
Asymmetric synthesis of 3-aminodihydrocoumarins via the chiral guanidine catalyzed cascade reaction of azlactones†
Sai
Ruan
 ,
Xiaobin
Lin
,
Lihua
Xie
,
Lili
Lin
,
Xiaoming
Feng
and
Xiaohua
Liu
*
,
Xiaobin
Lin
,
Lihua
Xie
,
Lili
Lin
,
Xiaoming
Feng
and
Xiaohua
Liu
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, People's Republic of China. E-mail: liuxh@scu.edu.cn
First published on 18th September 2017
Abstract
A highly efficient bifunctional guanidine catalyst was developed for the asymmetric cascade reaction between 2-nitrovinylphenols and azlactones. A wide variety of 3-aminodihydrocoumarin derivatives could be obtained in high yields (up to 99% yield) with excellent enantioselectivities (up to 99% ee) and diastereoselectivities (>19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr).
:1 dr).
Introduction
The asymmetric catalytic synthesis of substituted dihydrocoumarins has particular significance in modern organic chemistry.1 This is attributed to the fact that this functional structural motif frequently occurs in biologically and pharmacologically active compounds as well as natural products.2 During the past few years, many methods have been reported to generate these valuable molecules.3 For example, 3-aminodihydrocoumarin derivatives are used as antihypertensive agents and platelet aggregation inhibitors.4 New methods based on the reactions of alkylideneoxazolones5 and azlactones5e,6,7 are the representative routes for the construction of 3-aminodihydrocoumarins (Scheme 1). A chiral thiourea catalyzed cascade reaction between o-hydroxy aromatic aldimine A and alkylidene azlactones yielded chromeno[4,3-b]pyrrolidines (eqn (1)).5b A tandem Friedel–Crafts/transesterification process with phenol B gave access to aryl(benzoylamino)chromanones albeit in racemic version (eqn (2)).5c The formal asymmetric [4 + 2] cycloaddition of azlactones with o-quinone methides C generated from various precursors enabled the formation of 4-aryl/alkyl substituted aminochromanones, which has been realized separately by several groups (eqn (3)).7a–d In addition, enantioselective organocatalyzed nucleophilic addition/transesterification cascade reactions8 of azlactones with 2-hydroxyphenyl substituted electrophiles D benefited the formation of various 4-functionalized 3-aminochromanones (eqn (4)).7e,f Albrecht7g and Wang7h developed the diastereo- and enantioselective cascade reaction between o-hydroxychalcones and azlactones using cinchona-alkaloid-based squaramide or thiourea as the catalyst, respectively. Zhou and coworkers used L-tert-leucine derived squaramide for the related reaction of 2-(2-nitrovinyl)phenols7e to obtain 4-nitromethyl substituted chroman-2-ones. Excellent yield and stereoselectivity were achieved but the reaction is limited in the scope of azlactones.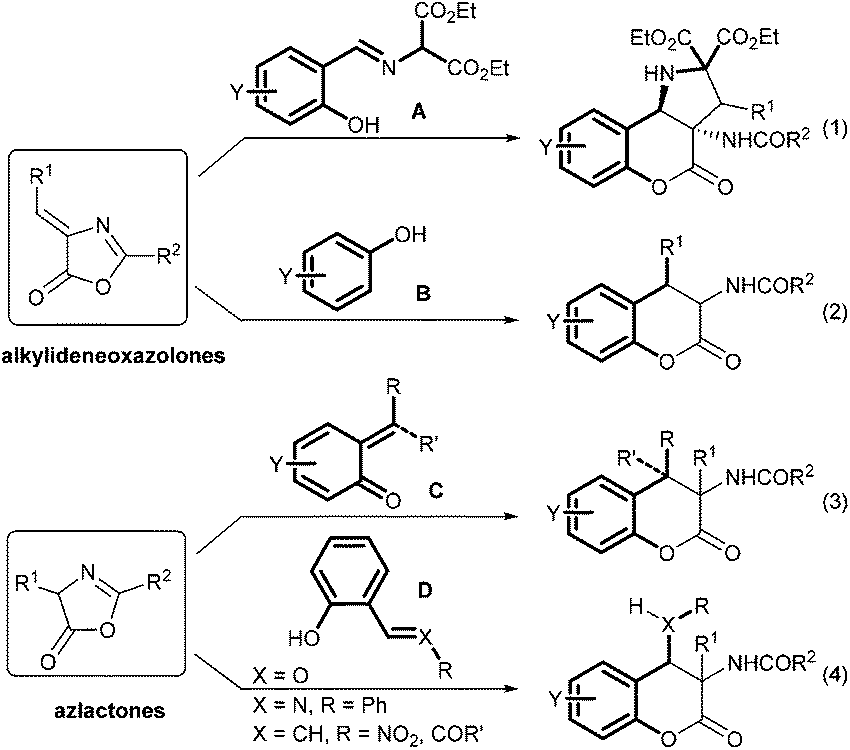 | ||
| Scheme 1 Representative synthesis routes for 3-aminodihydrocoumarin derivatives. | ||
In our efforts to develop new chiral bifunctional guanidine catalysts,9,10 we succeeded in several asymmetric transformations of azlactones, including a cascade reaction with o-hydroxy aldimines to obtain 3,4-diamino chroman-2-ones.7f Thus we sought to exploit chiral guanidine in the cascade reaction for the generation of 4-nitromethyl 3-aminochromanones. Herein, we report the highly diastereo- and enantioselective conjugate addition/lactonization between various 2-nitrovinylphenols11 and azlactones.
Results and discussion
In our initial research, we chose (E)-2-(2-nitrovinyl)phenol 1a and azlactone 2a as the model substrates, and the reaction was investigated at −30 °C in ethyl acetate with various chiral guanidine organocatalysts (Table 1). Firstly, bisguanidine BG-1 and its hemi-salt BG-1·HBArF47f,10c,d which were proved to be efficient for several asymmetric transformations of azlactones were used for the current cascade reaction. It was found that these two catalysts could promote the reaction to give the desired 3-aminodihydrocoumarin 3aa but with poor diastereo- and enantioselectivities (entries 1 and 2). Nevertheless, other bifunctional guanidines G-1 to G-5 which were derived from chiral amino acids and 1,2-diphenylethane-1,2-diamine but embellishing a sulfonamide unit showed excellent reactivity (94–99% yield) and diastereoselectivity (>19:1) (entries 3–7). The amino acid backbone had an obvious influence on the enantioselectivity of the reaction, and tetrahydroisoquinoline-based G-4 and G-5 gave higher results than guanidines G-1–G-3 derived from L-proline, L-pipecolic acid, and L-ramipril subsequently.12 Further adjustment of the sulfonamide substituent indicated that the installation of the 2,6-difluorobenzenesulfonamide group onto G-5 enhanced the enantioselection to 85% ee in comparison with the TsNH substituted G-4 (entry 7 vs. entry 6). When the solvent was changed from ethyl acetate to THF, the enantioselectivity of the reaction became 88% ee with the yield and dr maintained (entry 8). When the reaction was performed at −60 °C, the enantioselectivity greatly improved to 95% ee (entry 9). If the catalyst loading of G-5 was reduced to 5 mol%, the ee value of 3aa slightly declined but with maintained yield and diastereoselectivity (entry 10).
|
|
||||
|---|---|---|---|---|
| Entrya | Cat | Yieldb (%) | drc | eed (%) |
| a Unless otherwise noted, the reactions were carried out with guanidine (10 mol%), 1a (0.10 mmol) and 2a (1.2 equiv.) in AcOEt (1.0 mL) at −30 °C for 24 h. b Isolated yield. c Determined by NMR analysis. d Determined by UPC2 analysis. e THF (1.0 mL) instead of AcOEt. f At −60 °C for 72 h. g G-5 (5 mol%). | ||||
| 1 | BG-1 | 86 | 2.5:1 |
24/14 |
| 2 | BG-1·HBArF4 | 75 | 2:1 |
23/11 |
| 3 | G-1 | 98 | >19:1 |
47 |
| 4 | G-2 | 94 | >19:1 |
52 |
| 5e | G-3 | 97 | >19:1 |
41 |
| 6 | G-4 | 95 | >19:1 |
74 |
| 7 | G-5 | 99 | >19:1 |
85 |
| 8e | G-5 | 99 | >19:1 |
88 |
| 9f | G-5 | 99 | >19:1 |
95 |
| 10f,g | G-5 | 99 | >19:1 |
93 |

Having identified the optimized reaction conditions (Table 1, entry 9), we next investigated the scope of (E)-2-(2-nitrovinyl) phenols 1 which were employed to react with azlactone 2a. As shown in Table 2, a variety of substrates bearing electron-withdrawing and electron-donating substituted phenolic groups were experimented on. Both the position and electronic nature of the substituent affected the yield obviously. For halo-substituted nitroolefins 1b–1d, the yield decreased along with the increase in atomic volume but without affecting the enantioselectivity (entries 2–4). 5-Substituted nitroalkenes 1e–1g underwent the reaction in relatively lower yield compared with the others (entries 5–7). It was noteworthy that the hydroxyl group tolerated well in the reaction, and the transformation of the substrate 1g afforded the corresponding product 3ga in 80% yield and 96% ee (entry 7). 6-Substituted nitroolefins 1h–1n performed the reaction well to give the related dihydrocoumarins 3ha–3na in 80–99% yield and 91–95% ee (entries 8–14). It was obvious that C6-electron-withdrawing groups resulted in lower yield than the electron-donating ones. Particularly, substrate 1n bearing the sterically hindered tert-butyl group adjacent to the hydroxyl group participated in the cascade reaction with 99% yield and 95% ee (entry 14). In all cases, the diastereoselectivity of the reaction was as high as 19:1.
|
|
||||
|---|---|---|---|---|
| Entrya | 1: R1 | Yieldb (%) | drc | eed (%) |
| a Unless otherwise noted, the reactions were carried out with G-5 (10 mol%), 1 (0.10 mmol) and 2a (1.2 equiv.) in THF (1.0 mL) at −60 °C for 72 h. b Isolated yield. c Determined by NMR analysis. d Determined by UPC2 analysis. | ||||
| 1 | 1a: H | 99 (3aa) | >19:1 |
95 |
| 2 | 1b: 4-F | 99 (3ba) | >19:1 |
95 |
| 3 | 1c: 4-Cl | 90 (3ca) | >19:1 |
95 |
| 4 | 1d: 4-Br | 84 (3da) | >19:1 |
96 |
| 5 | 1e: 5-Me | 80 (3ea) | >19:1 |
92 |
| 6 | 1f: 5-Cl | 85 (3fa) | >19:1 |
92 |
| 7 | 1g: 5-OH, 6-Me | 80 (3ga) | >19:1 |
96 |
| 8 | 1h: 6-F | 83 (3ha) | >19:1 |
94 |
| 9 | 1i: 6-Cl | 82 (3ia) | >19:1 |
91 |
| 10 | 1j: 6-Br | 80 (3ja) | >19:1 |
91 |
| 11 | 1k: 6-Me | 92 (3ka) | >19:1 |
95 |
| 12 | 1l: 6-MeO | 99 (3la) | >19:1 |
92 |
| 13 | 1m: 6-EtO | 95 (3ma) | >19:1 |
93 |
| 14 | 1n: 6-t-Bu | 99 (3na) | >19:1 |
95 |
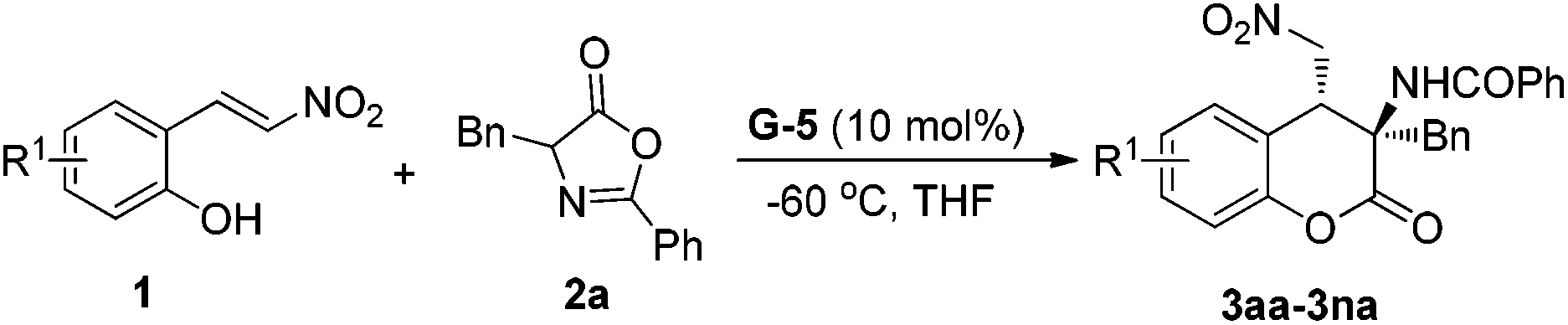
Encouraged by the above mentioned results, we continued to investigate the scope of azlactones 2 (Table 3). 2-Aromatic substituents of azlactones 2b–2g underwent the reaction resulting in good to excellent yields and satisfactory enantioselectivity (entries 1–6). The electron-deficient substituent at the para-position of the 2-aryl group weakened the reactivity in comparison with electron-donating ones, and cyano-substituted 2d gave the lowest yield of 63% with 93% ee but ethyl-substituted 2g afforded up to 99% yield with 98% ee (entry 6 vs. entry 3). Next, azlactones varied at the C4-position were subjected to the reaction (entries 8–11). We synthesized azlactones 2i–2l from alanine, leucine, 2-amino-4-phenylbutanoic acid, and tryptophan separately, and allowed them to react with 1a. These reactions proceeded smoothly to the desired products in very high yields and enantioselectivities. Particularly, the 3-indolylmethyl substituted one afforded the formation of functional dihydrocoumarin derivative 3al in almost optical purity and equivalent yield.
|
|
||||
|---|---|---|---|---|
| Entry | 2: R2/R3 | Yieldb (%) | drc | eed (%) |
| a Unless otherwise noted, the reactions were carried out with G-5 (10 mol%), 1a (0.10 mmol) and 2 (1.2 equiv.) in THF (1.0 mL) at −60 °C for 72 h. b Isolated yield. c Determined by NMR analysis. d Determined by UPC2 analysis. e The azlactones 2 (2.0 equiv.). | ||||
| 1 | 2b: 4-ClC6H4/Bn | 87 (3ab) | >19:1 |
95 |
| 2 | 2c: 4-BrC6H4/Bn | 87 (3ac) | >19:1 |
96 |
| 3 | 2d: 4-NCC6H4/Bn | 63 (3ad) | >19:1 |
93 |
| 4 | 2e: 4-MeC6H4/Bn | 99 (3ae) | >19:1 |
95 |
| 5 | 2f: 4-MeOC6H4/Bn | 99 (3af) | >19:1 |
95 |
| 6 | 2g: 4-EtC6H4/Bn | 99 (3ag) | >19:1 |
98 |
| 7 | 2h: Ph/4-ClC6H4CH2 | 99 (3ah) | >19:1 |
96 |
| 8e | 2i: Ph/Me | 94 (3ai) | >19:1 |
98 |
| 9e | 2j: Ph/i-Bu | 97 (3aj) | >19:1 |
97 |
| 10 | 2k: Ph/CH2CH2Ph | 97 (3ak) | >19:1 |
98 |
| 11 | 2l: Ph/3-Indolylmethyl | 99 (3al) | >19:1 |
99 |
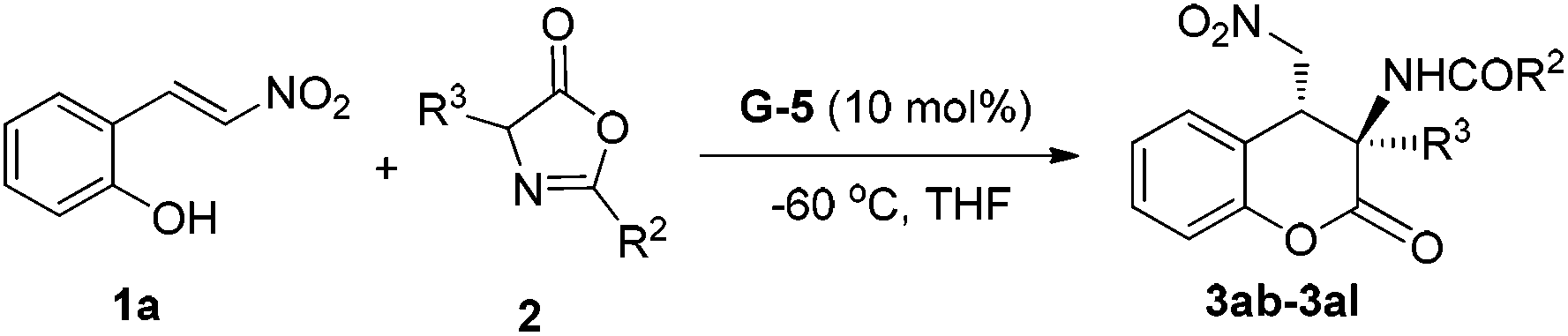
Taking into account the practical application of the catalyst system, the gram-scale synthesis of 3aa was performed. Under the optimal reaction conditions, 3 mmol (0.50 g) 1a and 3.6 mmol (0.91 g) azlactone 2a reacted well to afford the desired adduct 3aa in 84% yield (1.06 g) with 92% ee and >19:1 dr (Scheme 2). The absolute configuration of the product 3aa was determined to be (3R, 4R) by X-ray crystallography analysis, and the others were confirmed in comparison with the Cotton effect in the CD spectra analysis (see the ESI† for details).13 The chiral G-5 acts as a bifunctional catalyst such that the guanidine unit benefits the activation of azlactones and the sulfonamide group activates the nitroolefins via H-bonding. The diastereo- and enantioselective oxazolone–nitroalkene conjugate addition intermediate11f undergoes an intramolecular transesterification to give the final 4-nitromethyl 3-aminochromanone.
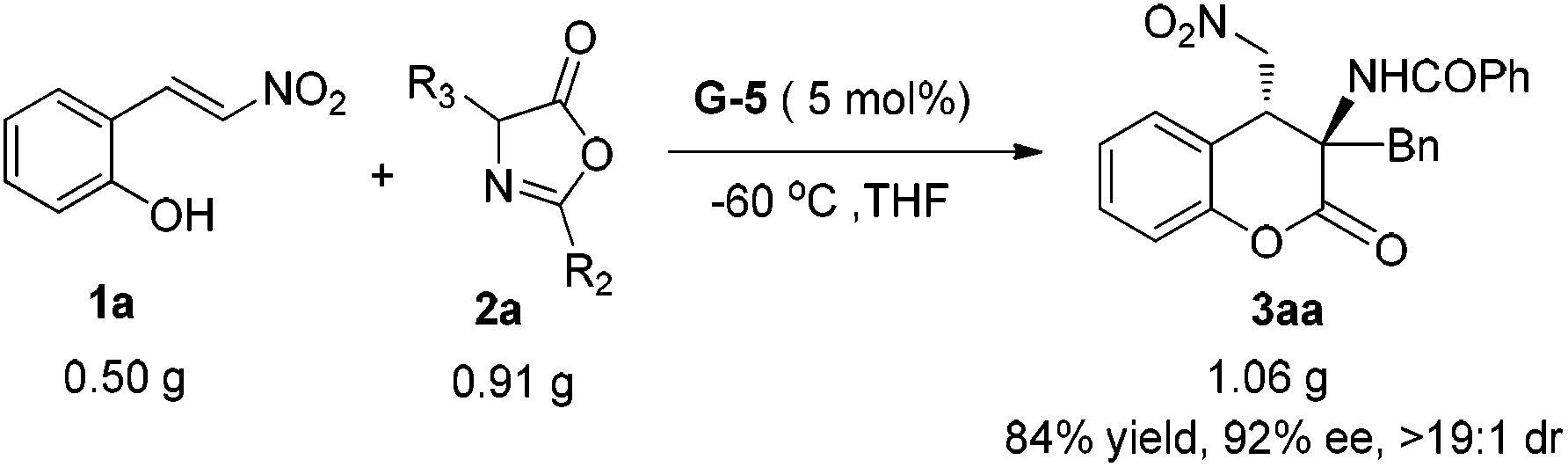 | ||
| Scheme 2 The gram-scale synthesis of 3aa. | ||
Conclusions
We have successfully developed an efficient method for the asymmetric synthesis of chiral 3,4-dihydrocoumarins in the presence of a bifunctional guanidine catalyst. A variety of azlactones and ortho-nitrovinylphenols were tolerated well, providing the corresponding 4-functionalized 3-aminochromanones in high yields and excellent diastereo- and enantioselectivities. Further applications of the bifunctional guanidines in asymmetric catalysis are ongoing in our laboratory.Experimental section
General procedure for the cascade reaction
2-Nitrovinylphenol 1 (0.10 mmol), azlactone 2 (1.2–2.0 equiv.) and the catalyst G-5 (10 mol%) were added into a test tube, followed by the addition of THF (1.0 mL). Then the mixture was stirred at −60 °C. After completion, the reaction mixture was purified by silica gel column chromatography (ethyl acetate/petroleum ether 1/4–1/2) to afford the desired products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China (No. 21625205 and 21332003) and the National Program for Support of Top-Notch Professionals for financial support.Notes and references
- (a) G. Chen, N. Tokunaga and T. Hayashi, Org. Lett., 2005, 7, 2285 CrossRef CAS PubMed; (b) L. A. Duffy, H. Matsubara, D. J. Procter and J. Am, Chem. Soc., 2008, 130, 1136 CrossRef CAS PubMed; (c) A. H. Rezayan, P. Azerang, S. Sardari and A. Sarvary, Chem. Biol. Drug Des., 2012, 80, 929 CrossRef CAS PubMed; (d) M. Marchetti, O. Piccolo and F. Ulgheri, J. Org. Chem., 2007, 72, 6056 CrossRef PubMed; (e) B. D. Gallagher, B. R. Taft and B. H. Lipshutz, Org. Lett., 2009, 11, 5374 CrossRef CAS PubMed.
- (a) D. M. X. Donnelly and G. M. Boland, Nat. Prod. Rep., 1995, 321 RSC; (b) J. Yang, G.-Y. Liu, F. Dai, X.-Y. Cao, Y. Kang, L.-M. Hu, J.-J. Tang, X.-Z. Li, X.-L. Jin and B. Zhou, Bioorg. Med. Chem. Lett., 2011, 21, 6420 CrossRef CAS PubMed; (c) C. A. Kontogiorgis and D. J. Hadjipavlou-Litina, J. Med. Chem., 2005, 48, 6400 CrossRef CAS PubMed; (d) B. Biswas, P. K. Sen and R. V. Venkateswaran, Tetrahedron, 2007, 63, 12026 CrossRef CAS; (e) E. Ballerini, L. Minuti, O. Piermatti and F. Pizzo, J. Org. Chem., 2009, 74, 4311 CrossRef CAS PubMed.
- For selected examples of the asymmetric synthesis of dihydrocoumarins, see: (a) J.-L. Li, S.-L. Zhou, B. Han, L. Wu and Y.-C. Chen, Chem. Commun., 2010, 46, 2665 RSC; (b) Y. L. Kuang, X. H. Liu, L. Chang, M. Wang, L. L. Lin and X. M. Feng, Org. Lett., 2011, 13, 3814 CrossRef CAS PubMed; (c) O. D. Engl, S. P. Fritz, A. Käslin and H. Wennemers, Org. Lett., 2014, 16, 5454 CrossRef CAS PubMed; (d) X.-H. Li, P. Fang, D. Chen and X.-L. Hou, Org. Chem. Front., 2014, 1, 969 RSC; (e) A. Lee and K. A. Scheidt, Chem. Commun., 2015, 51, 3407 RSC; (f) H. Lv, L. You and S. Ye, Adv. Synth. Catal., 2009, 351, 2822 CrossRef CAS.
- (a) M. Sletzinger, D. F. Reinhold and J. H. Markillie, U.S. Patent, 3161655, 1964 Search PubMed; (b) B. P. Roger, M. Masatern, R. J. Gerace and R. T. Edward, WO Patent WO, 931203, 1993 Search PubMed; (c) S. Robert, C. Bertolla, B. Masereel, J. M. Dogné and L. Pochet, J. Med. Chem., 2008, 51, 3077 CrossRef CAS PubMed; (d) D. Cabaret, S. A. Adediran, M. J. Garcia Gonzalez, R. F. Pratt and M. Wakselman, J. Org. Chem., 1999, 64, 713 CrossRef CAS PubMed.
- (a) T.-P. Gao, D. Liu, J.-B. Lin, X.-Q. Hu, Z.-Y. Wang and P.-F. Xu, Org. Chem. Front., 2016, 3, 598 RSC; (b) L. Tian, G.-Q. Xu, Y.-H. Li, Y.-M. Liang and P.-F. Xu, Chem. Commun., 2014, 50, 2428 RSC; (c) D. B. Chandrasekhar, S.-C. Tsay, K. P. Tapan and J.-R. Hwu, J. Org. Chem., 2017, 82, 5524 CrossRef CAS PubMed; (d) B.-D. Cui, J. Zuo, J.-Q. Zhao, M.-Q. Zhou, Z.-J. Wu, X.-M. Zhang and W.-C. Yuan, J. Org. Chem., 2014, 79, 5305 CrossRef CAS PubMed; (e) P. P. Castro, A. G. Carpanez and G. W. Amarante, Chem. – Eur. J., 2016, 22, 10294 CrossRef PubMed.
- For reviews of azlactones see: (a) J. S. Fisk, R. A. Mosey and J. J. Tepe, Chem. Soc. Rev., 2007, 36, 1432 RSC; (b) R. A. Mosey, J. S. Fisk and J. J. Tepe, Tetrahedron: Asymmetry, 2008, 19, 2755 CrossRef CAS; (c) A. R. Alba and R. Rios, Chem. – Asian J., 2011, 6, 720 CrossRef CAS PubMed.
- For selected examples of azlactones in the synthesis of dihydrocoumarins, see: (a) H. H. Hu, Y. B. Liu, J. Guo, L. L. Lin, Y. L. Xu, X. H. Liu and X. M. Feng, Chem. Commun., 2015, 51, 3835 RSC; (b) X.-Y. Yu, J.-R. Chen, Q. Wei, H.-G. Cheng, Z.-C. Liu and W.-J. Xiao, Chem. – Eur. J., 2016, 22, 6774 CrossRef CAS PubMed; (c) J. Zhou, M.-L. Wang, X. Gao, F. Jiang and Y.-G. Zhou, Chem. Commun., 2017, 53, 3531 RSC; (d) Y.-C. Zhang, Q.-N. Zhu, X. Yang, L.-J. Zhou and F. Shi, J. Org. Chem., 2016, 81, 1681 CrossRef CAS PubMed; (e) Y. Wang, J. Pan, R. Jiang, Y. Wang and Z. Zhou, Adv. Synth. Catal., 2016, 358, 195 CrossRef CAS; (f) S. X. Dong, X. H. Liu, Y. L. Zhang, L. L. Lin and X. M. Feng, Org. Lett., 2011, 13, 5060 CrossRef CAS PubMed; (g) J. Hejmanowska, A. Albrecht, J. Pieta and Ł. Albrechta, Adv. Synth. Catal., 2015, 357, 3843 CrossRef CAS; (h) S.-Y. Zhang, M. Lv, S.-J. Yin, N.-K. Li, J.-Q. Zhang and X.-W. Wang, Adv. Synth. Catal., 2016, 358, 143 CrossRef CAS; (i) S. X. Dong, X. H. Liu, X. H. Chen, F. Mei, Y. L. Zhang, B. Gao, L. L. Lin and X. M. Feng, J. Am. Chem. Soc., 2010, 132, 10650 CrossRef CAS PubMed.
- For selected reviews on organocatalyzed asymmetric cascade reactions, see: (a) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (b) X.-H. Yu and W. Wang, Org. Biomol. Chem., 2008, 6, 2037 RSC; (c) D. Enders, C. Grondal and M. R. M. Huettl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS PubMed; (d) J. H. Feng, L. L. Lin, K. R. Yu, X. H. Liu and X. M. Feng, Adv. Synth. Catal., 2015, 357, 1305 CrossRef CAS; (e) J. H. Feng, X. Fu, Z. L. Chen, L. L. Lin, X. H. Liu and X. M. Feng, Org. Lett., 2013, 15, 2640 CrossRef CAS PubMed; (f) J. H. Feng, X. Yuan, W. W. Luo, L. L. Lin, X. H. Liu and X. M. Feng, Chem. – Eur. J., 2016, 22, 15650 CrossRef CAS PubMed; (g) B. W. Ma, X. B. Lin, L. L. Lin, X. M. Feng and X. H. Liu, J. Org. Chem., 2017, 82, 701 CrossRef CAS PubMed.
- For reviews of chiral guanidine catalysts, see: (a) T. Ishikawa and T. Isobe, Chem. – Eur. J., 2002, 8, 552 CrossRef CAS PubMed; (b) D. Leow and C.-H. Tan, Chem. – Asian J., 2009, 4, 488 CrossRef CAS PubMed; (c) X. Fu and C.-H. Tan, Chem. Commun., 2011, 47, 8210 RSC; (d) X. H. Liu, L. L. Lin and X. M. Feng, Chem. Commun., 2009, 6145 RSC; (e) J. E. Taylor, S. D. Bull and J. M. Williams, Chem. Soc. Rev., 2012, 41, 2109 RSC.
- (a) Z. P. Yu, X. H. Liu, L. Zhou, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2009, 48, 5195 CrossRef CAS PubMed; (b) X. Xiao, X. H. Liu, S. X. Dong, Y. F. Cai, L. L. Lin and X. M. Feng, Chem. – Eur. J., 2012, 18, 159226 Search PubMed; (c) S. X. Dong, X. H. Liu, Y. Zhu, P. He, L. L. Lin and X. M. Feng, J. Am. Chem. Soc., 2013, 135, 10026 CrossRef CAS PubMed; (d) B. Fang, X. H. Liu, J. N. Zhao, Y. Tang, L. L. Lin and X. M. Feng, J. Org. Chem., 2015, 80, 3332 CrossRef CAS PubMed; (e) K. R. Yu, X. H. Liu, X. B. Lin, L. L. Lin and X. M. Feng, Chem. Commun., 2015, 51, 14897 RSC; (f) Q. G. Chen, Y. Tang, T. Y. Huang, X. H. Liu, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2016, 55, 5286 CrossRef CAS PubMed; (g) X. B. Lin, S. Ruan, Q. Yao, C. K. Yin, L. L. Lin, X. M. Feng and X. H. Liu, Org. Lett., 2016, 18, 3602 CrossRef CAS PubMed; (h) S. S. Guo, X. H. Liu, B. Shen, L. L. Lin and X. M. Feng, Org. Lett., 2016, 18, 5070 CrossRef CAS PubMed.
- (a) Z. Wu, X. Wang, F. Li, J. Wu and J. Wang, Org. Lett., 2015, 17, 3588 CrossRef CAS PubMed; (b) X. Zhang, S. Zhang and W. Wang, Angew. Chem., Int. Ed., 2010, 49, 1481 CrossRef CAS PubMed; (c) C.-F. Yao, Y.-J. Jang and M.-C. Yan, Tetrahedron Lett., 2003, 44, 3813 CrossRef CAS; (d) D.-F. Lu, Y.-J. Li and Y.-F. Gong, J. Org. Chem., 2010, 75, 6900 CrossRef CAS PubMed; (e) J. M. Andrés, J. Losada, A. Maestro, P. R. Ferrer and R. Pedrosa, J. Org. Chem., 2017, 82, 8444 CrossRef PubMed; (f) J. Alemán, A. Milelli, S. Cabrera, E. Reyes and K. A. Jørgensen, Chem. – Eur. J., 2008, 14, 10958 CrossRef PubMed.
- (a) Y. S. Chen, X. H. Liu, W. W. Luo, L. L. Lin and X. M. Feng, Synlett, 2017, 28, 966 CrossRef CAS; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC.
- CCDC 1560312 (3aa). For more data, see the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1560312. For ESI and crystallographic data in CIF or other electronic format, see DOI: 10.1039/c7qo00768j |
| This journal is © the Partner Organisations 2018 |