DOI:
10.1039/C7RA06570A
(Paper)
RSC Adv., 2018,
8, 606-618
Comparative study on La-promoted Ni/γ-Al2O3 for methane dry reforming – spray drying for enhanced nickel dispersion and strong metal–support interactions†
Received
12th June 2017
, Accepted 22nd November 2017
First published on 2nd January 2018
Abstract
Dry reforming of methane (DRM) enables an efficient utilization of two abundant greenhouse gases by converting them into syngas, a versatile feedstock for chemical synthesis. Aiming for high catalyst performance and enhanced coke resistance, different preparation techniques of La-promoted Ni/γ-Al2O3 catalysts for DRM were compared facilitating structure–performance correlations. The studied synthesis techniques comprehend incipient wetness impregnation and co-precipitation as well as alternative techniques such as spray drying. All catalysts were fully characterized before and after reaction by N2-physisorption, XRD, H2-TPR and STEM-EDX elemental mapping. Additionally, a thorough investigation of carbon deposits has been carried out by TGA/DSC and STEM-EDX, respectively. The different preparation techniques led generally to very different physical properties, structure, chemical species and anti-coking properties of the catalyst. However, some catalysts with similar physicochemical characteristics differed in catalytic performance and coking resistance. Superior catalytic performance could be reached for catalysts prepared by spray drying and related to excellent Ni dispersion, strong metal–support interaction and very low coke formation of only 2.7% of the catalyst weight. After 6 h time on stream only minor sintering occurred, with few Ni nanoparticles up to 10 nm.
1. Introduction
Concerns over CO2 emissions have substantially increased over the past few decades. CO2 is a very stable molecule and is considered to be among the main greenhouse gases leading to global warming.1 Currently, anthropogenic emissions of CO2 in the atmosphere are estimated to contribute around 60% of the total of all the anthropogenic gases.2 In the future they are expected to rise further due to the ongoing combustion of fossil fuels. Almost all CO2 produced from combustion processes is emitted into the atmosphere where it gradually accumulates.1 Besides CO2, CH4 is considered to be another major greenhouse gas.3,4 Therefore, there is much interest in the utilization of CO2 and CH4. A promising approach to carbon capture and utilization (CCU) presents dry reforming of methane (DRM) (eqn (1)). The process makes use of the two abundantly available greenhouse gases by converting them into syngas, a versatile feedstock for chemical synthesis. DRM has attracted considerable attention in recent years since it produces high purity syngas compared to coal and biomass.5,6 Different classes of catalysts have shown potential to deliver reasonable and economically feasible results for DRM.7–9 Even though noble metal catalysts exhibit superior activity and stability amongst others,10–14 their scarcity and price make them unattractive. On the other hand, nickel based catalysts are a commonly proposed alternative due to their high activity, low cost and the abundant supply of nickel compared to noble metals. The main challenge nickel based catalysts face is deactivation, occurring via sintering and carbon deposition.15–17 Carbon deposits are formed mainly via CH4 cracking (eqn (2)) and CO disproportion (Boudouard reaction) (eqn (3)).18 Sintering on the other hand, although found to have less direct influence on activity and stability of the catalyst compared to carbon deposition, significantly promotes coking.15,17 Many efforts have been made to improve the carbon deposition resistance of nickel based catalysts. Amongst them, rare earth metals such as La,19–21 Ce,22,23 and Zr,24 can reduce carbon deposition and improve sintering resistance. Lanthanides favor metal dispersion,25,26 and strengthen CO2 adsorption on the support.27 The presence of oxy-carbonates over La2O3 facilitates coke removal since they seem to act as a dynamic oxygen pool.28| | |
CO2 + CH4 ↔ 2CO + 2H2, ΔHo298 K = +247 kJ mol−1
| (1) |
| | |
CH4 ↔ C + 2H2, ΔHo298 K = +75 kJ mol−1
| (2) |
| | |
2CO ↔ CO2 + C, ΔHo298 K = −172 kJ mol−1
| (3) |
| | |
CO2 + H2 ↔ H2O + CO, ΔHo298 K = +41 kJ mol−1
| (4) |
Therefore, this study focusses on Ni on γ-Al2O3 promoted with lanthanum. La is commonly used to form perovskite-like structures. Whereas in this study, La is envisioned as a textural promoter to enhance nickel dispersion and to accelerate carbon deposits removal, respectively. Numerous studies, both experimental,29–31 and computational (DFT),31–34 suggest that a reduced Ni-nanoparticle size enhances activity and minimizes carbon formation. Especially a Ni particle size of less than 5 nm appears advantageous for coke-resistant catalysts.34 Despite intensive research in the field, the influence of preparation techniques on catalyst properties and the associated performance in DRM has been investigated rarely. In order to address these, we compared seven La-promoted Ni/γ-Al2O3 catalysts prepared by a wide range of different synthetic techniques, all aiming at obtaining catalysts with high specific surface area and high active metal dispersion. The methods comprehend incipient wetness impregnation and co-precipitation as they are commonly used in the literature providing decent catalytic results towards DRM. Spray drying was also utilized since the achieved Ni dispersion is excellent. To the best of our knowledge, spray drying has been rarely used for Ni-based catalysts in DRM. Additionally, we modified spray drying and co-precipitation by combining them with physical mixture and an ethanol suspension approach. In this way, we could also evaluate the influence of ethanol as a suspension medium on the properties of the catalyst. Furthermore, feed ratio of CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH4 = 2:1 was utilized since higher partial pressure of CO2 in the feed was found to suppress sintering and coke formation.35,36 We focused on structural and physico-chemical characterization of these materials before and after testing, as well as their activity and stability in DRM. Together with a throughout analysis of carbon deposits, a correlation between the impact of preparation techniques on catalyst properties and their catalytic performance and coking behavior became evident.
:CH4 = 2:1 was utilized since higher partial pressure of CO2 in the feed was found to suppress sintering and coke formation.35,36 We focused on structural and physico-chemical characterization of these materials before and after testing, as well as their activity and stability in DRM. Together with a throughout analysis of carbon deposits, a correlation between the impact of preparation techniques on catalyst properties and their catalytic performance and coking behavior became evident.
2. Experimental
2.1. Catalyst preparation
Seven different preparation techniques were applied to synthesize La (25 wt%)-promoted Ni (10 wt%)/γ-Al2O3 catalysts (Table 1). The catalyst denoted as IW3M was synthesized by the standalone preparation technique of incipient wetness impregnation, utilizing all 3 metals. 0.5 g of Ni(NO3)2·6H2O (Sigma Aldrich, ≥97.0) and 0.74 g of La(NO3)3·6H2O (Sigma Aldrich, ≥99.9%) were dissolved in distilled water to form a saturated solution which was impregnated to incipient wetness on 1.0 g of γ-Al2O3 powder (Sasol, ABET = 160 m2 g−1, VP ≥ 0.45 mL g−1). The obtained material was dried overnight at 80 °C and calcined at 800 °C in air for 8 h with a ramping rate of 10 K min−1. The same drying/calcination parameters were applied to all catalysts of the series.
Table 1 Catalyst preparation overview
| Catalyst |
Preparation technique |
Step 1 |
Step 2 |
Step 3 |
| Overnight at 80 °C. At 800 °C in air for 8 h (10 K min−1). |
| IW3M |
Incipient wetness impregnation |
Ni(NO3)2·6H2O and La(NO3)3·6H2O dissolved in water and impregnated on γ-Al2O3 |
Dried overnighta and calcinedb |
— |
| SD3M |
Spray drying |
Ni(NO3)2·6H2O, La(NO3)3·6H2O and Al(NO3)3·9H2O dissolved in water and then spray dried |
— |
| CP3M |
Co-precipitation |
Ni(NO3)2·6H2O, La(NO3)3·6H2O, Al(NO3)3·9H2O dissolved in water and mixed with NaOH(aq). Filtered and washed |
— |
| SD + SolidMix |
Spray drying/physical mixture |
Ni(NO3)2·6H2O and La(NO3)3·6H2O dissolved in water and then spray dried |
Physical mixture with γ-Al2O3 and calcinedb second time |
| SD + suspension |
Spray drying/ethanol suspension |
Ni(NO3)2·6H2O and La(NO3)3·6H2O dissolved in water and then spray dried |
Ethanol suspension with γ-Al2O3, drieda and calcinedb second time |
| CP + SolidMix |
Co-precipitation/physical mixture |
Ni(NO3)2·6H2O and La(NO3)3·6H2O dissolved in water and mixed with NaOH(aq). Filtered and washed |
Physical mixture with γ-Al2O3 and calcinedb second time |
| CP + suspension |
Co-precipitation/ethanol suspension |
Ni(NO3)2·6H2O and La(NO3)3·6H2O dissolved in water and mixed with NaOH(aq). Filtered and washed |
Ethanol suspension with γ-Al2O3, drieda and calcinedb second time |
The catalyst denoted as SD3M was synthesized by the standalone preparation technique of spray drying, utilizing a 3-metal solution. 0.5 g of Ni(NO3)2·6H2O, 0.74 g of La(NO3)3·6H2O and 7.36 g of Al(NO3)3·9H2O (Sigma Aldrich, ≥98.0) were dissolved in 230 mL distilled water to form a solution of 0.1 M concentration based on the total amount of metal nitrates. The solution was then fed with a constant rate of 3 mL min−1 into a spray dryer (BÜCHI Mini-Spray-Dryer B-290) with N2 as carrier gas at 4 mL min−1 and air as drying gas at 550 L min−1 at 140 °C. The obtained material was dried overnight and calcined.
The catalyst denoted as CP3M was synthesized by co-precipitation, utilizing a 3-metal solution. 0.5 g of Ni(NO3)2·6H2O, 0.74 g of La(NO3)3·6H2O and 7.36 g of Al(NO3)3·9H2O were dissolved in 250 mL distilled water and heated to 50 °C. 185 mL of 0.4 M NaOH were added and stirred for 30 min. The formed precipitate was cooled to room temperature, filtered and washed multiple times with distilled water. The obtained material was dried and calcined as already indicated.
The catalysts denoted as SD + SolidMix and SD + suspension were synthesized by applying the combined techniques of spray drying/physical mixture and spray drying/ethanol suspension, respectively. 2.0 g of Ni(NO3)2·6H2O and 2.96 g of La(NO3)3·6H2O were dissolved in 137 mL distilled water to form a solution of 0.1 M concentration based on the total amount of metal nitrates. The rest of the procedure is identical to the one applied to SD3M catalyst. After calcination, half of the collected material was physically mixed with 2 g of γ-Al2O3 to obtain SD + SolidMix catalysts, whereas the other half was suspended in ethanol with 2 g of γ-Al2O3 and after agitation for 30 minutes dried to produce the SD + suspension catalyst. Both materials were finally calcined.
The catalysts denoted as CP + SolidMix and CP + suspension were synthesized by combining co-precipitation and physical mixture, and co-precipitation- and ethanol suspension, respectively. 2.0 g of Ni(NO3)2·6H2O and 2.96 g of La(NO3)3·6H2O were dissolved in 67 mL of distilled water and heated to 50 °C. 225 mL of 0.2 M NaOH were added and stirred for 30 min. The formed precipitate was quenched to room temperature, filtered and washed multiple times with distilled water. The obtained material was dried overnight and calcined. Half of the collected material was physically mixed with 2 g of γ-Al2O3 to obtain the CP + SolidMix catalyst, while the other half was suspended in ethanol with 2 g of γ-Al2O3, agitated for 30 minutes and dried to obtain the CP + suspension catalyst. Both materials were finally calcined.
2.2. Catalyst characterization
The total surface area was determined by the BET method based on N2-physisorption capacity using a Micromeritics ASAP 2020 at 77.35 K. Prior to analysis, all the samples were degassed at 600 K under pure He flow. ICP-OES was performed on an ICP Spectroflame D by Spectro. The phase composition of the catalysts was evaluated by X-ray diffraction measurements (XRD) performed with a Siemens D5000 X-ray diffractometer with Cu Kα (λ = 0.15409 nm, 40 kV, 40 mA) radiation and a scanning step width of 0.02°. Temperature programmed reduction with H2 (H2-TPR) was performed on a TPDRO 1100 Porotec unit using thermal conductivity detection. Thermogravimetric analysis and differential scanning calorimetry (TGA/DSC) were performed on a Netzsch STA 409C/CD appliance. The temperature was increased starting from 25 °C to 1000 °C with a heating rate of 5 K min−1. The morphology of both fresh and spent catalysts was characterized by a transmission electron microscope (TEM) JEOL JEM-2200FS equipped with Schottky type field emission electron emitter, in column omega-type electron energy filter, objective lens aberrations corrector CETCOR (CEOS GmbH), a slow scan CCD camera Gatan for high resolution (HR) TEM, a scanning transmission electron mode (STEM) and an energy dispersive X-ray (EDX) spectrometer JED-2300 (Jeol). Surface chemistry was investigated by X-ray spectroscopy (XPS) using a JEOL JAMP 9500F equipped with a non-monochromatic Al Kα X-ray source. Fresh and spent IW3M and SD3M catalysts were carefully pressed onto Sn-foil to embed the powders in a conductive medium. Charge calibration was carried out by using the aliphatic C 1s signal at 285.0 eV. However, shifting the energy scale according to the signals of the dielectric powders lead to an off-set of signals stemming from the Sn-foil, which were identified by their energetic distance to the Sn 3d5/2-signal and a comparison with signals of a plain Sn-foil. Detailed spectra were recorded with a pass energy of 20 eV of the detector, 0.05 eV step size and 20 recording cycles. CASA XPS was used for data analysis where mixed Gaussian–Lorentzian (30–70%) signals and Shirley backgrounds were applied for data fitting.
2.3. Catalytic testing
A plug-flow fixed-bed quartz tube reactor, with an internal diameter of 6 mm inside a tubular furnace was used. The furnace was regulated by a K-type thermocouple attached to it. The height of the bed was kept constant at 1 cm. All catalysts were firstly reduced in situ in 90 mL min−1 pure H2 flow at 800 °C for 2 h with the exception of SD3M which was reduced for 3 h. All reactions were carried out at 800 °C with a gas hourly space velocity of 35000 h−1 and feed ratio CO2:CH4 of 2:1 under atmospheric pressure. 10% N2 (v/v) of the reactant flow was introduced as an internal standard to account for volume expansion during reaction. The outlet gases were analyzed by an online gas chromatographer (GC) Bruker 400 GC Series equipped with two thermal conductivity (TCD) and one flame ionization (FID) detectors. The outlet gases were first allowed to pass through a cold trap to remove the produced water and then through a dryer/absorber containing Drierite™ to remove humidity before entering the GC.
Conversions and selectivities were calculated through the equations
| |
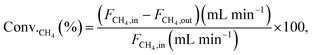 | (5) |
| |
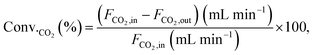 | (6) |
| |
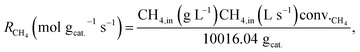 | (7) |
| |
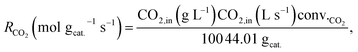 | (8) |
| |
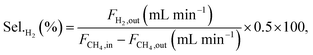 | (9) |
| |
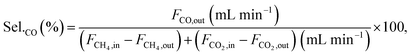 | (10) |
| |
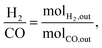 | (11) |
where Conv.
CH4 and conv.
CO2 are the conversions of methane and carbon dioxide as percentages, respectively, and Sel.
H2 and Sel.
CO are the selectivities to hydrogen and carbon monoxide as percentages, respectively.
FCH4,in,
FCH4,out and
FCO2,in,
FCO2,out are the inlet and outlet flows of methane and carbon dioxide (mL min
−1), respectively, whereas
FH2,out and
FCO,out are the respective flows of hydrogen and carbon monoxide at the reactor outlet (mL min
−1).
3. Results and discussion
3.1. Fresh catalyst characterization
Lanthanum (25 wt%) promoted nickel (10 wt%) catalysts supported on alumina were prepared using the standalone preparations techniques of incipient wetness impregnation (IW3M), co-precipitation (CP3M) and spray drying (SD3M), as well as the combined techniques of spray drying/physical mixture (SD + SolidMix), spray drying/ethanol suspension (SD + suspension), co-precipitation/physical mixture (CP + SolidMix) and co-precipitation/ethanol suspension (CP + suspension) of the different salts, respectively. All calcined catalysts exhibited specific surface areas around 98–138 m2 g−1 except the one of the catalyst prepared by spray drying (SD3M) that was only 9.8 m2 g−1 (Table 2).
Table 2 Specific surface area of all seven fresh calcined catalysts (BET method)
| |
IW3M |
SD3M |
CP3M |
SD + SolidMix |
SD + suspension |
CP + SolidMix |
CP + suspension |
| Surface area (m2 g−1) |
98.8 |
9.8 |
97.0 |
127.4 |
138.1 |
118.3 |
118.8 |
| Total pore volume (cm3 g−1) |
0.42 |
0.01 |
0.21 |
0.41 |
0.45 |
0.37 |
0.37 |
| Average pore size (Å) |
102 |
27 |
52 |
94 |
96 |
96 |
95 |
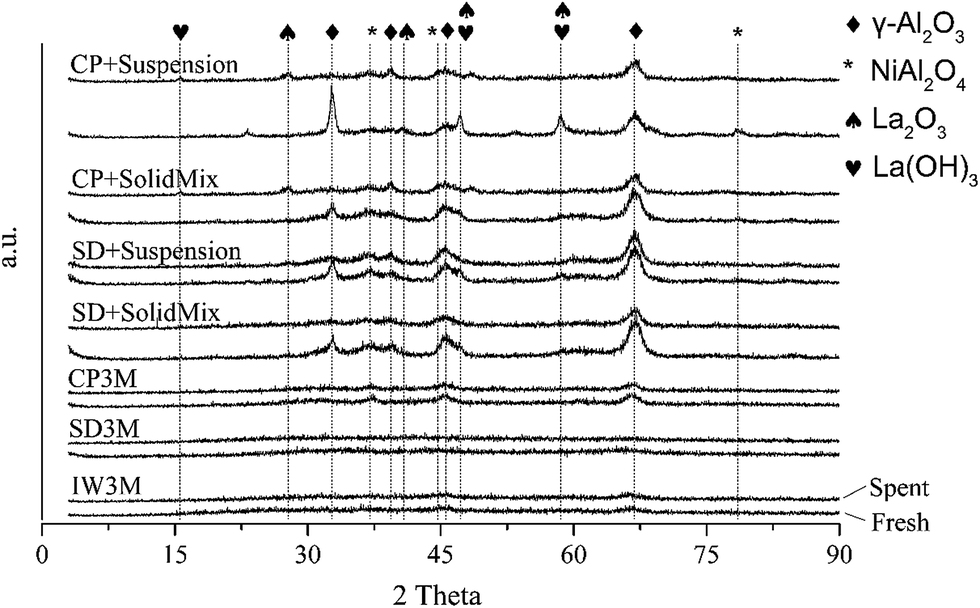
XRD measurements were also carried out (see ESI†) for all freshly calcined catalysts. IW3M, SD3M and CP3M exhibited very low crystallinity, which was not expected due to the harsh calcination conditions of 800 °C for 8 h. Literature reports suggest that γ-Al2O3 appears at temperatures between 350 and 1000 °C when formed from crystalline37 or amorphous38 precursors. More studies should be performed in order to understand better this observation. XRD patterns of these three catalysts showed reflections with relatively low intensity related to γ-Al2O3 as well as NiAl2O4, respectively. The other materials possessed a higher degree of crystallinity emphasized by more intense reflexes corresponding to γ-Al2O3 and NiAl2O4. More specifically, all four materials exhibited distinguishable reflexes of γ-Al2O3 (more pronounced at 32.7°, 39.5°, 45.5°, 60.6° and 66.9° 2-theta) and NiAl2O4 (at 37.2° and 78.5° 2-theta), respectively. For the catalysts prepared with the combined preparation techniques crystallinity of La2O3 and/or La(OH)3 was emphasized by reflexes found at 47.3° and 58.5° 2-theta. Furthermore, no reflections attributable to the NiLa2O3 perovskite-like structure were observed. This comes in good agreement with studies, reporting that high calcination temperature would favor perovskite-like structure formation but also an appropriate ratio of Ni and La is needed.39
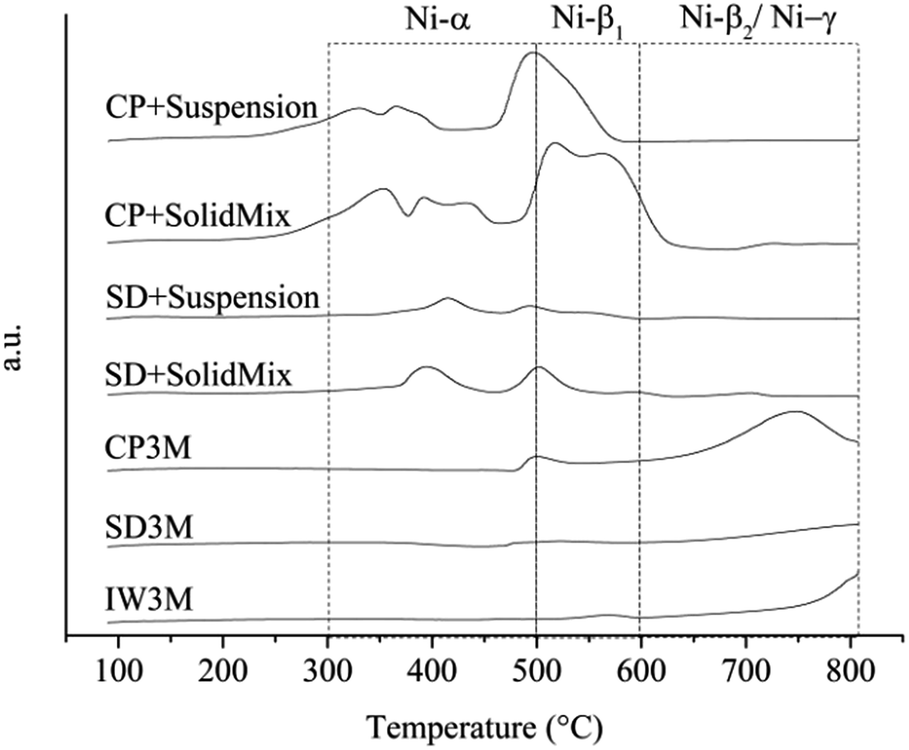
H2-TPR profiles of the calcined catalysts showed a great variation of the reducibility of the catalysts (Fig. 1). H2 uptake for each catalyst and temperature range was calculated and presented in Table 3. Concerning Ni-based catalysts supported on γ-Al2O3, 4 different species with increasing reduction temperature have been reported: Ni-α species [300–500 °C] that correspond to reducing bulk NiO with very low interaction with γ-Al2O3, Ni-β1 species [500–600 °C] that correspond to NiO interacting moderately with γ-Al2O3, Ni-β2 [600–900 °C] that correspond to non-stoichiometric spinel species with strong interaction with the support and finally Ni-γ species [600–900 °C] that correspond to bulk NiAl2O4 species.40–42 Signals corresponding to reduction of La species did not occur because of the high reduction temperature of lanthanates.43,44 The results obtained are in reasonable agreement with the reduction temperatures of the different Ni species, though some signals overlap and shift from the reported values. This observation can be attributed to the presence of the textural La-promoter. For catalysts IW3M and SD3M, reduction started at temperatures higher than 650 °C suggesting that only spinel species strongly interacting with the support and no separate NiO were available. We observed a comparable H2-TPR profile for CP3M; however, also small amounts of reducible NiO species potentially exhibiting a moderate support interaction appear. For SD + SolidMix, SD + suspension, CP + SolidMix and CP + suspension the acquired profiles differ significantly possessing two major peak regions; between 300 and 500 °C signals can be mainly attributed to Ni-α (bulk NiO species), whereas Ni-β1 species – smaller NiO domains moderately interacting with the γ-Al2O3 support – cause signal at temperature of 450 to 620 °C, respectively. Interestingly, these catalysts did not possess species causing high reduction temperatures of above 650 °C. Moreover, the H2-TPR profile of SD + SolidMix differs to some extent from SD + suspension although the NiO and La2O3 species were prepared for both cases in one step by spray drying. This could be attributed to the different preparation technique of introducing γ-Al2O3, physical mixture and ethanol for SolidMix and suspension, respectively.
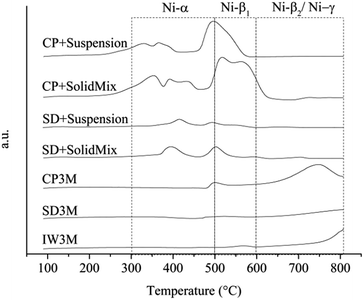 |
| | Fig. 1 H2-TPR profiles. | |
Table 3 H2-uptake of all seven fresh catalysts
| |
H2-uptake (mmol H2 gcat−1) |
| IW3M |
SD3M |
CP3M |
SD + SolidMix |
SD + suspension |
CP + SolidMix |
CP + suspension |
| Temperature (°C) |
300–500 |
0.49 |
0.28 |
0.20 |
1.82 |
1.56 |
3.47 |
3.14 |
| 500–600 |
0.39 |
0.23 |
0.50 |
0.47 |
0.32 |
4.02 |
1.87 |
| 600–800 |
4.58 |
1.78 |
4.41 |
0.04 |
0.13 |
0.28 |
0.16 |
| Total |
5.46 |
2.29 |
5.11 |
2.32 |
2.01 |
7.77 |
5.17 |
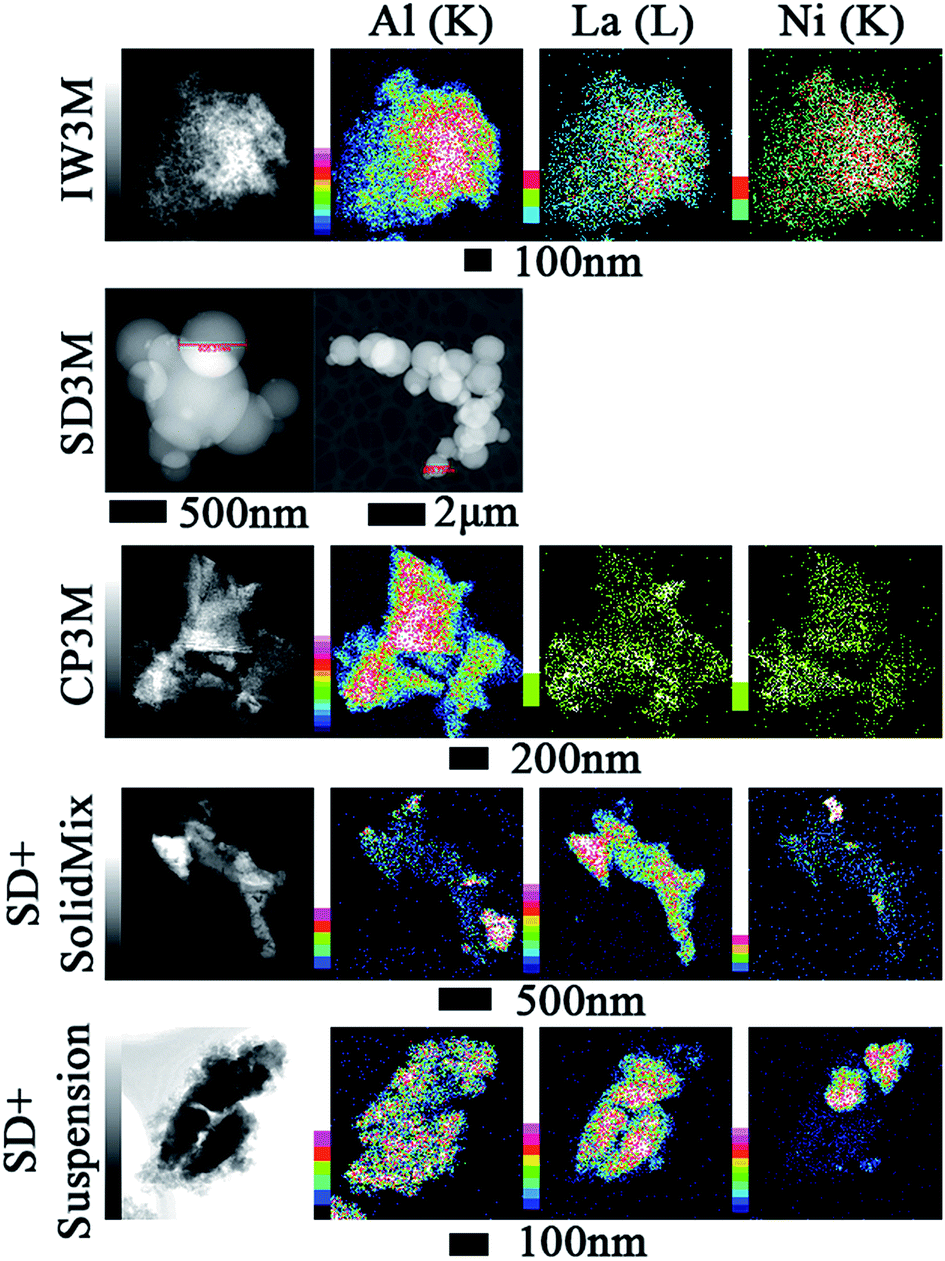
Elemental mapping (EDX) of the materials facilitated insights into element distribution. Fig. 2 provides an overview of Al, La and Ni for IW3M, CP3M, SD + SolidMix and SD + suspension, respectively. For most catalysts, Ni and La signals overlapped suggesting that La indeed acted as a spacer improving NiO dispersion. IW3M and CP3M exhibited a very good metal dispersion with no obvious nanoparticle formation for NiO. In contrast, for SD + SolidMix and SD + suspension, NiO nanoparticles of around 100 nm in size occurred. For SD3M only STEM images are provided, since only elemental line measurements were possible due to high charging and instability of the observed specimen (see ESI†). The utilized preparation technique of spray drying led to formation of spherical particles in the size range of 300 to 1500 nm with a good metal dispersion; NiO, La2O3 and Al2O3 were homogeneously dispersed throughout the formed spherical particles. Literature reports suggest that lower reducibility, namely strong metal–support interaction, could be a direct consequence of high metal dispersion and harsh calcination conditions (800 °C for 8 h).45 This is in good agreement with the H2-TPR profiles and EDX elemental mapping of the freshly calcined catalysts.
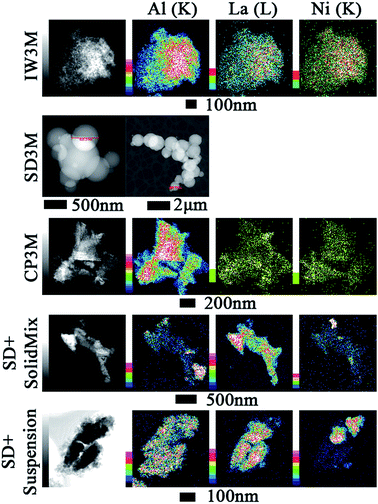 |
| | Fig. 2 EDX elemental mapping of the fresh calcined catalysts. Color scale, to the left of images, represents intensity of correspondent element characteristic X-ray line. | |
3.2. Dry reforming of methane
All materials were tested in DRM in an atmospheric fixed-bed reactor keeping the bed height constant. In order to account for the different catalyst weights, the reaction rates for CO2 and CH4 conversion were calculated. Conversion of CH4 for SD3M was close to equilibrium (>95%). Considering the approach to thermodynamic equilibrium for these values, the reaction rates of CH4 were corrected, by talking into account the reverse reaction as well:46,47| |
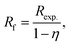 | (12) |
| |
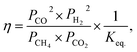 | (13) |
where, Rexp. is the net reaction rate based on unit catalyst weight (eqn (7)), Pi is the partial pressure of the i component and Keq. is the equilibrium constant (Keq. = 1577, 3 atm2 for the DRM under 1 atm at 800 °C (ref. 47)).
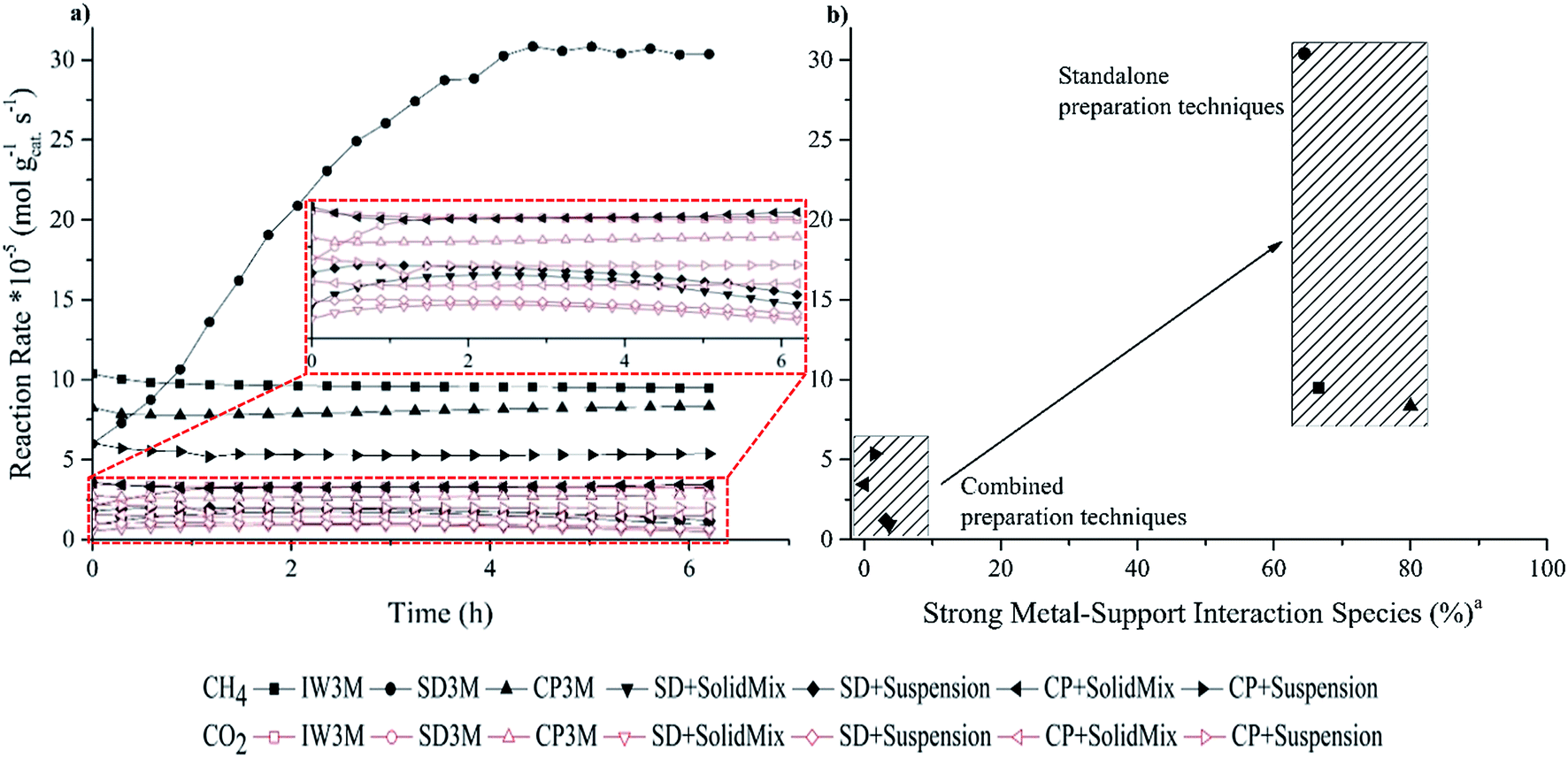
Fig. 3a shows reaction time courses. The standard error of the measurements (S.E.) was calculated to be 0.02 × 10−5 and 0.01 × 10−5 for the reaction rate of CO2 and CH4, respectively; the reaction rate for CH4 always exceeded the one of CO2 suggesting that reverse water–gas shift (RWGS) reaction (eqn (4)) was disfavored at the utilized reaction conditions (800 °C, CO2:CH4 = 2:1, 35000 h−1). Thermodynamic calculations, assuming ΔG° = 0, showed that 820 °C is the upper limiting temperature for RWGS and 640 °C the lower limiting for DRM.48 Comparing the yields of H2 and H2O derived from DRM and RWGS, respectively, at different temperatures it was found that the first was the dominant reaction at temperatures higher than 330 °C with a catalytic system that wasn't reported.49,50
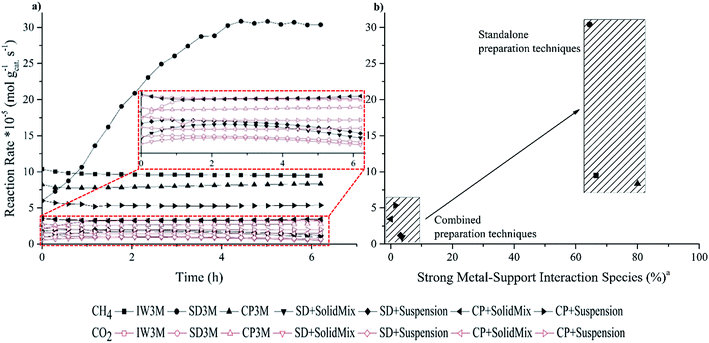 |
| | Fig. 3 (a) Reaction rates of CO2 and CH4 for the DRM (800 °C, CO2:CH4 = 2:1, 10% (v/v) N2, 35000 h−1) of all seven catalysts and (b) final reaction rates of CH4 at 6 h TOS related to the species with strong metal–support interaction. Standard errors (S.E.): S.E. (reaction rate of CH4) = 0.01 × 10−5 mol gcat−1 s−1 and S.E. (reaction rate of CO2) = 0.02 × 10−5 mol gcat−1 s−1. RCH4 of SD3M was corrected considering the approach to thermodynamic equilibrium. a . . | |
IW3M, SD3M and CP3M exhibited the best catalytic performance concerning both activity and stability throughout the 6 h time on stream (TOS). The final reaction rate of CH4 at 6 h TOS for SD3M was 30.8 × 10−5 mol gcat−1 s−1, surpassing the reaction rate of the other two by more than 3 times, 9.5 × 10−5 and 8.3 × 10−5 mol gcat−1 s−1 for IW3M and CP3M, respectively. In fact, IW3M and SD3M exhibited superior catalytic activity and stability compared to the other catalysts. However, IW3M deactivated over time, whereas SD3M showed stable activity for CO2 over the studied TOS and increasing activity over the first 4 h TOS for CH4. Concerning this initial activation of SD3M, incomplete reduction of the catalyst and subsequently further reduction by H2 derived from CH4 decomposition was ruled out, since the same observation was true for both 2 and 3 h of catalyst reduction before reaction. A possible explanation could be the structural change of the catalysts at 800 °C, revealing more active sites through Ni-nanoparticle migration, leading to higher activity. In the future, this finding will be further investigated. The four catalysts prepared by the combined techniques displayed inferior catalytic activity and stability. Focusing on the final reaction rates of CH4 at 6 h TOS, we identified the following ranking of all 7 catalysts:
SD3M > IW3M > CP3M > CP + suspension > CP + SolidMix > SD + suspension > SD + SolidMix.
Different preparation techniques have a strong influence on physico-chemical properties and performance of a catalyst.51,52 Mesoporous materials are commonly used due to their high surface area and pore volume as well as their uniform pore size leading to more accessible and exposed active sites.53 This comes in contradiction with our study, where the two worst performing catalysts, SD + SolidMix and SD + suspension, possessed the highest specific surface area with 127.4 and 138.1 m2 g−1, respectively. On the other hand, the specific surface area of SD3M was 14-fold lower than the latter with 9.8 m2 g−1, indicating that achieving high specific surface area was not crucial towards good catalytic performance. An explanation of the observed catalytic behavior of all studied catalysts can be their different reducibility. The first three catalysts, IW3M, SD3M and CP3M, displayed the lowest reducibility especially in the low temperature regions, which points at the very good dispersions of the active metal phase in these materials. This is in good agreement with the TEM investigations of the freshly calcined catalysts (Fig. 2) and literature reports suggesting that the stronger the metal–support interaction, the better the metal dispersion leading to enhanced activity and stability.45,54 However, the following contradictory point should be noted. Strong metal–support interactions causing low reducibility means that Ni2+ is not completely reduced to Ni0. The latter is considered to be the active site for DRM. Consequently, catalysts with less “reduced” active sites lead to higher activity. This is in line with literature reports, suggesting that mixture of Ni2+ and Ni0 could facilitate higher activity compared to samples only containing Ni0.45,55 The reaction rate of CO2 follows the same trend with the one of CH4 and varies from 0.5 to 3.5 × 10−5 mol gcat−1 s−1 among the different catalysts. It is reported that Ni0 formed by NiAl2O4 spinels, namely species with strong metal–support interaction, could accommodate the spill-over H derived by CH4 decomposition. With the assistance of spilt over H, CO2 dissociation rate could be enhanced leading to differences in CO2 activity.56
Despite identical calcination of all catalysts at 800 °C for 8 h, catalysts prepared by the combined techniques possessed significantly lower activity and stability. We relate this observation to phase separation between Ni- and La-oxides during this calcination step and subsequently to the Ni-nanoparticle size. With smaller nanoparticle size better activity and anti-coking properties were achieved.57–59 As confirmed by EDX mapping, individual domains of Ni-rich matter are easily distinguishable. These Ni-rich domains are reduced at lower temperatures (Fig. 1) and lead to formation of bigger agglomerates (Fig. 2) during DRM causing more pronounced coking. In line, higher reducibility suggests weaker metal–support interaction associated to the formation of bigger Ni nanoparticles due to sintering during DRM. The bigger the Ni nanoparticles the lower are the observed coke resistance and catalytic activity, respectively. In order to further point this relationship out, we plotted the final reaction rates of CH4 conversion over the amount of species with strong metal–support interaction (%) for all seven catalysts (Fig. 3b). The trend indicates that the higher the amount of strong metal–support interactions the higher catalytic activity. Two groups of catalyst preparation techniques can be distinguished: (a) standalone preparation techniques (IW3M, SD3M, CP3M) that exhibited superior catalytic results and (b) combined preparation techniques (SD + SolidMix, SD + suspension, CP + SolidMix, CP + suspension) with inferior catalytic results. In a comparative study between two Ce-promoted catalysts prepared by co-impregnation and sequential impregnation, the first was found to perform better towards DRM. This was attributed to closer interaction between promoter and active metal, increasing the redox properties of the catalyst.60 Similarly, for the catalysts prepared by the single preparation technique La, Ni and Al2O3 exhibited stronger interaction than the ones by the combined preparation techniques, leading to better activity and stability.
Selectivities of both H2 and CO (based on total carbon feed, CO2 + CH4) are presented in ESI.† The calculated standard error of the former is 0.17 and for the latter 0.35, respectively. Indeed, the selectivity of H2 varies significantly with the catalysts type. Catalysts prepared with combined preparation techniques exhibited the lowest values compared to IW3M, SD3M and CP3M. The difference in H2 selectivity could be attributed to RWGS and the Boudouard reaction. Reaction rate of CH4 was always higher than the one of CO2 (see ESI†). For the catalysts prepared by a single technique as well as SD + SolidMix and SD + suspension selectivity of hydrogen was higher than the one of CO, indicating that RWGS occurs at a greater extent than the Boudouard reaction. The opposite is suggested for CP + SolidMix and CP + suspension. For CO selectivity, all catalysts possessed stable values at around 50% with the only exception of SD + suspension, which started at 40.3%, increased slightly and then decreased to 42.4% at 6 h TOS. The utilized feed ratio of CO2:CH4 = 2:1 could provide a rationale for the stable value of CO selectivity at around 50% but further investigations are needed to better understand this observation.
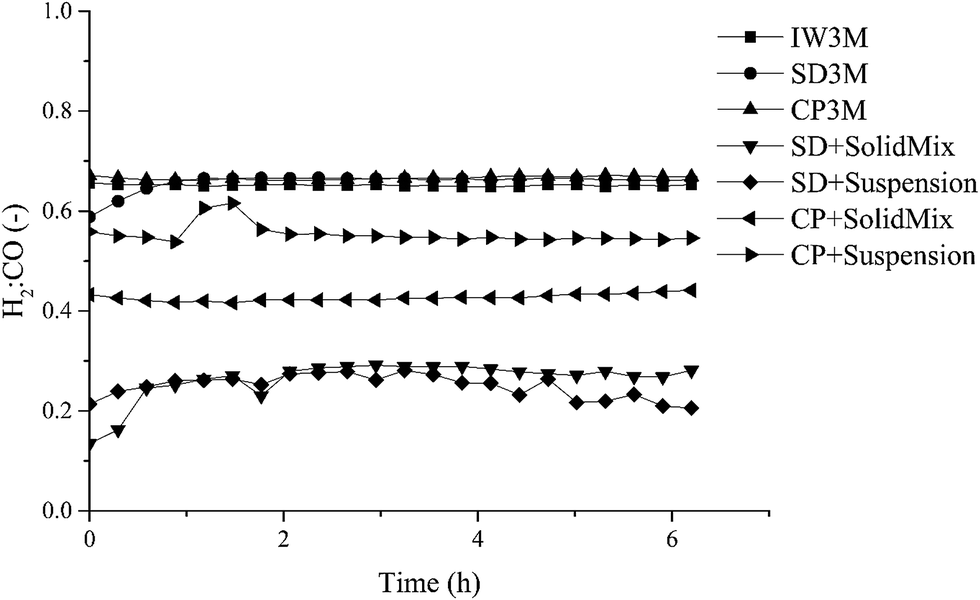
Focusing on syngas composition (H2:CO) (Fig. 4), catalysts prepared by the standalone techniques (IW3M, SD3M, CP3M) exhibited the highest values of ca. 0.65 as opposed to catalysts prepared by combined techniques (CP+*, SD+*). The latter showed relatively low values of the H2:CO ratio of about 0.20–0.55, indicating that the RWGS takes place at a higher extent. This is surprising, since RWGS is thermodynamically limited to 820 °C.48 Further studies will aim at an understanding of DRM and RWGS over these materials. H2:CO ratio in the absence of carbon deposits and C2+ should follow the formula;61
| |
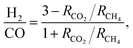 | (14) |
where
RCH4 and
RCO2 are the apparent rates of CH
4 and CO
2 conversion, respectively. For the single preparation technique catalysts, we found that the ratio based on stoichiometry was 3-fold greater than the experimental one. For the combined preparation technique catalysts, the stoichiometric-based ratio was 3.5- to 7-fold greater. The reason for this deviation from the stoichiometric ratio would be coke deposition which occurs on all catalysts.
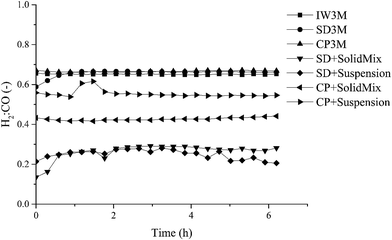 |
| | Fig. 4 H2:CO ratio of all seven catalysts. | |
3.3. Spent catalyst characterization
XRD analysis after catalytic reactions showed no change of crystallinity for IW3M, SD3M and CP3M compared to the fresh materials (Fig. 5). Interestingly, catalysts based on combined preparation techniques such as CP+* and SD+* exhibited lower crystallinity, especially of the γ-Al2O3 phase. This could be attributed to partial phase transition of γ-Al2O3 to α-Al2O3. In line, reflections related to γ-Al2O3 at 32.5° and 45.8° 2-theta possessed less intensity compared to the fresh materials. At the same time the reflex at 67.1° retained similar intensity suggesting a potential preferential orientation. Less intense NiAl2O4 reflections were found at 36.7° and 44.5° for all spent catalysts. A possible explanation can be the in situ reduction of NiAl2O4 domains to metallic Ni before reaction. It should be noted that no NiO reflections were identified for both fresh and spent catalysts although NiO species were detected by H2-TPR for all fresh catalysts. This indicates small NiO domains not detectable by XRD.
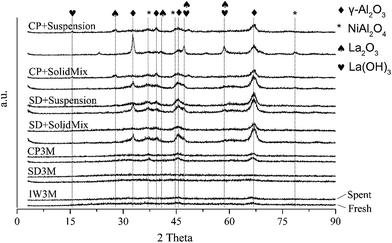 |
| | Fig. 5 XRD patterns of all fresh and spent catalysts. | |
TGA/DSC analyses in air enabled investigating carbon formation on all seven spent catalysts (Table 4 and see ESI†). Focusing on DSC, exothermic events corresponding to oxidation of different carbon species became evident at different temperatures. DSC signals at 300 to 500 °C and at 550 to 700 °C were related to amorphous and graphitic carbon oxidation, respectively.62–64
Table 4 TGA/DSC analysis of all seven spent catalysts
| |
IW3M |
SD3M |
CP3M |
SD + SolidMix |
SD + suspension |
CP + SolidMix |
CP + suspension |
| Temperature of DSC peaks. |
| Mass loss (%) |
3.9 |
2.7 |
14.4 |
6.4 |
6.0 |
0.1 |
5.1 |
| Temperature (°C)a |
300 |
650 |
300, 600 |
500 |
500 |
350 |
350, 600 |
| Carbon species |
Amorphous |
Graphitic |
Amorphous graphitic |
Amorphous |
Amorphous |
Amorphous |
Amorphous graphitic |
Firstly, amorphous carbon is formed and then, depending on the reaction conditions and the catalytic system, graphitization gradually occurs converting amorphous to graphitic carbon. All seven spent catalysts demonstrated the existence of at least some amount of amorphous carbon. The amount of graphitic carbon varies throughout the catalysts with CP3M exhibiting the highest amount of all seven spent catalysts. SD3M and SD + suspension presented moderate amounts, while the rest of the catalysts exhibited very low or no graphitic carbon. The weight loss due to carbon oxidation was also measured and translated to percentage of carbon deposits. The best performing catalysts, IW3M and SD3M, presented relatively low amounts of carbon at 3.9 and 2.7%, respectively. This is in good agreement with literature reports, suggesting that homogeneity of active sites, as it was confirmed by EDX elemental mapping, could sharply decrease carbon deposits.31 Carbon could be gasified either by O* derived by CO2 decomposition or by H2O produced by RWGS leading to less carbon deposits after reaction. The later is mainly met in steam reforming of methane but with the presence of the produced water during DRM, this is expected to lower the amount of carbon deposits for IW3M and SD3M.65,66
CP3M exhibited the highest amount of carbon of all seven spent catalysts with 14.4%. The catalysts with combined preparation techniques showed relatively low amounts of carbon deposits. CP + SolidMix had practically no carbon deposits, although it exhibited moderate reaction rates compared to the other catalysts indicating carbon free operation at the given reaction conditions. SD + SolidMix and SD + suspension possessed very low activity but relatively high amounts of coke deposits. A possible explanation could be that they started with higher activity that lead to coking and deactivated rapidly before sampling from the online GC.
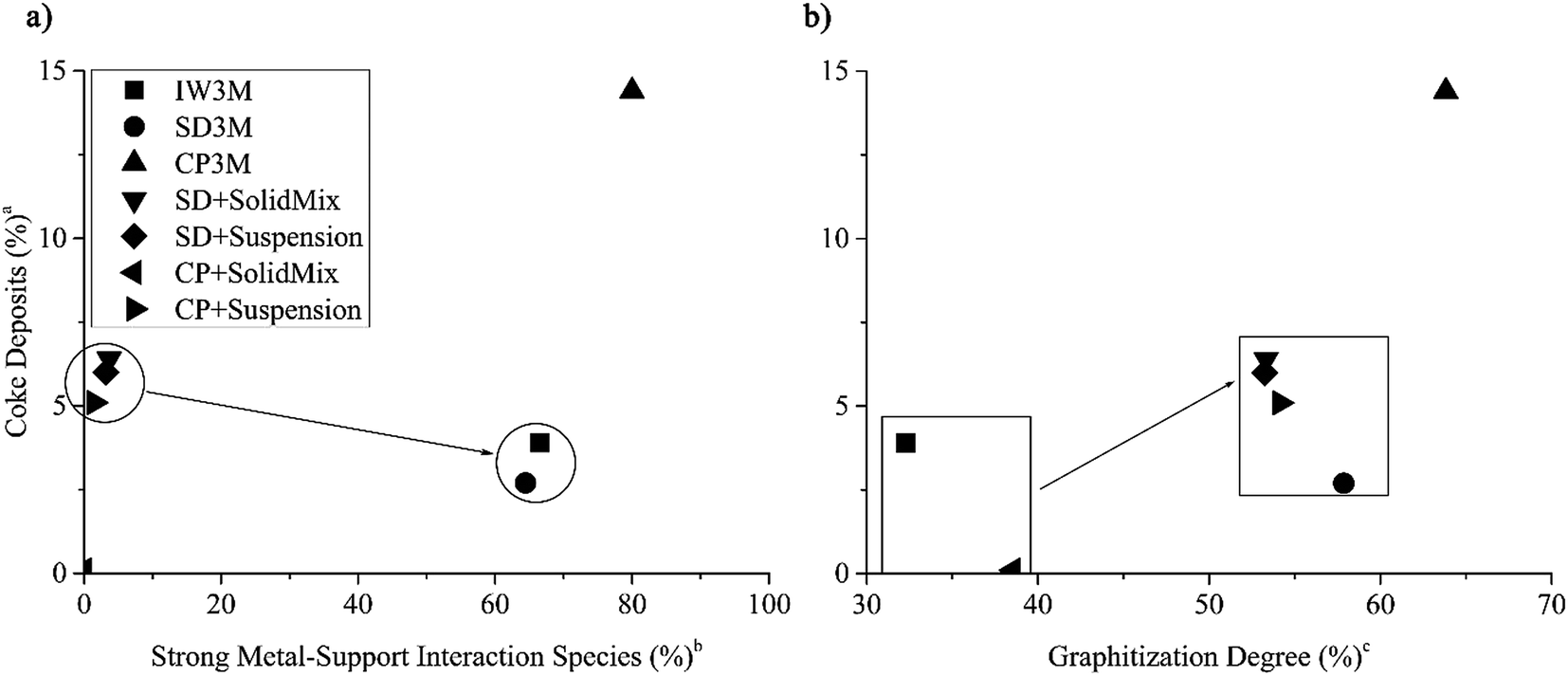
Further, investigating carbon deposits on all spent catalysts, we found that the higher the amount of strong metal–support interacting species on the fresh catalyst, found by H2-TPR analysis, the less the amount of coke deposits (%) (Fig. 6a). CP + SolidMix exhibited a minimal amount of coke deposits (%) as the reaction rates were also minimal. CP3M had at the same time significantly higher values of both. For the mentioned correlation, we considered these extreme points as outliers. Also, a trend of the nature of the carbon deposits could be identified; the higher the amount of coke deposits (%) the higher the graphitization degree (Fig. 6b). Again, CP3M was not considered for the correlation.
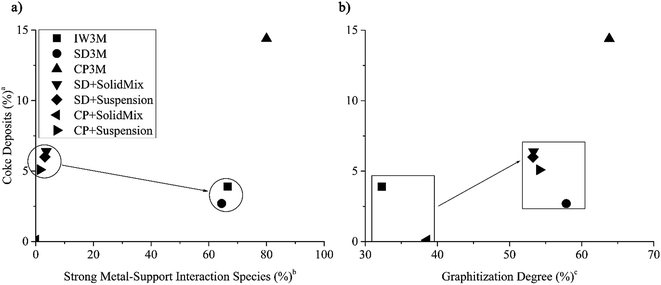 |
| | Fig. 6 (a) Amount of coke deposits in percentage in spent catalyst over the amount of species with strong metal–support interaction in the fresh catalyst in percentage and (b) amount of coke deposits in percentage over the graphitization degree in all seven spent catalysts. (CP3M has been excluded from the correlation). aCoke deposits (%) = 100%-TGA mass loss (%) at 1000 °C. b . c . c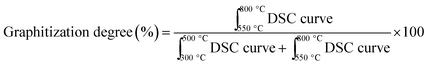 . . | |
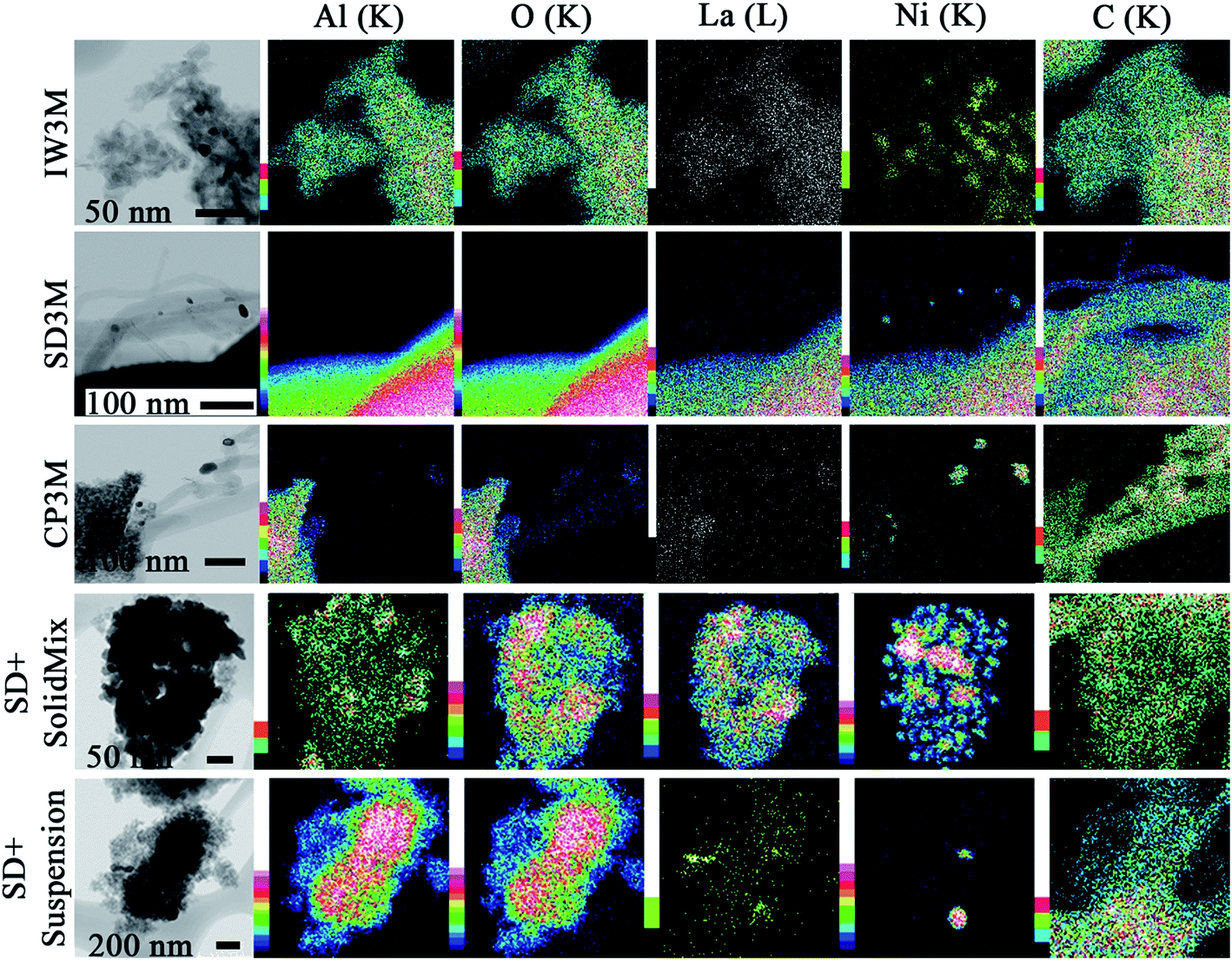
In Fig. 7 the results of the STEM-EDX elemental mapping are presented for five spent catalysts. It is clear that sintering occurs during DRM leading to bigger nanoparticles of Ni for IW3M and CP3M. For these two catalysts, the metal dispersion was very high with no distinguishable Ni particles before reaction. However, after reaction, we observed nanoparticles of 15–70 nm size. SD + SolidMix and SD + suspension displayed the same big Ni-rich domains as observed for the fresh catalysts. No carbon filaments formed for these two materials. Most probably, the nature of the preparation procedure played a crucial role for this behavior; lanthanum and nickel segregated during the first calcination step forming separate domains of NiO and La containing species. We assume that La is present in the form of La2O3. La2O3 in close proximity of the DRM active Ni-rich domains is known to possess high oxidation potential with respect to deposited surface carbon species.21 Therefore, no carbon nanotubes could be observed for SD + SolidMix and SD + suspension. In practice, for the mixed preparations, we obtained La2O3-supported Ni catalysts, where the third phase, γ-Al2O3, is considered to be only a spectator. SD3M exhibited no visible large scale sintering; only some small Ni particles (less than 10 nm in size) were found on the tips of the carbon filaments (Fig. 7). These particles are most probably detached from the support during reaction, suggesting that some small scale phase segregation occurred. This observation is in line with the suggested growth mechanism of carbon nanotubes, where carbon, derived mainly from CH4 dissociation, migrates through the Ni nanoparticle to precipitate at the nickel–support interface. This interfacial carbon built-up pushes the isolated Ni-particles away from the support leading eventually to its deactivation.67 Focusing on the carbon species formed, the STEM results are in reasonable agreement with the ones from TGA/DSC, where for CP3M and SD3M (the catalysts with the highest graphitization degree) carbon nanotubes were found. For the rest of the catalysts with low graphitization degree no carbon nanotubes were observed.
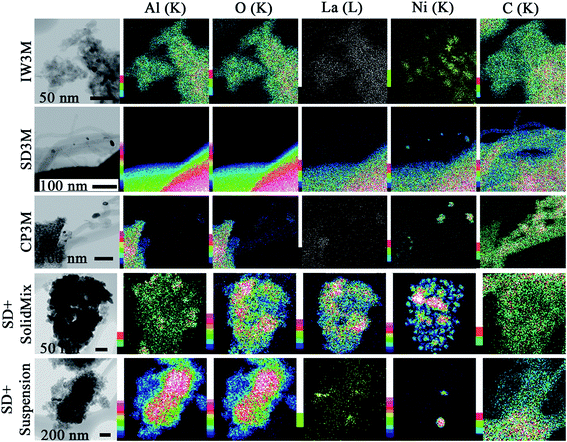 |
| | Fig. 7 EDX elemental mapping of the spent catalysts. Color scale, to the left of images, represents intensity of correspondent element characteristic X-ray line. | |
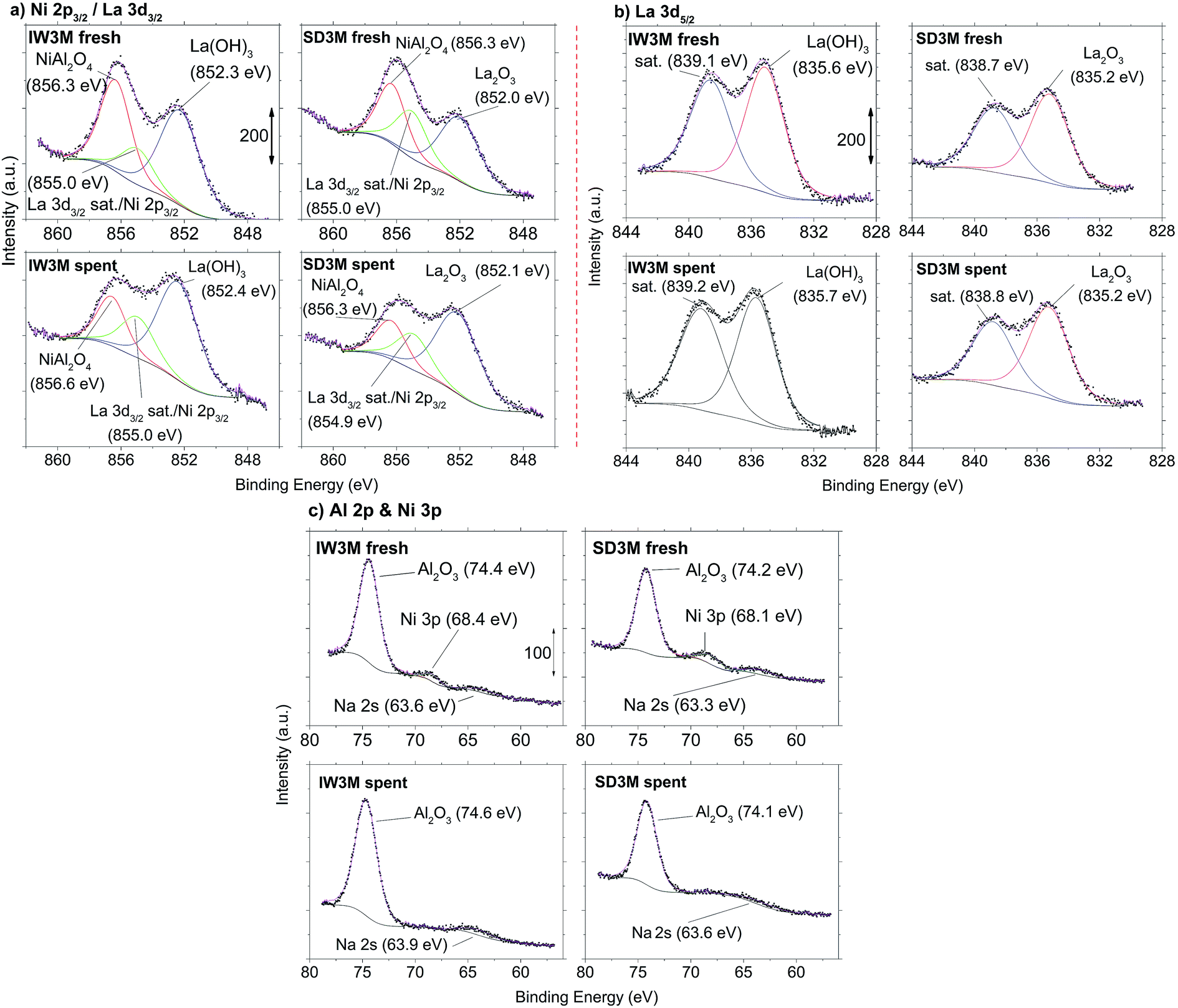
The surface chemistry of IW3M and SD3M, the two best performing catalysts, was investigated for both, fresh and spent catalysts by means of XPS analysis (Fig. 8). The most significant difference in surface chemistry between the two catalysts lies in the chemical state of La which was probed by the La 3d5/2 signal (Fig. 8b). Deconvoluted signals of IW3M, for both fresh and spent catalysts, are located at 835.6 eV and attributed to the formation of La(OH)3.68 In contrast, the main La 3d5/2-signal of the fresh and spent SD3M catalyst is positioned at 0.4 eV lower binding energy consistent with the formation of La2O3.68 The Ni 2p3/2 signal (Fig. 8a) overlaps with the La 3d3/2 signal. However, peaks at 856.3 and 856.6 eV clearly indicate the formation of NiAl2O4 for both the fresh and spent catalysts,69–72 while a signals 854.9–855.0 eV may originate from the La 3d3/2 satellite signal68 and/or the Ni 2p3/2-signal which could indicate the formation of NiO.70–72 While for all examined powders the XPS analysis remains inconclusive regarding the potential formation of NiO due to the overlap with the La satellite signal, the formation of NiAl2O4 is evident. Furthermore, the intensity of signals associated with the presence of NiAl2O4 is clearly reduced for the spent catalysts as compared to the fresh state. The Ni 3p signal detected at 68.4 eV and 68.1 eV (Fig. 8c) for the fresh catalysts IW3M and SD3M cannot be detected in the spent state for both materials, indicating a Ni depletion consistent with the concomitant reduction in signal associated with the presence of NiAl2O4. Finally, the Al 2p signals (Fig. 8c) detected at 74.1–74.6 eV for all samples investigated are attributed to the formation of Al2O3.69,72,73 It is reasonable to assume that the detected Na was incorporated during storage of the sample in air prior to the XPS analysis.
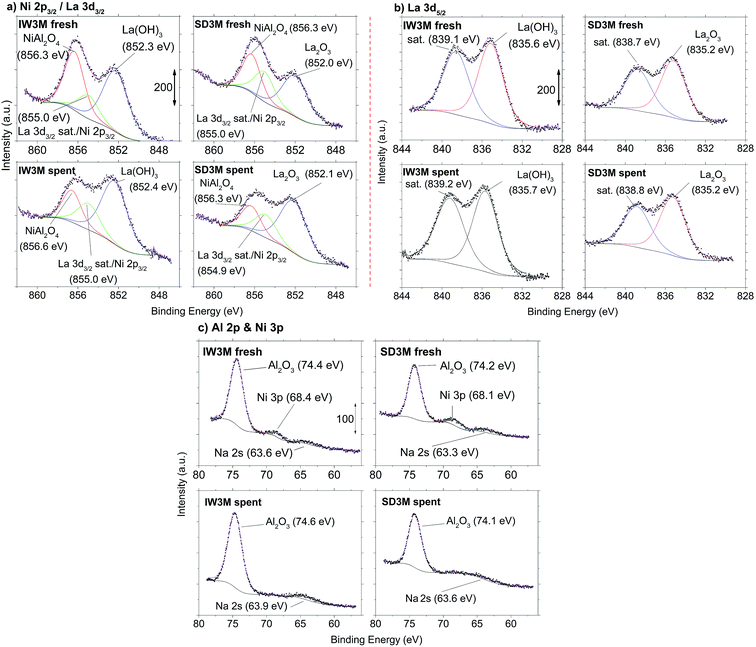 |
| | Fig. 8 XPS spectra of IW3M and SD3M prior (fresh) and after reaction (spent). (a) Ni 2p3/2- and La 3d3/2-signals (b) La 3d5/2-signals and (c) Al 2p- and Ni 3p-signals. | |
4. Summary and conclusion
La promoted Ni/γ-Al2O3 catalysts were synthesized by seven different preparation techniques. All catalysts were tested in 6 h catalytic experiments of methane dry reforming (DRM). Additionally, the materials were extensively characterised before and after reaction. Three catalysts were prepared in a single step with all constituents present: incipient wetness impregnation, spray drying, co-precipitation denoted as IW3M, SD3M and CP3M, respectively. Four other catalysts were synthesized by combined techniques: spray drying-physical mixture, spray drying-ethanol suspension, coprecipitation–physical mixture and coprecipitation–ethanol suspension, denoted as SD + SolidMix, SD + suspension, CP + SolidMix and CP + suspension, respectively. The total surface area of all catalysts measured by the BET method was in the same order of magnitude with the exception of SD3M which displayed much lower surface area. XRD patterns showed overall low crystallinity for all catalysts, despite calcination at 800 °C. Additionally, XRD analyses confirmed the presence of γ-Al2O3 and NiO for almost all calcined catalysts, and metallic Ni for some of the spent materials. Interestingly, lanthana was not found by XRD, suggesting a high dispersion of this phase. Most probably the same argument explains the difficulty to clearly distinguish the Ni-containing phases for some catalysts. H2-TPR profiles revealed generally four Ni species with low, moderate and strong metal–support interactions. IW3M, SD3M and CP3M exhibited mainly Ni species with strong metal–support interaction facilitating very good metal dispersions, with SD3M exhibiting the highest amount of strongly interacting Ni species. Catalytic results revealed that single-step preparation techniques provided catalysts exhibiting better reaction rates for both CO2 and CH4 conversions. SD3M showed superior activity and stability even outperforming IW3M. Selectivity to CO was stable at around 50% for all seven catalysts except SD + suspension displaying much lower CO selectivity. On the other hand, H2 selectivities were quite different among the examined catalysts, with SD3M displaying the highest. XRD analysis of the spent catalysts demonstrated an overall decrease of crystallinity after catalysis. Investigating further the carbon deposits formed, TGA/DSC analysis revealed different level of graphitization for all catalysts after reaction, with CP3M demonstrating the highest amount of graphitic and total carbon. Additionally, the presence of NiAl2O4 on the catalysts surface could be identified by XPS, while depletion of surface Ni was observed for spent catalysts. EDX elemental mapping of the spent catalysts showed that sintering occurred for IW3M and CP3M. SD3M retained its excellent metal dispersion after reaction, whereas only some small nanoparticles of Ni were detached from the support due to carbon nanotube growth. It exhibited the best catalytic results for DRM, addressing sintering which is considered to be one of the main deactivation mechanisms.
Catalyst prepared by spray drying, namely SD3M, showed superior catalytic results irrespective of its low specific surface area compared to materials prepared via more conventional techniques. This superior performance can be attributed to excellent Ni dispersion and strong metal–support interactions leading to very low formation of carbon deposits. These properties also facilitate very stable catalytic activity. In line, catalyst morphology remains intact in the course of the reaction for 6 h time-on-stream in DRM. Overall, spray drying appears promising to further improve the performance of catalysts for DRM. Future studies will focus on optimized spray drying conditions aiming for even higher nickel dispersion, further enhanced metal–support interactions and materials possessing high specific surface areas.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank K. Vaeβen for XRD, physisorption and TGA/DSC measurements. We acknowledge financial support by the project house Power2Fuel of RWTH Aachen University funded by the Excellence Initiative of the German federal and state government to promote science and research at German universities.
References
- J. R. H. Ross, Catal. Today, 2005, 100, 151–158 CrossRef CAS.
- M. Sohrabi, N. I. Kechut, M. Riazi, M. Jamiolahmady, S. Ireland and G. Robertson, Transp. Porous Media, 2011, 91, 101–121 CrossRef.
- H. Lei, S. Yang, L. Zu, Z. Wang and Y. Li, Energy Fuels, 2016, 30, 8922–8931 CrossRef CAS.
- M. Younas, M. Sohail, L. K. Leong, M. J. Bashir and S. Sumathi, Int. J. Environ. Sci. Technol., 2016, 13, 1839–1860 CrossRef CAS.
- J. H. Bitter, K. Seshan and J. A. Lercher, Top. Catal., 2000, 10, 295–305 CrossRef CAS.
- J. Galuszka, R. N. Pandey and S. Ahmed, Catal. Today, 1998, 46, 83–89 CrossRef CAS.
- M.-S. Fan, A. Z. Abdullah and S. Bhatia, ChemCatChem, 2009, 1, 192–208 CrossRef CAS.
- S. Kawi, Y. Kathiraser, J. Ni, U. Oemar, Z. Li and E. T. Saw, ChemSusChem, 2015, 8, 3556–3575 CrossRef CAS PubMed.
- S. Li and J. Gong, Chem. Soc. Rev., 2014, 43, 7245–7256 RSC.
- C. Carrara, J. Múnera, E. A. Lombardo and L. M. Cornaglia, Top. Catal., 2008, 51, 98–106 CrossRef CAS.
- P. Ferreira-Aparicio, C. Márquez-Alvarez, I. Rodríguez-Ramos, Y. Schuurman, A. Guerrero-Ruiz and C. Mirodatos, J. Catal., 1999, 184, 202–212 CrossRef CAS.
- J. Kehres, J. G. Jakobsen, J. W. Andreasen, J. B. Wagner, H. Liu, A. Molenbroek, J. Sehested, I. Chorkendorff and T. Vegge, J. Phys. Chem. C, 2012, 116, 21407–21415 CAS.
- K. Sutthiumporn and S. Kawi, Int. J. Hydrogen Energy, 2011, 36, 14435–14446 CrossRef CAS.
- Z. L. Zhang, X. E. Verykios, S. M. MacDonald and S. Affrossman, J. Phys. Chem., 1996, 100, 744–754 CrossRef CAS.
- P. Chen, Z. Hou, X. Zheng and T. Yashima, React. Kinet. Catal. Lett., 2005, 86, 51–58 CrossRef CAS.
- Y. K. Han, C.-I. Ahn, J.-W. Bae, A. R. Kim and G. Y. Han, Ind. Eng. Chem. Res., 2013, 52, 13288–13296 CrossRef CAS.
- Z. Xu, Y. M. Li, J. Y. Zhang, L. Chang, R. Q. Zhou and Z. T. Duan, Appl. Catal., A, 2001, 210, 45–53 CrossRef CAS.
- J. Zhang, H. Wang and A. K. Dalai, J. Catal., 2007, 249, 300–310 CrossRef CAS.
- G. S. Gallego, C. Batiot-Dupeyrat, J. Barrault, E. Florez and F. Mondragon, Appl. Catal., A, 2008, 334, 251–258 CrossRef CAS.
- G. S. Gallego, F. Mondragón, J. Barrault, J.-M. Tatibouët and C. Batiot-Dupeyrat, Appl. Catal., A, 2006, 311, 164–171 CrossRef CAS.
- J.-K. Xu, K.-W. Ren, X.-L. Wang, W. Zhou, X.-M. Pan and J.-X. Ma, Acta Phys.-Chim. Sin., 2008, 24, 1568–1572 CAS.
- S. Damyanova and J. M. C. Bueno, Appl. Catal., A, 2003, 253, 135–150 CrossRef CAS.
- M. Schmal, M. M. V. M. Souza, V. V. Alegre, M. A. P. da Silva, D. V. César and C. A. C. Perez, Catal. Today, 2006, 118, 392–401 CrossRef CAS.
- H.-S. Roh, H. S. Potdar and K.-W. Jun, Catal. Today, 2004, 93–95, 39–44 CrossRef CAS.
- R. Martínez, E. Romero, C. Guimon and R. Bilbao, Appl. Catal., A, 2004, 274, 139–149 CrossRef.
- Å. Slagtern, U. Olsbye, R. Blom, I. M. Dahl and H. Fjellvåg, Appl. Catal., A, 1997, 165, 379–390 CrossRef.
- B. S. Liu and C. T. Au, Appl. Catal., A, 2003, 244, 181–195 CrossRef CAS.
- X. E. Verykios, Int. J. Hydrogen Energy, 2003, 28, 1045–1063 CAS.
- S. Tang, L. Ji, J. Lin, H. C. Zeng, K. L. Tan and K. Li, J. Catal., 2000, 194, 424–430 CrossRef CAS.
- H. Y. Wang and E. Ruckenstein, Carbon, 2002, 40, 1911–1917 CrossRef CAS.
- J.-g. Wang, C.-j. Liu, Y.-p. Zhang, K.-l. Yu, X.-l. Zhu and F. He, Catal. Today, 2004, 89, 183–191 CrossRef CAS.
- H. A. Aleksandrov, N. Pegios, R. Palkovits, K. Simeonov and G. N. Vayssilov, Catal. Sci. Technol., 2017, 7(15), 3339–3347 CAS.
- Y.-x. Pan, C.-j. Liu, T. S. Wiltowski and Q. Ge, Catal. Today, 2009, 147, 68–76 CrossRef CAS.
- R. Zhao, S. J. Lee, I. H. Son, H. Lee and A. Soon, Appl. Surf. Sci., 2013, 265, 339–345 CrossRef CAS.
- K. Mette, S. Kühl, H. Düdder, K. Kähler, A. Tarasov, M. Muhler and M. Behrens, ChemCatChem, 2014, 6, 100–104 CrossRef CAS.
- J. W. Han, C. Kim, J. S. Park and H. Lee, ChemSusChem, 2014, 7, 451–456 CrossRef CAS PubMed.
- B. A. Latella and B. H. O'Connor, J. Am. Ceram. Soc., 1997, 80, 2941–2944 CrossRef CAS.
- T. C. Chou, D. Adamson, J. Mardinly and T. G. Nieh, Thin Solid Films, 1991, 205, 131–139 CrossRef CAS.
- S. M. de Lima, A. M. da Silva, L. O. O. da Costa, J. M. Assaf, G. Jacobs, B. H. Davis, L. V. Mattos and F. B. Noronha, Appl. Catal., A, 2010, 377, 181–190 CrossRef CAS.
- F. Bentaleb and E. Marceau, Microporous Mesoporous Mater., 2012, 156, 40–44 CrossRef CAS.
- J. M. Rynkowski, T. Paryjczak and M. Lenik, Appl. Catal., A, 1993, 106, 73–82 CrossRef CAS.
- R. Yang, X. Li, J. Wu, X. Zhang, Z. Zhang, Y. Cheng and J. Guo, Appl. Catal., A, 2009, 368, 105–112 CrossRef CAS.
- B. Valle, B. Aramburu, A. Remiro, J. Bilbao and A. G. Gayubo, Appl. Catal., B, 2014, 147, 402–410 CrossRef CAS.
- X. Wang, Y. Wei, J. Zhang, H. Xu and W. Li, React. Kinet. Catal. Lett., 2006, 89, 97–104 CrossRef CAS.
- S. Damyanova, B. Pawelec, K. Arishtirova and J. L. G. Fierro, Int. J. Hydrogen Energy, 2012, 37, 15966–15975 CrossRef CAS.
- J. Wei and E. Iglesia, J. Catal., 2004, 225, 116–127 CrossRef CAS.
- L. N. Bobrova, A. S. Bobin, N. V. Mezentseva, V. A. Sadykov, J. W. Thybaut and G. B. Marin, Appl. Catal., B, 2016, 182, 513–524 CrossRef CAS.
- S. Wang, G. Lu and G. J. Millar, Energy Fuels, 1996, 10, 896–904 CrossRef CAS.
- M. C. Bradford and M. A. Vannice, J. Catal., 1998, 173, 157–171 CrossRef CAS.
- M. C. J. Bradford and M. A. Vannice, Catal. Rev.: Sci. Eng., 1999, 41, 1–42 CAS.
- F. Menegazzo, M. Signoretto, F. Pinna, P. Canton and N. Pernicone, Appl. Catal., A, 2012, 439, 80–87 CrossRef.
- D.-W. Jeong, W.-J. Jang, J.-O. Shim, H.-S. Roh, I. H. Son and S. J. Lee, Int. J. Hydrogen Energy, 2013, 38, 13649–13654 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- Y.-g. Chen and J. Ren, Catal. Lett., 1994, 29, 39–48 CrossRef CAS.
- R. Wang, H. Xu, X. Liu, Q. Ge and W. Li, Appl. Catal., A, 2006, 305, 204–210 CrossRef CAS.
- Z. X. Cheng, X. G. Zhao, J. L. Li and Q. M. Zhu, Appl. Catal., A, 2001, 205, 31–36 CrossRef CAS.
- A. G. Bhavani, W. Y. Kim, J. Y. Kim and J. S. Lee, Appl. Catal., A, 2013, 450, 63–72 CrossRef CAS.
- L. Sun, Y. Tan, Q. Zhang, H. Xie, F. Song and Y. Han, Int. J. Hydrogen Energy, 2013, 38, 1892–1900 CrossRef CAS.
- V. Y. Bychkov, Y. P. Tyulenin, A. A. Firsova, E. A. Shafranovsky, A. Y. Gorenberg and V. N. Korchak, Appl. Catal., A, 2013, 453, 71–79 CrossRef CAS.
- Ş. Özkara-Aydınoğlu, E. Özensoy and A. E. Aksoylu, Int. J. Hydrogen Energy, 2009, 34, 9711–9722 CrossRef.
- M. C. J. Bradford and M. A. Vannice, J. Catal., 1999, 183, 69–75 CrossRef CAS.
- S. de Llobet, J. L. Pinilla, R. Moliner and I. Suelves, Fuel, 2015, 139, 71–78 CrossRef CAS.
- L. Mo, K. K. M. Leong and S. Kawi, Catal. Sci. Technol., 2014, 4, 2107–2114 CAS.
- X. Yu, N. Wang, W. Chu and M. Liu, Chem. Eng. J., 2012, 209, 623–632 CrossRef CAS.
- S. Wang, G. Q. Lu and G. J. Millar, Energy Fuels, 1996, 10, 896–904 CrossRef CAS.
- L. Maier, B. Schädel, K. Herrera Delgado, S. Tischer and O. Deutschmann, Top. Catal., 2011, 54, 845 CrossRef CAS.
- H. Liu, C. Guan, X. Li, L. Cheng, J. Zhao, N. Xue and W. Ding, ChemCatChem, 2013, 5, 3904–3909 CrossRef CAS.
- M. Sunding, K. Hadidi, S. Diplas, O. Løvvik, T. Norby and A. Gunnæs, J. Electron Spectrosc. Relat. Phenom., 2011, 184, 399–409 CrossRef CAS.
- P. Bolt, E. Ten Grotenhuis, J. Geus and F. Habraken, Surf. Sci., 1995, 329, 227–240 CrossRef CAS.
- S. Velu, K. Suzuki, M. Vijayaraj, S. Barman and C. S. Gopinath, Appl. Catal., B, 2005, 55, 287–299 CrossRef CAS.
- I. Pettiti, D. Gazzoli, P. Benito, G. Fornasari and A. Vaccari, RSC Adv., 2015, 5, 82282–82291 RSC.
- N. Pegios, G. Schroer, K. Rahimi, R. Palkovits and K. Simeonov, Catal. Sci. Technol., 2016, 6, 6372–6380 CAS.
- M. Giza, P. Thissen and G. Grundmeier, Langmuir, 2008, 24, 8688–8694 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06570a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
K. Simeonova
*a and
K. Simeonova
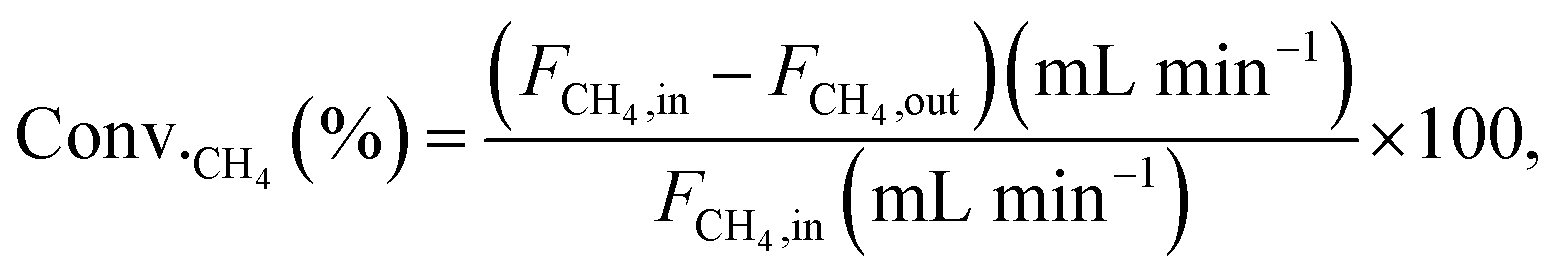
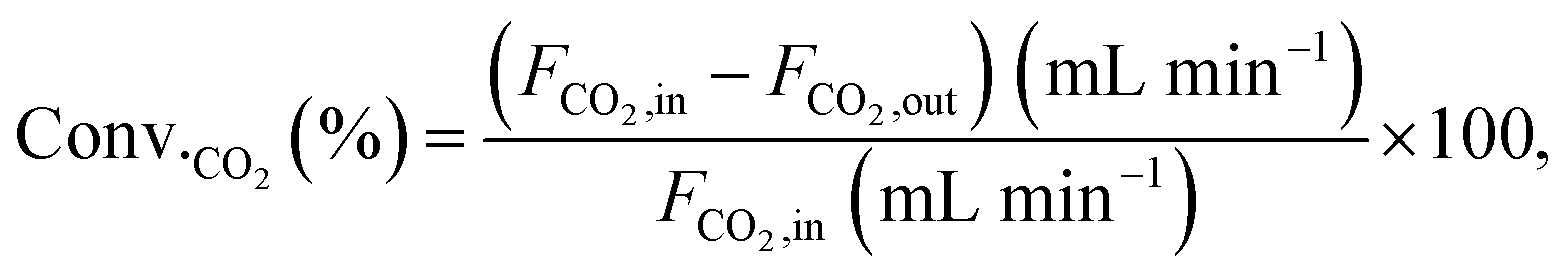
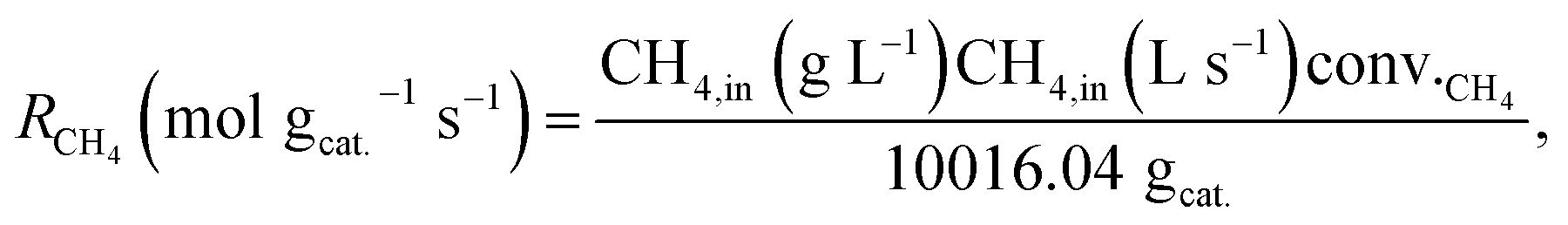
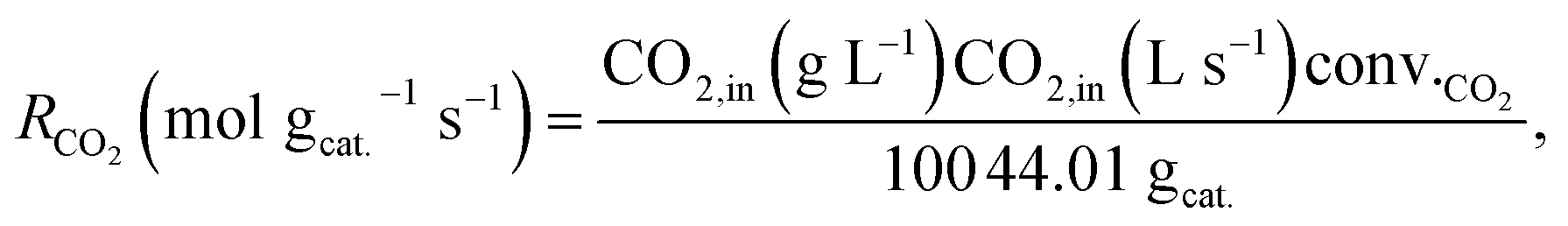
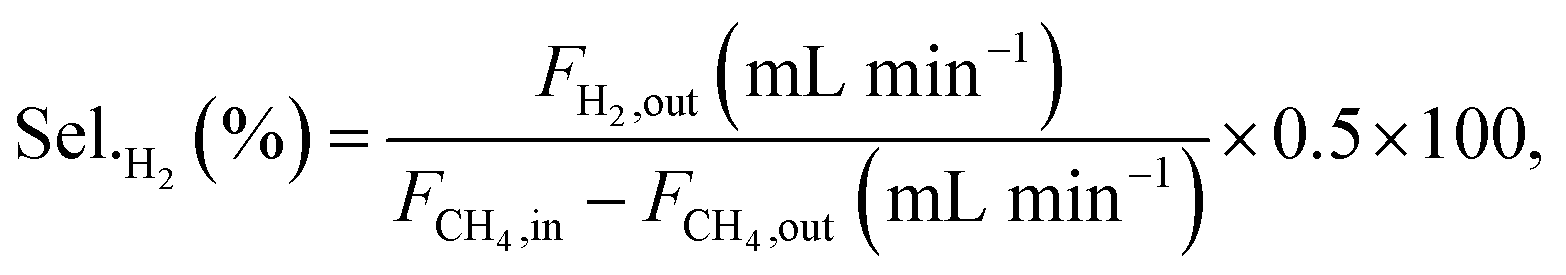
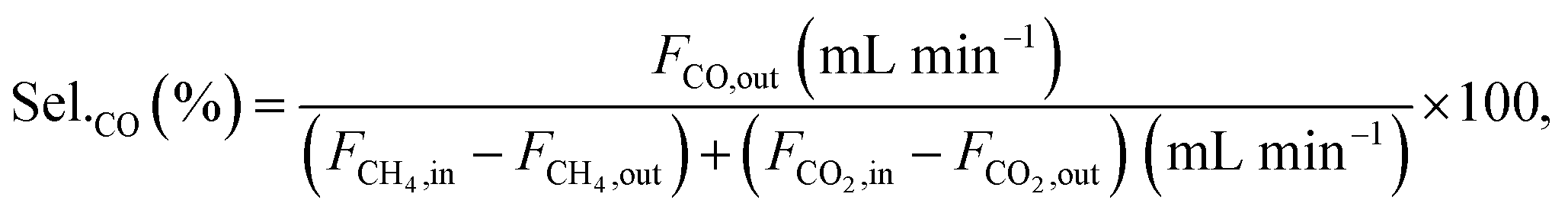
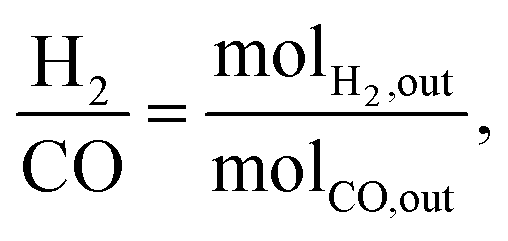

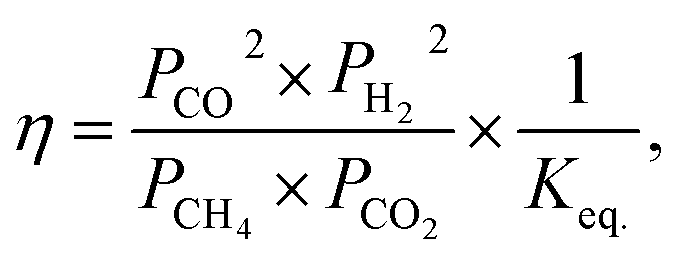
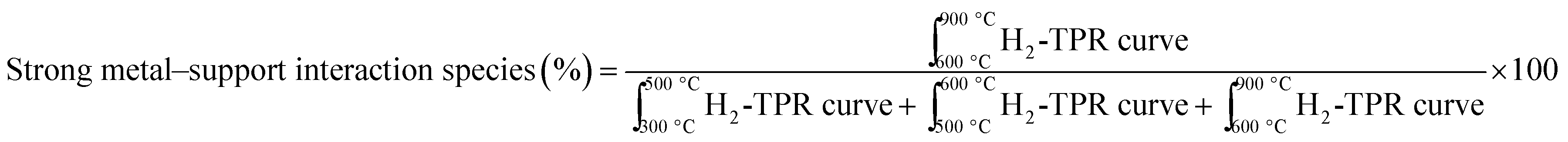 .
.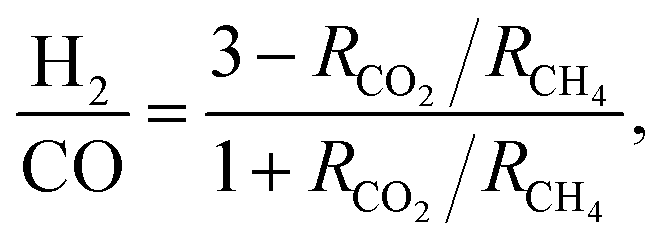
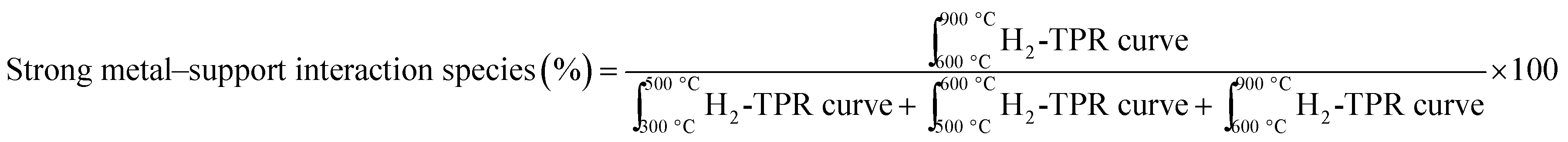 . c
. c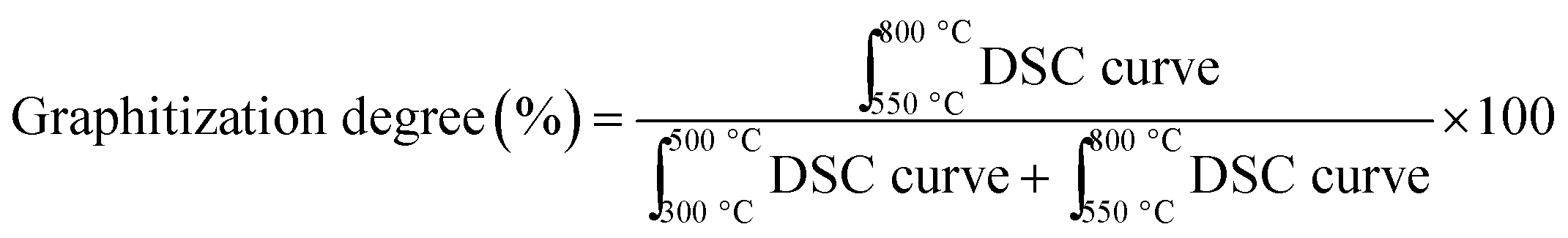 .
.