
Cu-Catalyzed tertiary alkylation of α-(trifluoromethyl)styrenes with tertiary alkylmagnesium reagents†
Wenpeng
Dai
,
Yingyin
Lin
,
Yan
Wan
and
Song
Cao
 *
*
Shanghai Key Laboratory of Chemical Biology, School of Pharmacy, East China University of Science and Technology (ECUST), Shanghai 200237, China. E-mail: scao@ecust.edu.cn
First published on 18th September 2017
Abstract
A novel and efficient method for the synthesis of 3-tertiary alkylated 1,1-difluorostyrene derivatives via Cu-catalyzed alkylation of α-(trifluoromethyl)styrenes with tertiary alkylmagnesium reagents at room temperature was developed.
Introduction
The modification of organic molecules by the incorporation of a tert-butyl group is widely used in drug discovery and agrochemistry because its introduction often alters the orientation of the alkyl side chain, lipophilicity and the binding affinity of the parent compounds (Fig. 1).1 For example, tebufenozide is a famous synthetic nonsteroidal ecdysone agonist (Fig. 1, bottom right). However, replacing the tert-butyl group by other alkyl groups resulted in a dramatic decrease in the insecticidal activity.2 Therefore, the development of new methodologies for preparing tertiary alkylated compounds has become a subject of great interest.3 Among various approaches for the incorporation of a tert-butyl group into the target molecules, the transition metal-catalyzed cross-coupling of sterically hindered tertiary alkyl nucleophiles or electrophiles is considered to be quite challenging because of the steric hindrance of the tertiary alkyl group. Furthermore, these tertiary alkylations were frequently accompanied by the isomerization of the tertiary alkyl group, hydrodehalogenation and β-H elimination.4 Recently, significant advances in tertiary alkylation reactions have been achieved through copper,5 nickel,6 cobalt7 and silver-catalyzed coupling.8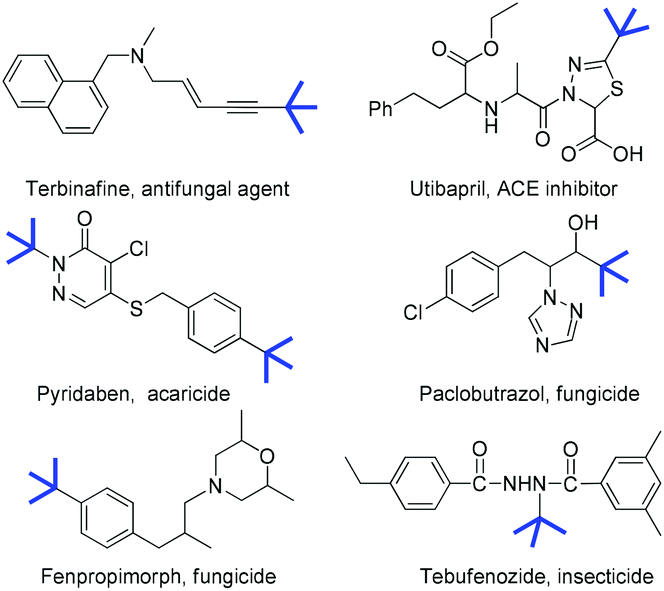 | ||
| Fig. 1 Examples of drugs and pesticides bearing a tert-butyl group. | ||
gem-Difluoroalkene derivatives are synthetically interesting and versatile fluorinated building blocks.9 The active difluoromethylidene (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2) moiety provides an entry to the synthesis of structurally diverse useful compounds through nucleophilic vinylic substitution (SNV),10 coupling reaction,11 cyclization12 and hydrodefluorination.13 Due to the high synthetic utility of gem-difluoroalkene in organic synthesis, numerous methods have been developed for the synthesis of gem-difluoroalkenes, including Wittig and Julia-type difluoromethylenation of carbonyl compounds, β-elimination of functionalized difluoromethyl compounds and gem-difluoroolefination of diazo compounds.14 The SN2′ reaction of trifluoromethyl alkene derivatives with carbon and heteroatom nucleophiles such as organolithiums, Grignard reagents, N-lithiated amines and silyl lithium reagents is a traditional method for the synthesis of functionalized 1,1-difluoroalkenes.15 Generally, primary alkyl, alkenyl, aryl, and even secondary alkyl Grignard or lithium reagents are good substrates that react with α-trifluoromethylstyrenes to afford SN2′ products.16 However, the reaction of sterically bulky tertiary alkyl Grignard or lithium reagents with trifluoromethylalkenes has been scarcely reported. For example, in 1995, Rock and co-workers reported the reaction of trifluoromethylated alkenes with t-BuLi to furnish 3-tert-butyl substituted 1,1-difluoroalkenes (Scheme 1a).17 Although the reaction proceeded smoothly and provided the corresponding product in high yields, the reaction should be performed at a very low temperature (−78 °C). Furthermore, this method has obvious limitation due to the poor tolerance of functional groups such as carbonyl, ester and nitrile groups towards a strong base t-BuLi. Therefore, the development of an efficient and mild alternative method for the incorporation of a tert-butyl group into trifluoromethylated alkenes without the use of a strong base is highly desirable. In this paper, we reported a mild and practical synthetic method for the preparation of 3-tertiary alkylated 1,1-difluorostyrene derivatives by Cu-catalyzed tertiary alkylation of α-(trifluoromethyl)styrenes with tertiary alkylmagnesium reagents at room temperature (Scheme 1b).
CF2) moiety provides an entry to the synthesis of structurally diverse useful compounds through nucleophilic vinylic substitution (SNV),10 coupling reaction,11 cyclization12 and hydrodefluorination.13 Due to the high synthetic utility of gem-difluoroalkene in organic synthesis, numerous methods have been developed for the synthesis of gem-difluoroalkenes, including Wittig and Julia-type difluoromethylenation of carbonyl compounds, β-elimination of functionalized difluoromethyl compounds and gem-difluoroolefination of diazo compounds.14 The SN2′ reaction of trifluoromethyl alkene derivatives with carbon and heteroatom nucleophiles such as organolithiums, Grignard reagents, N-lithiated amines and silyl lithium reagents is a traditional method for the synthesis of functionalized 1,1-difluoroalkenes.15 Generally, primary alkyl, alkenyl, aryl, and even secondary alkyl Grignard or lithium reagents are good substrates that react with α-trifluoromethylstyrenes to afford SN2′ products.16 However, the reaction of sterically bulky tertiary alkyl Grignard or lithium reagents with trifluoromethylalkenes has been scarcely reported. For example, in 1995, Rock and co-workers reported the reaction of trifluoromethylated alkenes with t-BuLi to furnish 3-tert-butyl substituted 1,1-difluoroalkenes (Scheme 1a).17 Although the reaction proceeded smoothly and provided the corresponding product in high yields, the reaction should be performed at a very low temperature (−78 °C). Furthermore, this method has obvious limitation due to the poor tolerance of functional groups such as carbonyl, ester and nitrile groups towards a strong base t-BuLi. Therefore, the development of an efficient and mild alternative method for the incorporation of a tert-butyl group into trifluoromethylated alkenes without the use of a strong base is highly desirable. In this paper, we reported a mild and practical synthetic method for the preparation of 3-tertiary alkylated 1,1-difluorostyrene derivatives by Cu-catalyzed tertiary alkylation of α-(trifluoromethyl)styrenes with tertiary alkylmagnesium reagents at room temperature (Scheme 1b).
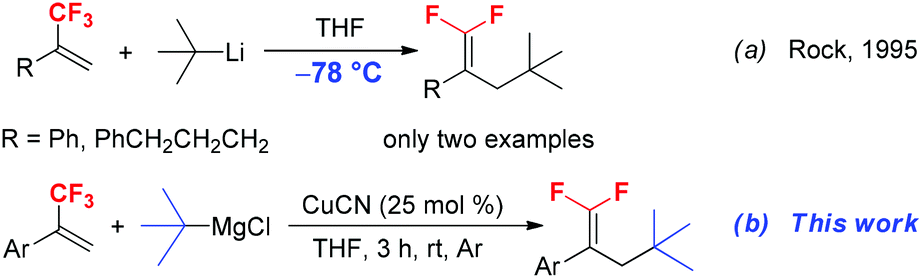 | ||
| Scheme 1 Reaction of trifluoromethyl alkenes with t-BuLi or t-BuMgCl. | ||
Results and discussion
We used 4-(3,3,3-trifluoroprop-1-en-2-yl)-1,1′-biphenyl 1a and t-BuMgCl 2a as model substrates to optimize the reaction conditions, and the results are shown in Table 1. Initially, the effect of different catalysts was evaluated. Among the various catalysts examined, CuCN was the most suitable for the reaction, providing 3aa in 97% yield (entry 10). When other cuprous or cupric salts, Pd(PPh3)4 and NiCl2(dppe) were used as catalysts, yields of the expected products were significantly decreased (entries 2–9). In the absence of a catalyst, only a small amount of the desired product 3aa was formed (entry 1). A brief examination of the solvents showed that THF was the optimal solvent for this reaction and the use of other solvents such as 1,4-dioxane, DME (1,2-dimethoxyethane), Et2O and toluene resulted in lower yields (entries 11–14). Optimization of the amounts of CuCN and t-BuMgCl 2a revealed that 0.25 equivalents of CuCN and 1.5 equivalents of t-BuMgCl 2a gave the best results. Lowering or increasing the amounts of CuCN and 2a significantly decreased the product yields (entries 15–18).|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | 2a (equiv.) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (1.0 mmol), solvent (3 mL), rt, Ar. b Yields determined by GC analysis and based on 1a. | ||||
| 1 | None | 1.5 | THF | 10 |
| 2 | Pd(PPh3)4 (25) | 1.5 | THF | 41 |
| 3 | NiCl2(dppe) (25) | 1.5 | THF | 43 |
| 4 | Cu(OAc)2 (25) | 1.5 | THF | 18 |
| 5 | CuCl2 (25) | 1.5 | THF | 19 |
| 6 | CuCl (25) | 1.5 | THF | 20 |
| 7 | CuBr (25) | 1.5 | THF | 24 |
| 8 | CuOAc (25) | 1.5 | THF | 62 |
| 9 | CuI (25) | 1.5 | THF | 68 |
| 10 | CuCN (25) | 1.5 | THF | 97 |
| 11 | CuCN (25) | 1.5 | Dioxane | 1 |
| 12 | CuCN (25) | 1.5 | DME | 2 |
| 13 | CuCN (25) | 1.5 | Et2O | 43 |
| 14 | CuCN (25) | 1.5 | Toluene | 53 |
| 15 | CuCN (25) | 1.2 | THF | 80 |
| 16 | CuCN (25) | 2.0 | THF | 92 |
| 17 | CuCN (20) | 1.5 | THF | 85 |
| 18 | CuCN (30) | 1.5 | THF | 86 |
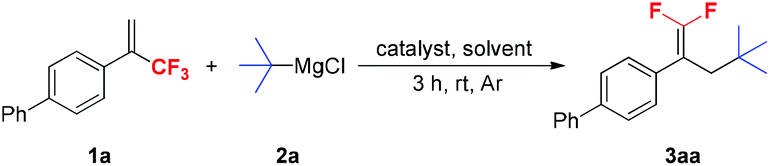
With the optimal reaction conditions in hand, various trifluoromethyl alkenes were subjected to the examination of the generality of this new method (Scheme 2). This reaction exhibited good functional group compatibility, and various functional groups such as methoxy, dimethoxy and tert-butyl (1c–e), and electron-withdrawing groups, such as carbonyl, ester, chloro, bromo and cyano (1h–l) were well-tolerated. Both electron-deficient and -rich trifluoromethyl alkenes could be converted into their corresponding 3-tertiary alkylated 1,1-difluorostyrene derivatives in moderate to high yields. The presence of Cl, Br and CN groups in the products is very useful for further synthetic transformations. N-(3-(3,3,3-Trifluoroprop-1-en-2-yl)phenyl)acetamide 1g was a poor substrate and only 55% yield of the expected product was furnished. The reaction of benzothiophene substrate 1m with t-BuMgCl 2a also proceeded smoothly and gave 3ma in 84% yield. Interestingly, when (E)-(3-(trifluoromethyl)buta-1,3-dien-1-yl)benzene 1n was used as a substrate, no desired product was observed and ditertiary alkylated monofluoroalkene was generated as the sole product (3na).5c,18 Further increasing the amount of t-BuMgCl 2a to 4.0 equivalents provided the optimum yield of 3na (88%). This reaction is presumed to proceed via two consecutive C–F substitutions, SN2′ and SNV reactions. Notably, when other substrates reacted with 4.0 equivalents of t-BuMgCl 2a, no ditertiary alkylated product was detected, probably due to the steric hindrance between the second introduced bulky tert-butyl group and the aryl ring.
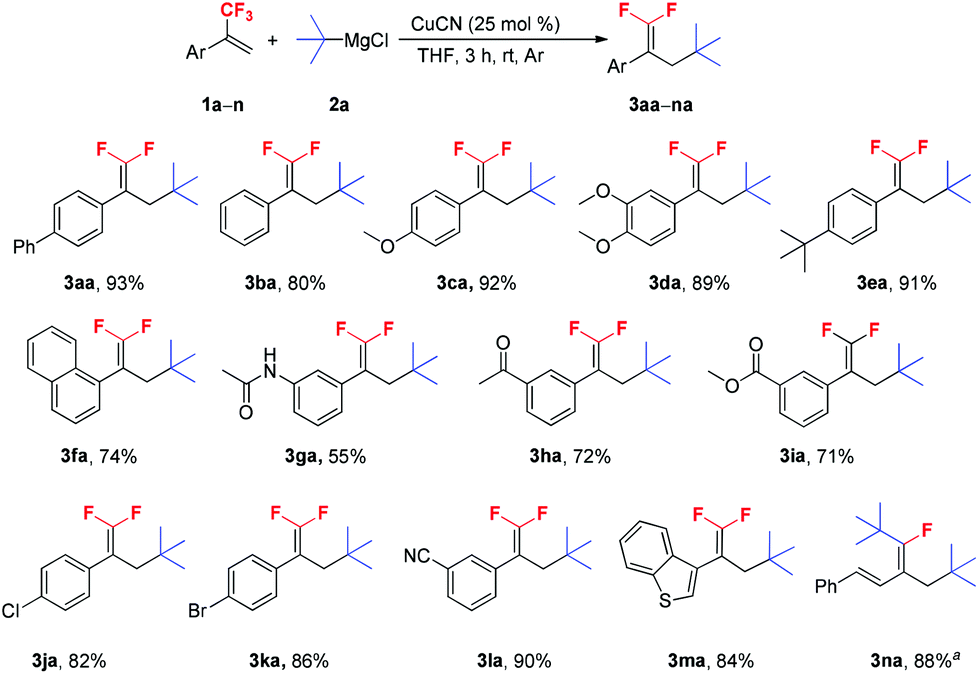 | ||
Scheme 2 Scope of α-(trifluoromethyl)styrenes. Reaction conditions: 1a–n (1.0 mmol), 2a (1.5 mmol), CuCN (0.25 mmol), THF (3 mL), rt, 3 h, Ar. Isolated yield. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2a (4.0 mmol). 2a (4.0 mmol). | ||
To further verify the scope of the new reaction, the reactions between several trifluoromethyl alkenes with tert-pentylmagnesium chloride 2b were investigated (Scheme 3). In most cases, the Cu-catalyzed tertiary alkylation reactions also proceeded smoothly and afforded the expected products in moderate to high yields. It should be noted that when secondary and primary alkyl Grignard reagents were subjected to the optimized reaction conditions, a mixture of mono-, di- and trialkylated substituted byproducts was obtained.
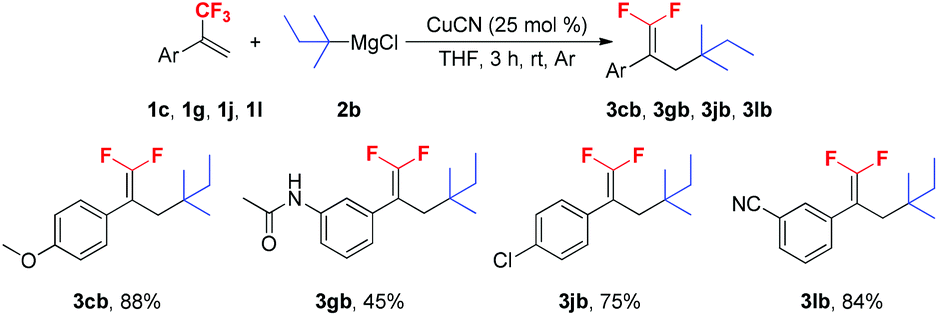 | ||
| Scheme 3 Reactions of trifluoromethyl alkenes with tert-pentylmagnesium chloride 2b. Reaction conditions: 1c, 1g, 1j, 1l (1.0 mmol), 2b (1.5 mmol), CuCN (0.25 mmol), THF (3 mL), rt, 3 h, Ar. Isolated yield. | ||
According to our results and the reported literature studies,16b,18,19 a plausible reaction mechanism is proposed in Scheme 4. Initially, the in situ-generated RCu reacts with tertiary alkyl Grignard reagents to afford dialkylcuprate magnesium halide (R2CuMgX, R = tertiary alkyl group). Subsequently, the SN2′ alkylation of trifluoromethylated alkenes with ditertiary alkyl cuprate (R2CuMgX) provides allyl cuprate I. Finally, the elimination of RCu from intermediate I results in the formation of tertiary alkylated products.
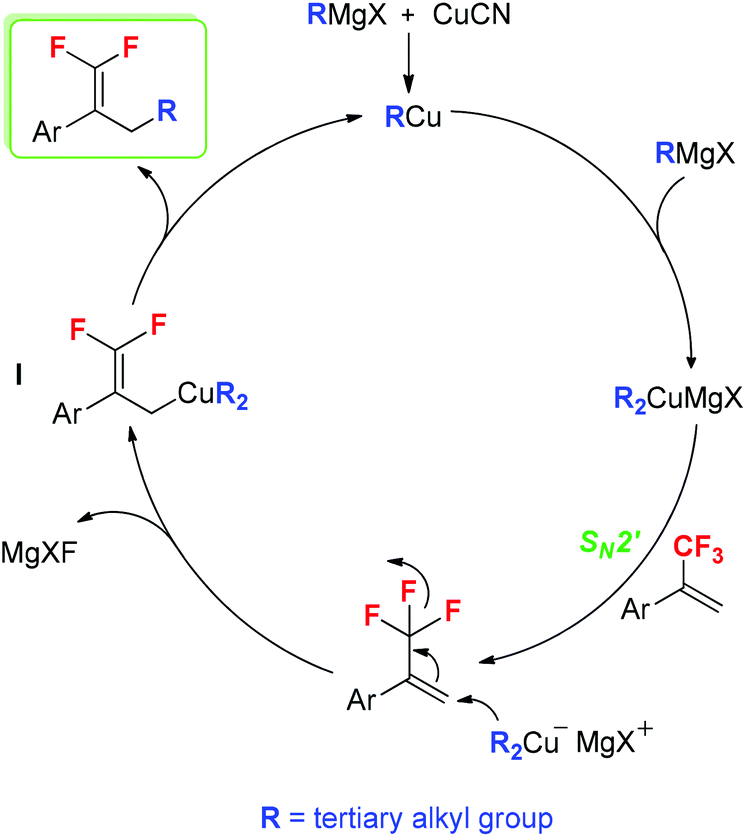 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In summary, we have developed a mild and easy-to-handle Cu-catalyzed tertiary alkylation of α-(trifluoromethyl)styrenes with tertiary alkylmagnesium reagents. The SN2′ alkylation reaction proceeds smoothly at room temperature and affords 3-tertiary alkylated 1,1-difluorostyrene derivatives in moderate to high yields. This method exhibits good functional-group compatibility. The present reaction may serve as an attractive method for the synthesis of 1,1-difluoroalkene bearing a sterically bulky tert-butyl group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (Grant No. 21472043 and 21272070).Notes and references
- (a) V. M. Gokhale and V. M. Kulkarni, J. Med. Chem., 1999, 42, 5348 CrossRef CAS PubMed; (b) H. Buikema, Y. M. Pinto, P. P. van Geel, G. Rooks, C. D. J. de Langen, P. A. de Graeff and W. H. van Gilst, J. Cardiovasc. Pharmacol., 1997, 29, 684 CrossRef CAS PubMed; (c) J. Boyd, A. Saksena, J. B. Patrone, H. N. Williams, N. Boggs, H. Le and M. Theodore, Chem. Res. Toxicol., 2011, 24, 1242 CrossRef CAS PubMed; (d) B. Sugavanam, Pestic. Sci., 1984, 15, 296 CrossRef CAS; (e) S. A. Forsyth, H. Q. N. Gunaratne, C. Hardacre, A. McKeown and D. W. Rooney, Org. Process Res. Dev., 2006, 10, 94 CrossRef CAS.
- (a) C. Mao, Q. Wang, R. Huang, F. Bi, L. Chen, Y. Liu and J. Shang, J. Agric. Food Chem., 2004, 52, 6737 CrossRef CAS PubMed; (b) Y. Nakagawa, K. Hattori, B. Shimizu, M. Akamatsu, H. Miyagawa and T. Ueno, Pestic. Sci., 1998, 53, 267 CrossRef CAS.
- (a) Z. S. Han, M. A. Herbage, H. P. R. Mangunuru, Y. Xu, L. Zhang, J. T. Reeves, J. D. Sieber, Z. Li, P. DeCroos, Y. Zhang, G. Li, N. Li, S. Ma, N. Grinberg, X. Wang, N. Goyal, D. Krishnamurthy, B. Lu, J. J. Song, G. Wang and C. H. Senanayake, Angew. Chem., Int. Ed., 2013, 52, 6713 CrossRef CAS PubMed; (b) J. K. Matsui, D. N. Primer and G. A. Molander, Chem. Sci., 2017, 8, 3512 RSC; (c) A. Joshi-Pangu and M. R. Biscoe, Synlett, 2012, 1103 CAS; (d) H. Dvořáková, D. Dvořák and A. Holý, Tetrahedron Lett., 1996, 37, 1285 CrossRef; (e) S. L. Zultanski and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 624 CrossRef CAS PubMed; (f) C. Sämann, V. Dhayalan, P. R. Schreiner and P. Knochel, Org. Lett., 2014, 16, 2418 CrossRef PubMed; (g) C. Zhao, X. Jia, X. Wang and H. Gong, J. Am. Chem. Soc., 2014, 136, 17645 CrossRef CAS PubMed; (h) X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562 CrossRef CAS PubMed.
- (a) K. Tamao, Y. Kiso, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 9268 CrossRef CAS; (b) T. Hayashi, M. Konishi, K. Yokota and M. Kumada, Chem. Lett., 1980, 767 CrossRef CAS; (c) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674 CrossRef CAS PubMed; (d) W. M. Czaplik, S. Grupe, M. Mayer and A. J. von Wangelin, Chem. Commun., 2010, 46, 6350 RSC; (e) M. R. Netherton and G. C. Fu, Adv. Synth. Catal., 2004, 346, 1525 CrossRef CAS; (f) C. Lohre, T. Dröge, C. Wang and F. Glorius, Chem. – Eur. J., 2011, 17, 6052 CrossRef CAS PubMed.
- (a) L. Hintermann, L. Xiao and A. Labonne, Angew. Chem., Int. Ed., 2008, 47, 8246 CrossRef CAS PubMed; (b) P. Ren, L. Stern and X. Hu, Angew. Chem., Int. Ed., 2012, 51, 9110 CrossRef CAS PubMed; (c) W. Dai, H. Shi, X. Zhao and S. Cao, Org. Lett., 2016, 18, 4284 CrossRef CAS PubMed.
- A. Joshi-Pangu, C. Wang and M. R. Biscoe, J. Am. Chem. Soc., 2011, 133, 8478 CrossRef CAS PubMed.
- T. Iwasaki, H. Takagawa, S. P. Singh, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2013, 135, 9604 CrossRef CAS PubMed.
- Y. Mitamura, Y. Asada, K. Murakami, H. Someya, H. Yorimitsu and K. Oshima, Chem. – Asian J., 2010, 5, 1487 CAS.
- (a) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS PubMed; (b) D. L. Orsi and R. A. Altman, Chem. Commun., 2017, 53, 7168 RSC; (c) J. Zhang, C. Xu, W. Wu and S. Cao, Chem. – Eur. J., 2016, 22, 9902 CrossRef CAS PubMed; (d) J. Zhang, W. Dai, Q. Liu and S. Cao, Org. Lett., 2017, 19, 3283 CrossRef CAS PubMed.
- (a) T. Fujita, M. Takazawa, K. Sugiyama, N. Suzuki and J. Ichikawa, Org. Lett., 2017, 19, 588 CrossRef CAS PubMed; (b) Z. Cong, Y. Li, L. Chen, F. Xing, G. Du, C. Gu and L. He, Org. Biomol. Chem., 2017, 15, 3863 RSC.
- (a) W. Dai, J. Xiao, G. Jin, J. Wu and S. Cao, J. Org. Chem., 2014, 79, 10537 CrossRef CAS PubMed; (b) M. Yokota, D. Fujita and J. Ichikawa, Org. Lett., 2007, 9, 4639 CrossRef CAS PubMed; (c) L. Kong, X. Zhou and X. Li, Org. Lett., 2016, 18, 6320 CrossRef CAS PubMed; (d) M. Ohashi, T. Kambara, T. Hatanaka, H. Saijo, R. Doi and S. Ogoshi, J. Am. Chem. Soc., 2011, 133, 3256 CrossRef CAS PubMed; (e) P. Tian, C. Feng and T. Loh, Nat. Commun., 2015, 7472 CrossRef PubMed.
- (a) E. Ausekle, S. A. Ejaz, S. U. Khan, P. Ehlers, A. Villinger, J. Lecka, J. Sévigny, J. Iqbal and P. Langer, Org. Biomol. Chem., 2016, 14, 11402 RSC; (b) B. Zhang, X. Zhang, J. Hao and C. Yang, Org. Lett., 2017, 19, 1780 CrossRef CAS PubMed; (c) X. Zhang, W. Dai, W. Wu and S. Cao, Org. Lett., 2015, 17, 2708 CrossRef CAS PubMed; (d) X. Zhang, M. Wu, J. Zhang and S. Cao, Org. Biomol. Chem., 2017, 15, 2436 RSC; (e) N. Suzuki, T. Fujita and J. Ichikawa, Org. Lett., 2015, 17, 4984 CrossRef CAS PubMed.
- (a) J. Wu, J. Xiao, W. Dai and S. Cao, RSC Adv., 2015, 5, 34498 RSC; (b) G. Landelle, M. Turcotte-Savard, L. Angers and J. Paquin, Org. Lett., 2011, 13, 1568 CrossRef CAS PubMed.
- X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375 CrossRef CAS.
- (a) G. Chelucci, Chem. Rev., 2012, 112, 1344 CrossRef CAS PubMed; (b) T. Kitazume and T. Ohnogi, Synthesis, 1988, 614 CrossRef CAS; (c) J. Ichikawa, Y. Ishibashi and H. Fukui, Tetrahedron Lett., 2003, 44, 707 CrossRef CAS; (d) Y. Huang and T. Hayashi, J. Am. Chem. Soc., 2016, 138, 12340 CrossRef CAS PubMed.
- (a) S. Watanabe, K. Sugahara, T. Fujita and M. Sakamoto, J. Fluorine Chem., 1993, 62, 201 CrossRef CAS; (b) T. Kitamme, T. Ohnogi, H. Miyauchi and T. Yamazaki, J. Org. Chem., 1989, 54, 5630 CrossRef; (c) F. Tellier, M. Baudry and R. Sauvêtre, Tetrahedron Lett., 1997, 38, 5989 CrossRef CAS; (d) T. Fuchikami, Y. Shibata and Y. Suzuki, Tetrahedron Lett., 1986, 27, 3173 CrossRef CAS; (e) K. Funabiki, K. Sawa, K. Shibata and M. Matsui, Synlett, 2002, 1134 CrossRef CAS; (f) H. M. Park, T. Uegaki, T. Konno, T. Ishihara and H. Yamanaka, Tetrahedron Lett., 1999, 40, 2985 CrossRef CAS.
- J. Bégué, D. Bonnet-Delpon and M. H. Rock, Tetrahedron Lett., 1995, 36, 5003 Search PubMed.
- J. Bégué, D. Bonnet-Delpon and M. H. Rock, J. Chem. Soc., Perkin Trans. 1, 1996, 1409 RSC.
- (a) K. Fuchibe, M. Takahashi and J. Ichikawa, Angew. Chem., Int. Ed., 2012, 51, 12059 CrossRef CAS PubMed; (b) S. Yamada, K. Shimoji, T. Takahashi, T. Konno and T. Ishihara, Chem. – Asian J., 2010, 5, 1846 CrossRef CAS PubMed; (c) G. Cahiez, O. Gager and J. Buendia, Angew. Chem., Int. Ed., 2010, 49, 1278 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qo00716g |
| This journal is © the Partner Organisations 2018 |