
Iridium catalyzed fragmentation/cyclization of N-butynyl 4,4-dimethylisoxazolidine-3,5-diones: a unique access to multiply substituted pyrroles†
Mei-Hua
Shen
 *,
Xin-Tao
Ren
,
Ying-Peng
Pan
and
Hua-Dong
Xu
*,
Xin-Tao
Ren
,
Ying-Peng
Pan
and
Hua-Dong
Xu
School of Pharmaceutical Engineering and Life Science, Changzhou University, Changzhou, Jiangsu Province 213164, China. E-mail: shenmh@cczu.edu.cn
First published on 18th September 2017
Abstract
A unique protocol for the synthesis of substituted and fused pyrroles has been developed through the iridium catalyzed fragmentation/cyclization of related N-butynyl 4,4-dimethylisoxazolidine-3,5-diones in hot ethanol. Both the substitution pattern and electronic properties can affect the reaction rate and yield.
The oxidative N–O single bond in hydroxylamine makes this unique moiety a powerful and convenient handle for a variety of organic transformations. The heterolytic cleavage of this bond serves as the driving force for the prestigious Beckmann rearrangement;1 it also plays a critical role in Bode's decarboxylative condensation of N-alkylhydroxylamines and α-ketoacids (KAHA ligation).2 Recently, N,O-dicarbonylated reagents A have been applied by Xu's group to achieve iron catalyzed amino-oxygenation/amino-halogenation of olefins where the N–O bond breaking event is the original power for the oxidizing process.3 Installation of an electron-withdrawing group on the O atom further polarizes the N–O bond making B a class of feasible electrophilic amination agents which have been applied in the amination of various carbon nucleophiles.4 The N–O bond of hydroxylamine is also activated when incorporated in a three-membered oxaziridine ring5 and reagents of type C have been successfully used in olefin aminohydroxylation,6 C–H amination7 and amination of arylmetals.8 Recently, cyclic motifs D (1,4,2-dioxazol-5-one, 1,3,2,4-dioxa-thiazole-2-oxide, 5,5-dimethyl-1,4,2-dioxazole and 1,2,4-oxadiazol-5-one) with a labile N–O bond have been used for metal catalyzed aryl C–H amidations9 and the synthesis of azaromatic amines.10 This progress in the chemistry of hydroxylamine derivatives aroused our interest in the related structure E (4,4-dimethylisoxazolidine-3,5-dione) featuring a cyclic N,O-diacylated hydroxylamine and we disclosed herein a unique protocol for the synthesis of substituted pyrroles (Fig. 1).
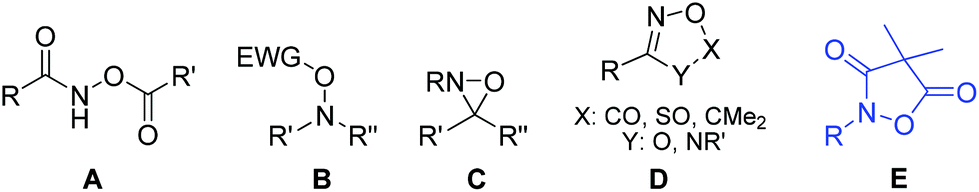 | ||
| Fig. 1 Reagents bearing the N–O component. | ||
At the outset, several N-alkyl-N,O-biacylated hydroxylamines E (R = alkyl group), which were prepared conveniently from the reaction of the corresponding aminohydroxide with 2,2-dimethylmalonyl dichloride, were subjected to extensive testing and the preliminary results indicated that these cyclic structures are exceptionally robust under both thermal and catalytic transition-metal conditions. Then our attention was turned to substrate 1a with a pending alkyl motif, and substantial conditions screening was carried out, i.e.1a was reacted in the presence of various catalytic transition metal complexes. The complexes of copper, iron, silver, nickel, manganese, rhodium, and ruthenium all failed to effect any change on the substrate (entries 1–19, Table S1†) in hot toluene; while several palladium and gold complexes led to complete decomposition of 1a (entries 20–23, Table S1†) to mess. Eventually, to our delight, when a solution of 1a in toluene was heated in the presence of 2 mol% [CpIrCl2]2 for 15 hours in a sealed tube, partial conversion of the starting materials was observed and the newly formed compound was isolated and determined as 1-isobutyryl-2-phenyl pyrrole 2a (entry 1, Table 1). This acylated pyrrole could be viewed as a product of the fragmentation/cyclization cascade of the starting materials. A higher yield was obtained with [Ir(COD)Cl]2 in place of [CpIrCl2]2 as the catalyst under the same conditions (entry 2). The best yield of 49% was obtained at 140 °C during the course of temperature screening for the oil bath from 110 to 150 °C (entries 3–6). The reaction didn't occur when 1,4-dioxane, acetonitrile and dimethylformide were used as reaction media (entries 7–9); halogenated solvents dichloroethane and chloroform were inferior to toluene with regard to the yield (entries 10 and 11). Interestingly, alcohols were also feasible reaction solvents and ethanol proved to be the best as the highest yield of 62% was achieved in this solvent (entries 12–15). The transformation didn't occur in water (entry 16). Neither halving nor doubling the catalyst loading of [Ir(COD)Cl]2 could improve the reaction outcome (entries 17 and 18). Substituting the Cl ligand with the OMe group gave rise to a complex [Ir(COD)OMe]2 which showed comparable catalytic performance (entry 19).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp (°C) | Yieldb |
| a Reaction conditions: 1a (0.2 mmol) and catalyst (2 mol%) in solvent (4 mL) were heated in a sealed tube under N2 for 15 h. b Isolated yield. c 1 mol% of [Ir(COD)Cl]2. d 4 mol% of [Ir(COD)Cl]2. | ||||
| 1 | [CpIrCl2]2 | Toluene | 110 | 20% |
| 2 | [Ir(COD)Cl]2 | Toluene | 110 | 32% |
| 3 | [Ir(COD)Cl]2 | Toluene | 120 | 38% |
| 4 | [Ir(COD)Cl]2 | Toluene | 130 | 44% |
| 5 | [Ir(COD)Cl]2 | Toluene | 140 | 49% |
| 6 | [Ir(COD)Cl]2 | Toluene | 150 | 45% |
| 7 | [Ir(COD)Cl]2 | 1,4-Dioxane | 140 | Trace |
| 8 | [Ir(COD)Cl]2 | MeCN | 140 | NR |
| 9 | [Ir(COD)Cl]2 | DMF | 140 | NR |
| 10 | [Ir(COD)Cl]2 | DCE | 140 | 42% |
| 11 | [Ir(COD)Cl]2 | CHCl3 | 140 | 25% |
| 12 | [Ir(COD)Cl]2 | MeOH | 140 | 32% |
| 13 | [Ir(COD)Cl]2 | EtOH | 140 | 62% |
| 14 | [Ir(COD)Cl]2 | iPrOH | 140 | 50% |
| 15 | [Ir(COD)Cl]2 | t BuOH | 140 | 20% |
| 16 | [Ir(COD)Cl]2 | H2O | 140 | NR |
| 17c | [Ir(COD)Cl]2 | EtOH | 140 | 55% |
| 18d | [Ir(COD)Cl]2 | EtOH | 140 | 52% |
| 19 | [Ir(COD)OMe]2 | EtOH | 140 | 58% |

The pyrrole scaffold is widely present in natural and synthetic compounds and many of them play essential roles in biological processes.11 Recently, pyrrole has become a hot structural platform for the development of therapeutic agents.12 Therefore an efficient protocol for selective synthesis of pyrrole derivatives is highly appreciated despite many existing methods.13 Since the current findings presaged a promising new method for pyrrole synthesis, we were prompted to carry out further studies to reveal its potential and limitations.
For this purpose, a series of arylbutynyl dimethylisoxazolidinediones 1 have been prepared and subjected to the optimal conditions to collect more information about this reaction (Table 2). Substrates with p/m methyl or p/m methoxyl goups on the phenyl ring all gave the corresponding 2-aryl pyrroles 2b–2e in 50–60% isolated yields after treatment with these conditions. With an o-methoxyl group, the reaction of related 4,4-dimethylisoxazolidine-3,5-dione delivered the pyrrole product 2f in a lower yield (42%). Electron-withdrawing substituents such as F and Cl are beneficial for this reaction giving about 10% increment in yields (2g and 2h). The ester group was compatible with these reaction conditions very well and 2i was obtained in 63% yield. 2-Thiophenyl pyrrole 2j could also be prepared via this protocol very efficiently.
| a Reaction conditions: 1 (0.2 mmol) and [Ir(COD)Cl]2 (2 mol%) in EtOH (4 mL) were heated at 140 °C in a sealed tube under N2 for 15 h. |
|---|

|
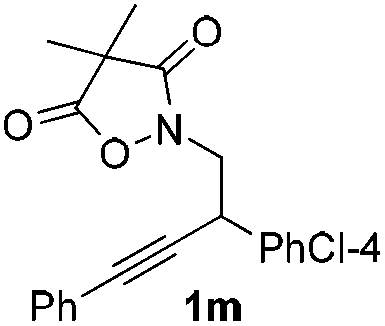
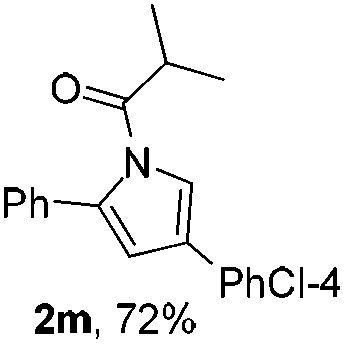
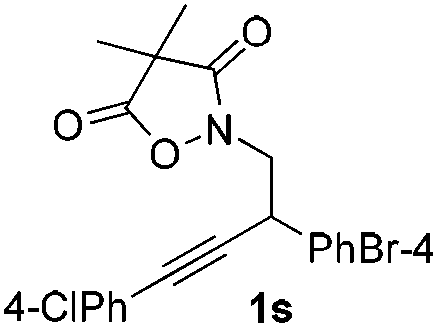
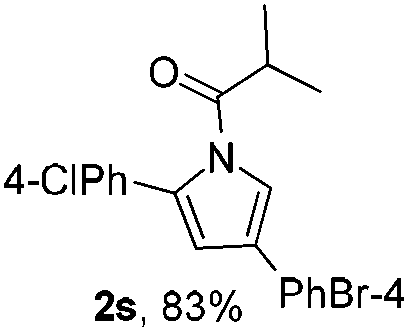
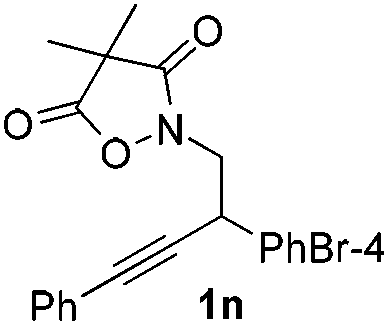
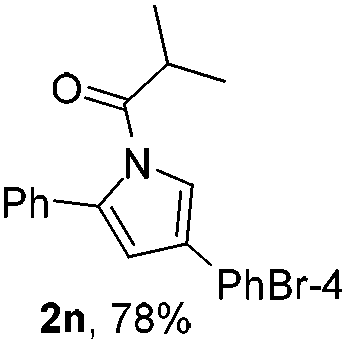
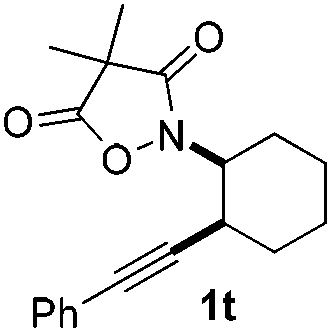
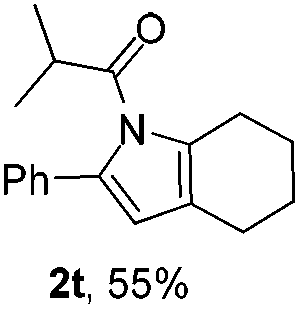
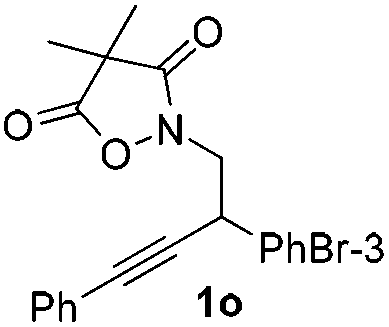
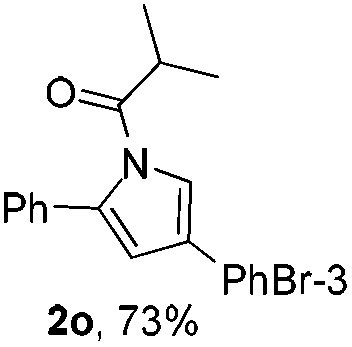
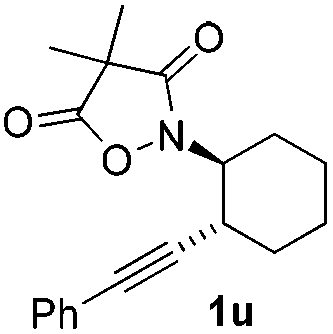
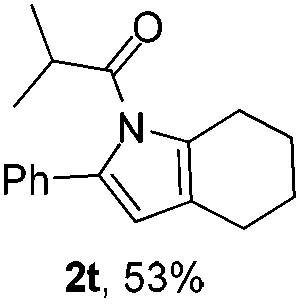
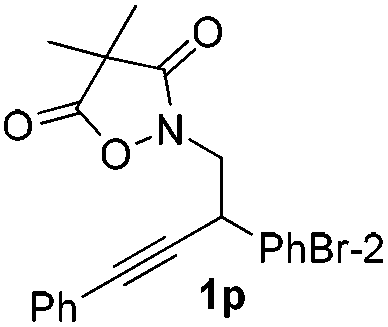
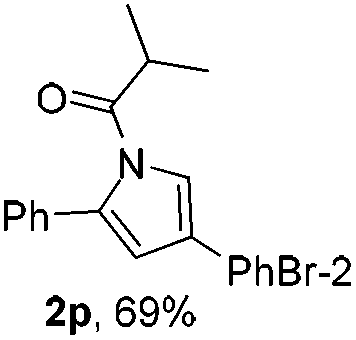
Next we decided to add one more aryl group at the propargylic C-2 of the butynyl moiety to prepare a number of 2,4-diarylbutynyl dimethylisoxazolidinediones (1k–1s). Interestingly, this alteration on the substrates accelerates the reaction rate remarkably as the reaction completed in 5 hours instead of completion in 15 hours for the substrates given in Table 2 and 2,4-diaryl pyrroles 2k–2s were collected in better yields (entries 1–9, Table 3). Generally speaking, the yields of 2,4-diaryl pyrroles are arguably higher than those of 2-aryl pyrroles and halogen substitutions on both aryl motifs were beneficial for this reaction which was typically illustrated by the isolated yield of 2s as high as 83% (entry 9). Incorporation of a cyclohexanyl ring onto the alkynyl segment resulted in a pair of stereoisomers 1t and 1u. Intriguingly, both cis- and trans-isomers underwent the fragmentation/cyclization sequence smoothly to give the same ring-fused pyrrole 2t in moderate yields though a longer reaction time was necessary for full conversion (entries 10 and 11, Table 3). Through this method, pyrroles with diverse aromatic rings at C2 and C3 could be prepared at will via arbitrary choice of aryl substituents in the corresponding starting materials.
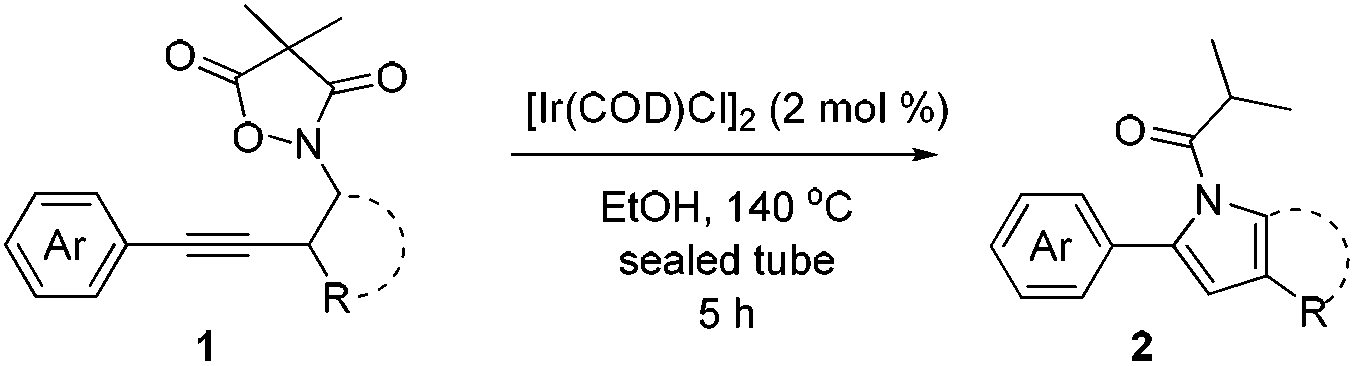

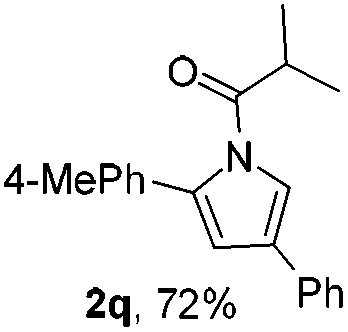
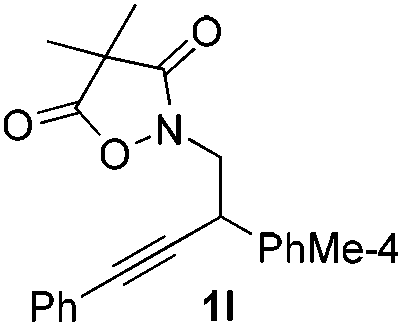
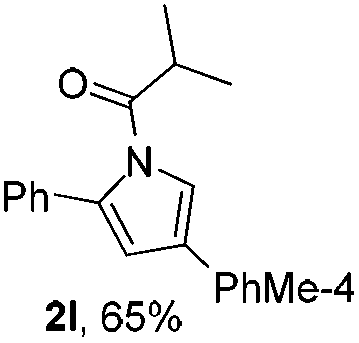
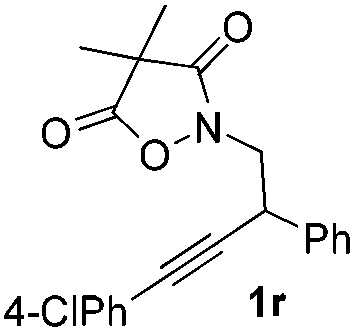
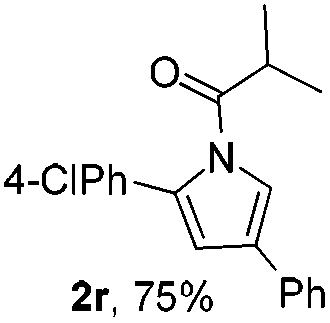
In the course of our study, it was found that some types of substrates (Fig. 2) didn't undergo this reaction. Pyridinyl alkyne 1v was almost recovered thoroughly, even after being heated to 150 °C for 15 hours, probably due to the strong coordinating property of the pyridine ring that might shut down the catalytic cycle. The triple bond was also totally unreactive for this reaction when a strong electron deficient nitro group was installed in 1w. A conjugated aromatic ring on the alkyne seems necessary since 1x also showed no reactivity under standard conditions. In sharp contrast to the isomeric substrates 1t/1u, a homologous pair of 1y/1z, namely both trans- and cis-stereomers, remained totally intact under exactly the same conditions. This discrepancy in reactivity might derive from the greater strain caused by the cyclopentane ring over the cyclohexane ring which impeded the cyclization step.
 | ||
| Fig. 2 Ineffective substrates. | ||
To collect more information about the reaction details, deuterated substrate d2-1k was prepared and subjected to the optimal reaction conditions, giving no deuterated product (eqn (a), Scheme 1). Meanwhile, the reaction of 1s in d4-MeOH produced the deuterated product d3-2s (eqn (b), Scheme 1). Interestingly, a mixture of 2s and [Ir(COD)Cl]2 in d4-MeOH was heated for 5 hours resulting in half deuteration on the pyrrole ring to afford d2-2s with the α-proton of the isobutyryl group untouched (eqn (c), Scheme 1). Based on these findings, a mechanism is proposed as shown in Fig. 3. The iridium dimer I could break into monomer II through solvolysis; then ligand exchange with alkyne 1 followed by oxidative addition of the Ir(I) center into the pending N–O bond would give rise to Ir(III) complex III. Under thermal conditions, III could undergo CO2 extrusion, alcohol bridged proton transfer and deprotonative metalation of the propargylic group to form allenyl Ir(III) complex IV. Then a 1,3-hydride shift would afford V and the subsequent reductive elimination would deliver the pyrrole product 2 and regenerate the active catalyst II for the next catalytic cycle.
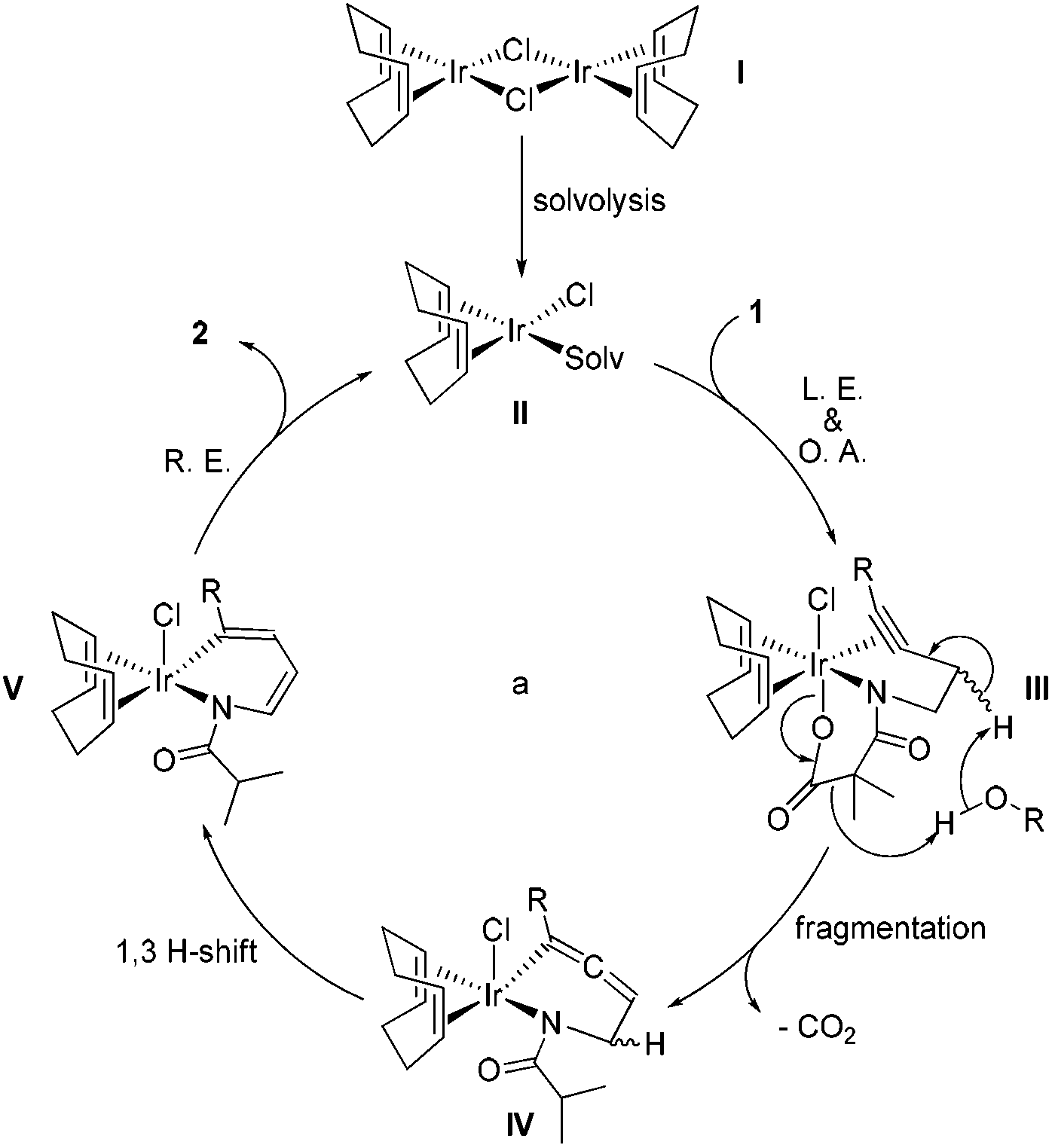 | ||
| Fig. 3 Possible mechanism for the pyrrole synthesis. | ||
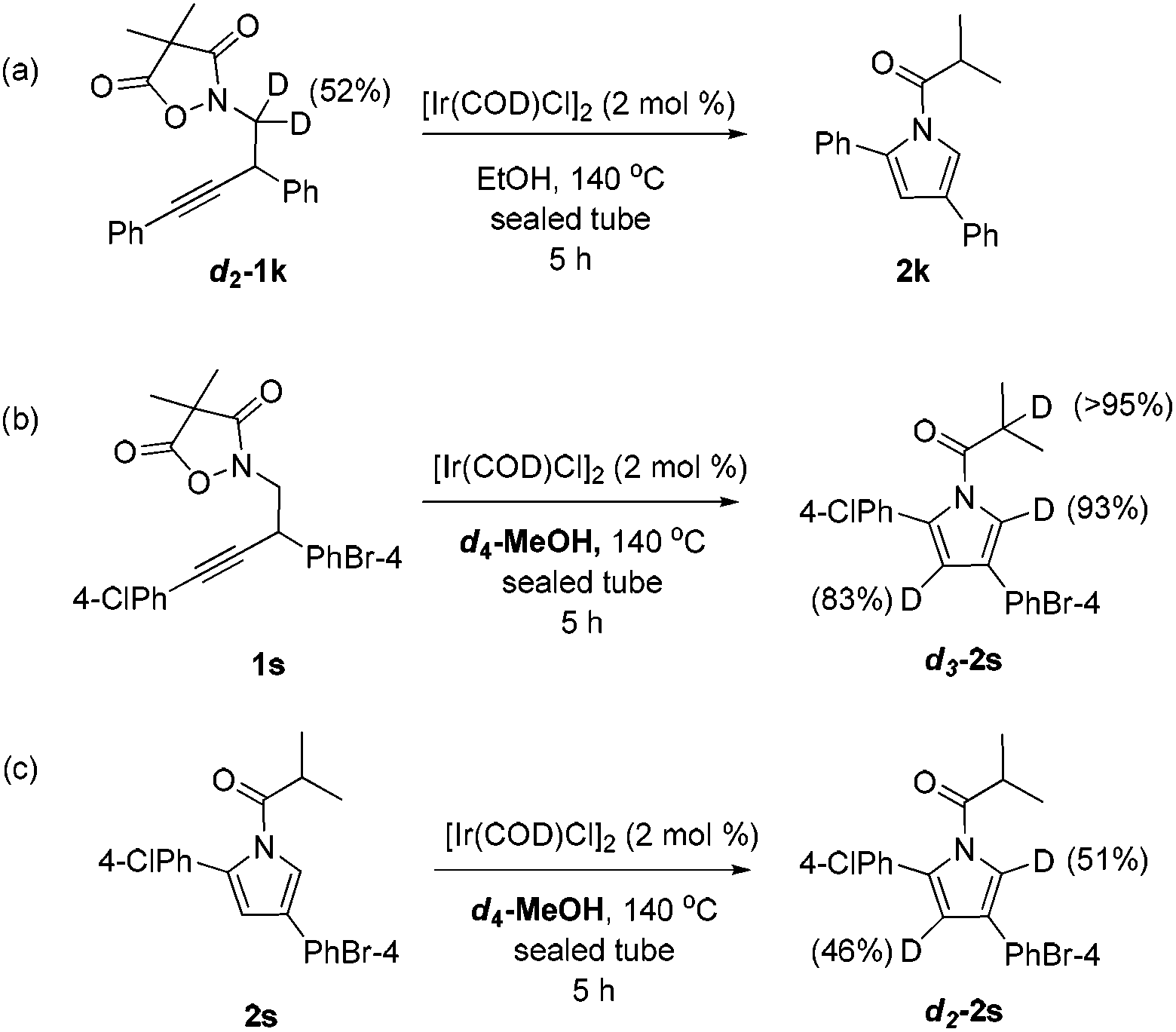 | ||
| Scheme 1 Experiments with a deuterated substrate or solvent. | ||
Conclusions
In summary, [Ir(COD)Cl]2 has been identified as a capable catalyst to successfully promote the fragmentation of the otherwise robust 4,4-dimethylisoxazolidine-3,5-dione moiety. Subsequent reactions with a pendent alkyne group eventually led to the construction of the corresponding 2-aryl or 2,4-diaryl pyrroles. Thus a new method for the synthesis of multiply substituted pyrroles has been developed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21402014 and 21672027), the QingLan Project of Jiangsu Province (2016) and the Six-Talent-Peaks Program of Jiangsu (2016) for financial support.Notes and references
- Comprehensive Organic Name Reactions and Reagents, ed. Z. Wang, John Wiley & Sons, Inc., 2010 Search PubMed.
- (a) J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248 CrossRef CAS PubMed; (b) N. Carrillo, E. A. Davalos, J. A. Russak and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 1452 CrossRef CAS PubMed.
- (a) G.-S. Liu, Y.-Q. Zhang, Y.-A. Yuan and H. Xu, J. Am. Chem. Soc., 2013, 135, 3343 CrossRef CAS PubMed; (b) D.-F. Lu, G.-S. Liu, C.-L. Zhu, B. Yuan and H. Xu, Org. Lett., 2014, 16, 2912 CrossRef CAS PubMed; (c) D.-F. Lu, C.-L. Zhu, Z.-X. Jia and H. Xu, J. Am. Chem. Soc., 2014, 136, 13186 CrossRef CAS PubMed; (d) D.-F. Lu, C.-L. Zhu, J. D. Sears and H. Xu, J. Am. Chem. Soc., 2016, 138, 11360 CrossRef CAS PubMed; (e) C.-L. Zhu, J.-S. Tian, Z.-Y. Gu, G.-W. Xing and H. Xu, Chem. Sci., 2015, 6, 3044 RSC.
- (a) X. Dong, Q. Liu, Y. Dong and H. Liu, Chem. – Eur. J., 2017, 23, 2481 CrossRef CAS PubMed; (b) M. T. Pirnot, Y.-M. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48 CrossRef CAS PubMed; (c) C. E. Hendrick and Q. Wang, J. Org. Chem., 2017, 82, 839 CrossRef CAS PubMed; (d) A. M. Berman and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 5680 CrossRef CAS PubMed; (e) E. J. Yoo, S. Ma, T.-S. Mei, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7652 CrossRef CAS PubMed; (f) N. Matsuda, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2011, 13, 2860 CrossRef CAS PubMed; (g) R. P. Rucker, A. M. Whittaker, H. Dang and G. Lalic, J. Am. Chem. Soc., 2012, 134, 6571 CrossRef CAS PubMed; (h) R. P. Rucker, A. M. Whittaker, H. Dang and G. Lalic, Angew. Chem., Int. Ed., 2012, 51, 3953 CrossRef CAS PubMed; (i) N. Matsuda, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 3642 CrossRef CAS PubMed; (j) N. Matsuda, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 11827 CrossRef CAS PubMed; (k) Z. Dong and G. Dong, J. Am. Chem. Soc., 2013, 135, 18350 CrossRef CAS PubMed; (l) S. Zhu, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 15746 CrossRef CAS PubMed; (m) N. Matsuda, K. Hiranutero, T. Satoh and M. Miura, J. Am. Chem. Soc., 2013, 135, 4934 CrossRef CAS PubMed; (n) S. L. McDonald, C. E. Hendrick and Q. Wang, Angew. Chem., Int. Ed., 2014, 53, 4667 CrossRef CAS PubMed; (o) X. Yan, X. Yang and C. Xi, Catal. Sci. Technol., 2014, 4, 4169 RSC; (p) J. L. Jat, M. P. Paudyal, H. Gao, Q.-L. Xu, M. Yousufuddin, D. Devarajan, D. H. Ess, L. Kurti and J. R. Falck, Science, 2014, 343, 61 CrossRef CAS PubMed; (q) S. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2014, 136, 15913 CrossRef CAS PubMed; (r) A. Parra, L. Amenós, M. Guisán-Ceinos, A. López, J. L. García Ruano and M. Tortosa, J. Am. Chem. Soc., 2014, 136, 15833 CrossRef CAS PubMed; (s) S. L. McDonald and Q. Wang, Angew. Chem., Int. Ed., 2014, 53, 1867 CrossRef CAS PubMed; (t) K. Shen and Q. Wang, Chem. Sci., 2015, 6, 4279 RSC; (u) D. Niu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 9716 CrossRef CAS PubMed; (v) Y. Yang, S.-L. Shi, D. Niu, P. Liu and S. L. Buchwald, Science, 2015, 349, 62 CrossRef CAS PubMed; (w) K. Ohmatsu, Y. Ando, T. Nakashima and T. Ooi, Chem, 2016, 1, 802 CrossRef CAS; (x) S. A. Shuler, G. Yin, S. B. Krause, C. M. Vesper and D. A. Watson, J. Am. Chem. Soc., 2016, 138, 13830 CrossRef CAS PubMed; (y) B. N. Hemric, K. Shen and Q. Wang, J. Am. Chem. Soc., 2016, 138, 5813 CrossRef CAS PubMed; (z) M. P. Paudyal, A. M. Adebesin, J. R. Falck, S. R. Burt, D. H. Ess, Z. Ma and L. Kurti, Science, 2016, 353, 1144 CrossRef CAS PubMed; (a a) J. Liu, K. Wu, T. Shen, Y. Liang, M. Zou, Y. Zhu, X. Li, X. Li and N. Jiao, Chem. – Eur. J., 2017, 23, 563 CrossRef CAS PubMed.
- K. S. Williamson, D. J. Michaelis and T. P. Yoon, Chem. Rev., 2014, 114, 8016 CrossRef CAS PubMed.
- (a) T. Benkovics, J. Du, I. A. Guzei and T. P. Yoon, J. Org. Chem., 2009, 74, 5545 CrossRef CAS PubMed; (b) D. J. Michaelis, M. A. Ischay and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 6610 CrossRef CAS PubMed; (c) D. J. Michaelis, C. J. Shaffer and T. P. Yoon, J. Am. Chem. Soc., 2007, 129, 1866 CrossRef CAS PubMed; (d) K. S. Williamson and T. P. Yoon, J. Am. Chem. Soc., 2012, 134, 12370 CrossRef CAS PubMed; (e) T. Benkovics, I. A. Guzei and T. P. Yoon, Angew. Chem., Int. Ed., 2010, 49, 9153 CrossRef CAS PubMed.
- C. P. Allen, T. Benkovics, A. K. Turek and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 12560 CrossRef CAS PubMed.
- (a) H. Gao, Z. Zhou, D.-H. Kwon, J. Coombs, S. Jones, N. E. Behnke, D. H. Ess and L. Kürti, Nat. Chem., 2016, 9, 681 CrossRef PubMed; (b) C. Zhu, G. Li, D. H. Ess, J. R. Falck and L. Kurti, J. Am. Chem. Soc., 2012, 134, 18253 CrossRef CAS PubMed.
- (a) J. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 14103 CrossRef CAS PubMed; (b) Y. Park, S. Jee, J. G. Kim and S. Chang, Org. Process Res. Dev., 2015, 19, 1024 CrossRef CAS; (c) Y. Park, K. T. Park, J. G. Kim and S. Chang, J. Am. Chem. Soc., 2015, 137, 4534 CrossRef CAS PubMed; (d) R. Mei, J. Loup and L. Ackermann, ACS Catal., 2016, 6, 793 CrossRef CAS; (e) Y. Liang, Y.-F. Liang, C. Tang, Y. Yuan and N. Jiao, Chem. – Eur. J., 2015, 21, 16395 CrossRef CAS PubMed.
- (a) X. Yu, K. Chen, F. Yang, S. Zha and J. Zhu, Org. Lett., 2016, 18, 5412 CrossRef CAS PubMed; (b) X. Yu, K. Chen, Q. Wang, S. Guo, S. Zha and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 5222 CrossRef CAS PubMed.
- (a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed; (b) I. Bauer and H.-J. Knoelker, Top. Curr. Chem., 2012, 309, 203 CrossRef CAS PubMed; (c) H. Fan, J. Peng, M. T. Hamann and J.-F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS PubMed; (d) J. C. P. Reyes and D. Romo, Angew. Chem., Int. Ed., 2012, 51, 6870 CrossRef CAS PubMed.
- S. S. Gholap, Eur. J. Med. Chem., 2016, 110, 13 CrossRef CAS PubMed.
- For selected reviews, see: (a) E. Vessally, RSC Adv., 2016, 6, 18619 RSC; (b) V. Estevez, M. Villacampa and J. C. Menendez, Chem. Soc. Rev., 2014, 43, 4633 RSC; (c) F. J. Leeper and J. M. Kelly, Org. Prep. Proced. Int., 2013, 45, 171 CrossRef CAS; (d) V. F. Ferreira, M. C. B. V. de Souza, A. C. Cunha, L. O. R. Pereira and M. L. G. Ferreira, Org. Prep. Proced. Int., 2001, 33, 411 CrossRef CAS. For examples of pyrrole synthesis via transition metal catalyzed N–O bond cleavage of ketoximes, see: (e) W. Du, M.-N. Zhao, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Chem. Commun., 2014, 50, 7437 RSC; (f) H.-Y. Wang, D. S. Mueller, R. M. Sachwani, R. Kapadia, H. N. Londino and L. L. Anderson, J. Org. Chem., 2011, 76, 3203 CrossRef CAS PubMed; (g) H.-Y. Wang, D. S. Mueller, R. M. Sachwani, H. N. Londino and L. L. Anderson, Org. Lett., 2010, 12, 2290 CrossRef CAS PubMed; (h) A. Fürstner, K. Radkowski and H. Peters, Angew. Chem., Int. Ed., 2005, 44, 2777 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The details of experiments, compound characterization and spectroscopic data. See DOI: 10.1039/c7qo00698e |
| This journal is © the Partner Organisations 2018 |