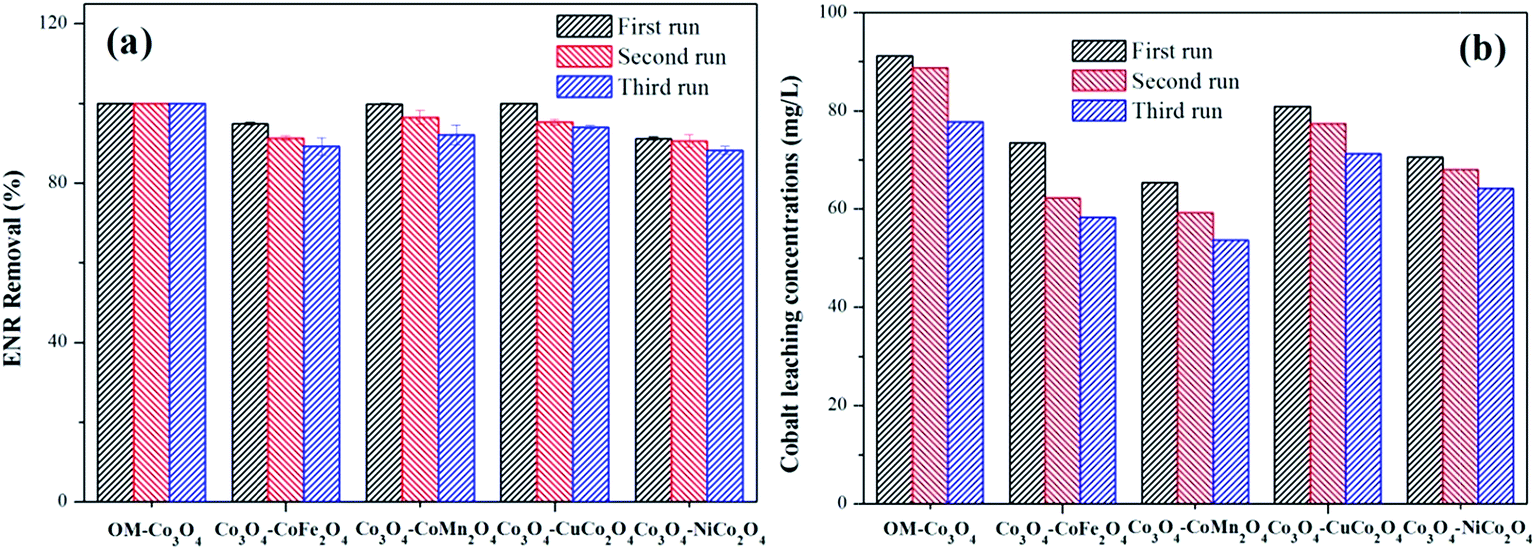 | ||
| Fig. 9 (a) Reusability of different Co3O4-composite spinels as catalyst for the degradation of ENR; (b) cobalt leaching concentrations in different Co3O4-composite spinels/PMS systems. Experimental condition: [ENR] = 10 mg L−1, [catalyst] = 0.1 g L−1, [PMS] = 1 mM, pH0 = 6, T = 25 °C, reaction time = 25 min. | ||
4. Conclusion
Co3O4-composite spinels were successfully synthesized through doping transition metals (Fe, Mn, Cu and Ni) to ordered mesoporous Co3O4. The obtained Co3O4-composite spinels all showed outstanding catalytic activity toward PMS. Co3O4–CuCo2O4 exhibited the highest catalytic performance in PMS solution, followed by Co3O4–CoMn2O4, Co3O4–CoFe2O4 and Co3O4–NiCo2O4. ENR degradation would be retarded in strong acidic and alkaline conditions, the improvement of reaction temperature could significantly accelerate ENR decomposition. Sulfate radical was confirmed to be the primary reactive species in Co3O4-composite spinels activated PMS processes and hydroxyl radical was also involved in these processes. The synergistic effect between two metals in Co3O4-composite spinels was the vital reason for the high catalytic reactivity. Co3O4-composite spinels displayed satisfactory reusability and doping different transition metals into OM-Co3O4 can effectively control the cobalt leaching. In consideration of cost and toxicity, Co3O4-composite spinels might have great potential in pollution control than OM-Co3O4.Conflicts of interest
No conflict of interest exists in the submission of this manuscript and manuscript is approved by all authors for publication.Acknowledgements
The authors are grateful for the financial support of this study by the National Natural Science Foundation of China (No. 51508509), China Postdoctoral Science Foundation (No. 2015M581936), Natural Science Foundation of Zhejiang Province (No. LY18E080036) and State Key Laboratory of Pollution Control and Resource Reuse Foundation (No. PCRRF16017).References
- J. Zhou, J. Ma, L. W. Chen, X. C. Li, Y. H. Guan, P. C. Xie and C. Pan, Environ. Sci. Technol., 2013, 47, 11685–11691 CrossRef PubMed.
- P. D. Hu and M. C. Long, Appl. Catal., B, 2016, 181, 103–117 CrossRef CAS.
- Y. B. Wang, X. Zhao, D. Cao, Y. Wang and Y. F. Zhu, Appl. Catal., B, 2017, 211, 79–88 CrossRef CAS.
- Y. M. Ren, L. Q. Lin, J. Ma, J. Yang, J. Feng and Z. J. Fan, Appl. Catal., B, 2015, 165, 572–578 CrossRef CAS.
- C. Q. Tan, N. Y. Gao, D. F. Fu, J. Deng and L. Deng, Sep. Purif. Technol., 2017, 175, 47–57 CrossRef CAS.
- C. Q. Tan, D. F. Fu, N. Y. Gao, Q. D. Qin, Y. Xu and H. M. Xiang, J. Photochem. Photobiol., A, 2017, 332, 406–412 CrossRef CAS.
- C. Q. Tan, Y. J. Dong, D. F. Fu, N. Y. Gao, J. X. Ma and X. Y. Liu, Chem. Eng. J., 2018, 334, 1006–1015 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- G. P. Anipsitakis, E. Stathatos and D. D. Dionysiou, J. Phys. Chem. B, 2005, 109, 13052–13055 CrossRef CAS PubMed.
- X. Y. Chen, J. W. Chen, X. L. Qiao, D. G. Wang and X. Y. Cai, Appl. Catal., B, 2008, 80, 116–121 CrossRef CAS.
- J. Y. Pu, J. Q. Wan, Y. Wang and Y. W. Ma, RSC Adv., 2016, 6, 91791–91797 RSC.
- S. N. Su, W. L. Guo, Y. Q. Leng, C. L. Yi and Z. M. Ma, J. Hazard. Mater., 2013, 244–245, 736–742 CrossRef CAS PubMed.
- J. Deng, Y. S. Shao, N. Y. Gao, C. Q. Tan, S. Q. Zhou and X. H. Hu, J. Hazard. Mater., 2013, 262, 836–844 CrossRef CAS PubMed.
- Y. J. Yao, Y. M. Cai, G. D. Wu, F. Y. Wei, X. Y. Li, H. Chen and S. B. Wang, J. Hazard. Mater., 2015, 296, 128–137 CrossRef CAS PubMed.
- Y. Feng, J. H. Liu, D. L. Wu, Z. Y. Zhou, Y. Deng, T. Zhang and K. M. Shih, Chem. Eng. J., 2015, 280, 514–524 CrossRef CAS.
- J. Deng, S. F. Feng, K. J. Zhang, J. Li, H. Y. Wang, T. Q. Zhang and X. Y. Ma, Chem. Eng. J., 2017, 308, 505–515 CrossRef CAS.
- R. C. Wei, F. Ge, M. Chen and R. Wang, J. Environ. Qual., 2012, 41, 1481–1486 CrossRef CAS PubMed.
- S. Waclawek, K. Grubel and M. Cernik, Spectrochim. Acta, Part A, 2015, 149, 928–933 CrossRef CAS PubMed.
- T. Grewe, X. H. Deng, C. Weidenthaler, F. Schuth and H. Tuysuz, Chem. Mater., 2013, 25, 4926–4935 CrossRef CAS.
- J. Wu, Y. Xue, X. Yan, W. S. Yan, Q. M. Cheng and Y. Xie, Nano Res., 2012, 5, 521–530 CrossRef CAS.
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- R. A. Floyd and L. M. Soong, Biochem. Biophys. Res. Commun., 1977, 74, 79–84 CrossRef CAS PubMed.
- S. V. Verstraeten, S. Lucangioli and M. Galleano, Inorg. Chim. Acta, 2009, 362, 2305–2310 CrossRef CAS.
- X. L. Zhang, M. B. Feng, L. S. Wang, R. J. Qu and Z. Y. Wang, Chem. Eng. J., 2017, 307, 95–104 CrossRef CAS.
- J. Deng, S. F. Feng, X. Y. Ma, C. Q. Tan, H. Y. Wang, S. Q. Zhou, T. Q. Zhang and J. Li, Sep. Purif. Technol., 2016, 167, 181–189 CrossRef CAS.
- C. Q. Tan, N. Y. Gao, Y. Deng, J. Deng and S. Q. Zhou, J. Hazard. Mater., 2014, 276, 452–460 CrossRef CAS PubMed.
- W. D. Oh, Z. L. Dong, Z. T. Hu and T. T. Lim, J. Mater. Chem. A, 2015, 3, 22208–22217 CAS.
- Y. Feng, P. H. Lee, D. Wu and K. Shih, Water Res., 2017, 120, 12–21 CrossRef CAS PubMed.
- P. H. Shi, R. J. Su, S. B. Zhu, M. C. Zhu, D. X. Li and S. H. Xu, J. Hazard. Mater., 2012, 229–230, 331–339 CrossRef CAS PubMed.
- J. Deng, Y. J. Ge, C. Q. Tan, H. Y. Wang, Q. S. Li, S. Q. Zhou and K. J. Zhang, Chem. Eng. J., 2017, 330, 1390–1400 CrossRef CAS.
- L. J. Xu and J. L. Wang, Environ. Sci. Technol., 2012, 46, 10145–10153 CrossRef CAS PubMed.
- Y. Feng, D. L. Wu, Y. Deng, T. Zhang and K. Shih, Environ. Sci. Technol., 2016, 50, 3119–3127 CrossRef CAS PubMed.
- Z. F. Huang, H. W. Bao, Y. Y. Yao, W. Y. Lu and W. X. Chen, Appl. Catal., B, 2014, 154–155, 36–43 CrossRef CAS.
- Y. B. Deng, L. H. Zhu, N. Wang and H. Q. Tang, Appl. Catal., B, 2013, 129, 153–162 CrossRef.
- M. E. Lindsey and M. A. Tarr, Environ. Sci. Technol., 2000, 34, 444–449 CrossRef CAS.
- H. M. Sun, X. J. Yang, L. J. Zhao, T. H. Xu and J. S. Lian, J. Mater. Chem. A, 2016, 4, 9455–9465 CAS.
- Y. L. Nie, C. Hu, J. H. Qu and X. Zhao, Appl. Catal., B, 2009, 87, 30–36 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |