
Stimuli-responsive perallyloxycucurbit[6]uril-based nanoparticles for selective drug delivery in melanoma cells†
Qian
Cheng‡
,
Shengke
Li‡
,
Chen
Sun
,
Ludan
Yue
 and
Ruibing
Wang
*
and
Ruibing
Wang
*
State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Taipa, Macau SAR, China. E-mail: rwang@umac.mo
First published on 29th September 2018
Abstract
Perallyloxycucurbit[6]uril nanoparticles were prepared for the first time via an emulsion method, and upon UVA-light irradiation they exhibited selective payload release in melanoma cells.
Cucurbit[n]urils (CB[n], n = 5–8) are a family of synthetic macrocycles, composed of n glycoluril units inter-connected by 2n methylene groups, forming a macrocyclic cavity accessible via two identical carbonyl-rimmed portals.1 Among the family of CB[n], CB[7] and its derivatives have attracted significant attention for their potential applications in pharmaceutical sciences and drug delivery,2 whereas CB[6] and its derivatives have been rarely studied directly as drug carriers, partly due to the small cavity size of CB[6] that cannot accommodate many common drug molecules. However, CB[6] is often the dominant product of the CB[n] synthesis, and its functionalization is often more achievable.3 For instance, Kim et al. reported the perhydroxylation of CB[n], which mainly worked for CB[6].3a Further derivatization of perhydoxyCB[6] led to (allyloxy)12CB[6] (AO12CB[6]), a derivative of CB[6] with 12 reactive allyloxy groups at the periphery,3a which allowed further laborious functionalization forming nanoparticles (NPs) for a variety of biomedical applications including drug delivery and bio-imaging.4 The preparation of NPs directly from AO12CB[6] without additional functionalization has not been considered possible or practical for drug delivery and thus has never been reported. In fact, very classical thiol–ene click conjugation can take place between the allyloxy groups of AO12CB[6] and glutathione (GSH) (that is typically enriched inside cancer cells)5 upon light irradiation. Thus, AO12CB[6] based materials may become responsive to light in the presence of GSH. On the other hand, melanoma, as one of the most deadly cancers in the world, still lacks effective therapies.6 Systemic chemotherapy, due to its poor selectivity, often leads to significant side effects and resistance.7 Therefore, the selective delivery of chemotherapeutic agents into melanoma cells is particularly needed for the effective treatment of this type of cancer. In light of particular medical needs, the special feature of melanoma (e.g. accessible to sunlight and over-expression of GSH inside the cells), and the light-triggered reaction between allyloxy and GSH, herein we report novel, light-triggered GSH-responsive NPs directly formed from AO12CB[6] using an emulsion method, which can be used for specifically delivering chemotherapeutic drugs into melanoma cells (Scheme 1). The NPs exhibited uniform and stable morphology in an aqueous solution, high drug loading efficiency, effective cellular uptake and selective drug release in cancer cells upon Ultraviolet A (UVA) irradiation. To the best of our knowledge, this is the first GSH-responsive nanomedicine platform prepared via an emulsion method directly from AO12CB[6] without the need for further ingenious design and laborious functionalizations.4a,b,8
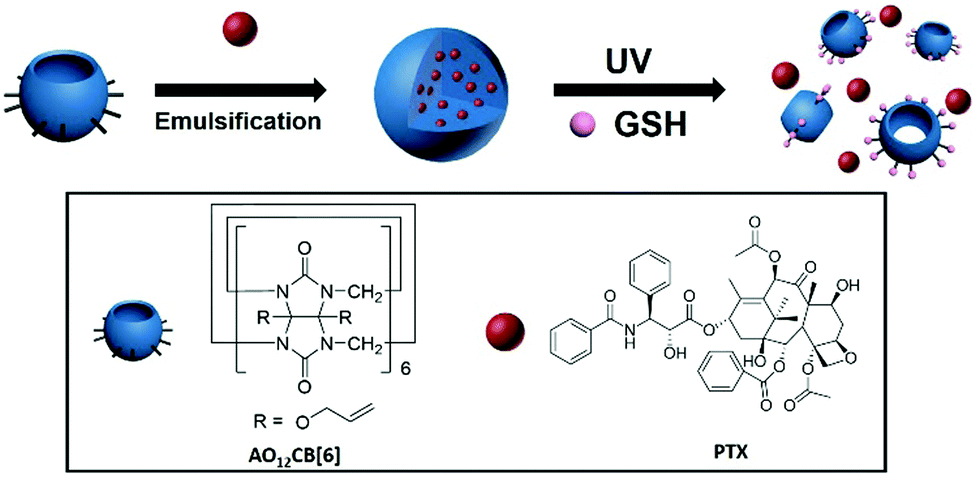 | ||
| Scheme 1 Scheme of the preparation of the AO12CB[6] NPs and their stimuli-responsive drug release. | ||
Firstly, the emulsion method was unprecedentedly employed to fabricate AO12CB[6] NPs, attributed to the lipophilicity of AO12CB[6]. Briefly described here, 1 mL of dichloromethane (DCM) solution containing 20 mg AO12CB[6] was added to 4 mL of 1.0 wt% aqueous solution of polyvinyl alcohol for emulsification. After sonication for 1 h, the NPs were collected by centrifuge and were subsequently characterized by transmission electron microscopy (TEM), scanning electron microscopy (SEM) and dynamic light scattering (DLS). As shown in Fig. 1(a), the NPs in aqueous solution exhibited a distinct Tyndall effect, indicating their colloidal nature. DLS measurements yielded a mean diameter of 220 nm with a narrow size distribution. Both the TEM (Fig. 1(b)) and SEM (Fig. 1(c)) images revealed the fine solid spherical morphology of the NPs with ca. 180 nm mean diameter. The nuances of the size measurements by these methods were mainly attributed to different experimental conditions (an aqueous solution for DLS vs. a solid state for TEM and SEM). For more intuitive observations, the NPs were loaded with a fluorescent dye, Cy5, generating red-fluorescent nano-dots that were observable under a confocal laser scanning microscope (CLSM) (Fig. 1(d)). In addition, the stability of the AO12CB[6] NPs in an aqueous solution was investigated by measuring their sizes by DLS over time under ambient conditions. The results (Table S1, ESI†) showed a reasonably good stability profile of the NPs after storage under ambient conditions for 14 days.
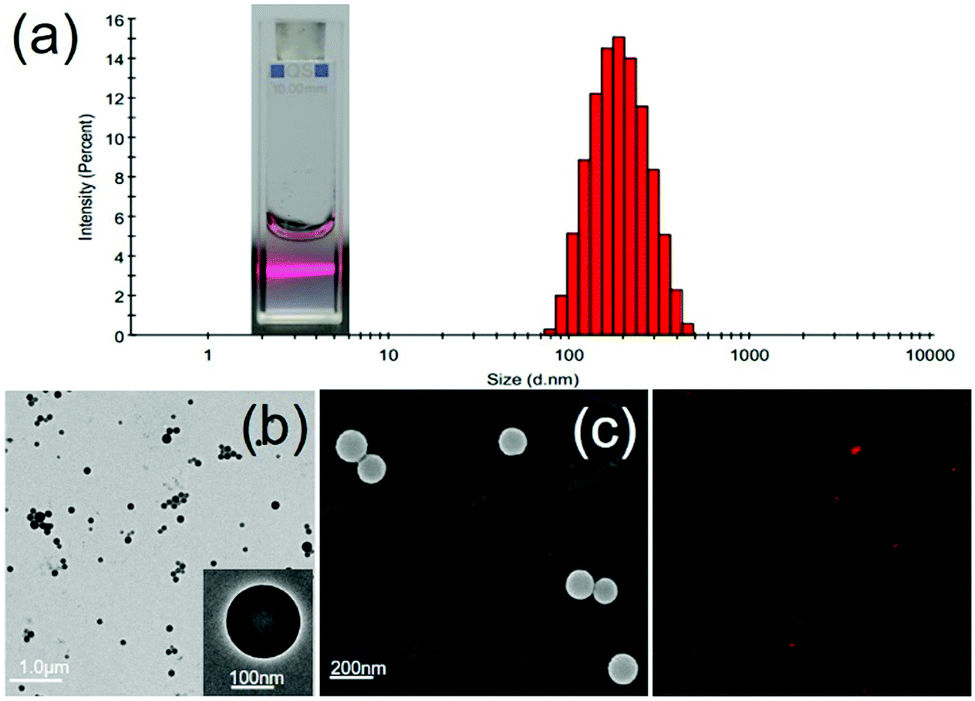 | ||
| Fig. 1 (a) Size and size-distribution of the AO12CB[6] NPs in an aqueous solution, measured by DLS. Inset: Tyndall effect of the NPs in an aqueous solution. (b) Representative TEM image of the AO12CB[6] NPs. Inset: Enlarged image showing an individual NP. (c) Representative SEM image of the AO12CB[6] NPs. (d) CLSM image of the Cy5 loaded NPs. | ||
To investigate intracellular drug delivery of the NPs, paclitaxel (PTX) was employed as a representative anticancer drug because of its poor water solubility and low specificity that often leads to severe side effects in its clinical applications.9 The preparation of PTX loaded NPs (PTX-NPs) was conducted using the same method as described above, by dissolving both PTX and AO12CB[6] together in DCM. The drug encapsulation efficiency (DEE) and drug loading efficiency (DLE) of the PTX loaded NPs reached up to 91.0% and 34.7%, respectively (Table S2, ESI†), suggesting a significant potential for practical application of the NPs as drug carriers.
It is well known that GSH concentration is significantly higher in cancer cells, e.g. melanoma cells (10 mM), in contrast to that in normal cells (1 mM).5 Thus GSH has been frequently employed as a stimuli to induce specific payload release in cancer cells.10 As previously discussed, the allyloxy group of AO12CB[6] would react with GSH upon UV-light (or sunlight) irradiation,11 leading to the disruption of the integrity of the NPs, thereby releasing the therapeutic payload, PTX. Very importantly, previous studies have shown that mammalian cells experience negligible damage upon UVA exposure for up to 2 h,12 and UVA light is abundant in sunlight as it is not absorbed by the ozone layer, thus we chose 365 nm UVA light as the irradiation light source to trigger the reaction of AO12CB[6] and GSH in this study. Firstly, the sensitivity of the AO12CB[6] NPs to a high concentration of GSH upon UVA irradiation (365 nm) was investigated in the absence and presence of 10 mM GSH, respectively. As shown in Fig. S1 (ESI†), upon UVA irradiation, the particle size of the AO12CB[6] NPs in the presence of 10 mM GSH increased significantly in a time dependent manner, similar to the behaviours of several other previously reported GSH-responsive NPs.13 In contrast, very moderate changes in size were observed for the NPs irradiated under UVA light without GSH. And negligible changes in size were observed for the NPs in the absence or in the presence of 10 mM GSH without UVA irradiation, suggesting a good stability of the AO12CB[6] NPs in the absence of either stimuli (GSH or UVA irradiation), both of which are typically encountered in melanoma. Subsequently, the drug release profile of PTX-NPs in the absence and presence of 1 and 10 mM GSH (respectively representing the GSH conditions inside non-cancerous and melanoma cells) under UVA irradiation was evaluated using HPLC. As shown in Fig. 2, the PTX loaded NPs exhibited excellent stability in the absence of GSH, with less than 10% accumulative drug release upon UVA irradiation for up to 2.5 h. When the GSH concentration was increased to 1 mM, the PTX-NPs exhibited a relatively slow drug release kinetics under UVA irradiation for 2.5 h, with approximately 35% accumulated drug release. In contrast, when the concentration of GSH reached 10 mM, the NPs exhibited rapid drug release under UVA irradiation, and the accumulative release of PTX reached ca. 70%, suggesting a successful, sunlight-triggerable GSH-responsive payload release as a result of the morphological changes of the AO12CB[6] NPs due to the reaction between allyloxy groups and GSH under UVA.
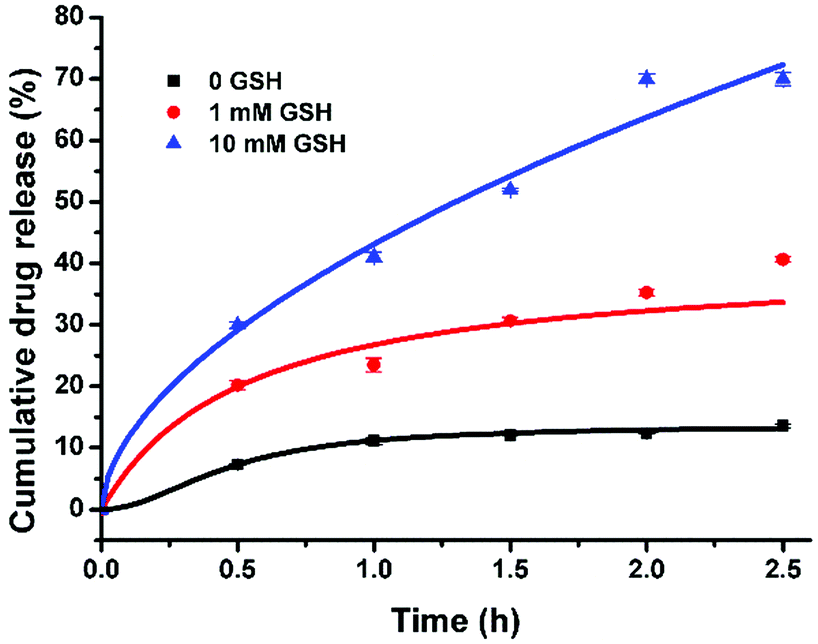 | ||
| Fig. 2 UVA irradiation time-dependent PTX release profile of the PTX loaded AO12CB[6] NPs at different GSH concentrations (n = 3). | ||
Prior to our investigations of the NPs for drug delivery applications in vitro, we examined their cytotoxicity profile, via MTT assays, against murine macrophage cell line RAW 264.7 and murine melanoma cell line B16, respectively. As shown in Fig. S2 (ESI†), upon exposure to up to 200 μg mL−1 NPs for 36 h, both cell lines still remained highly viable, suggesting a good biocompatibility profile of the blank AO12CB[6] NPs. To further examine the potential of the AO12CB[6] NPs for intracellular drug delivery into melanoma cells, the cellular uptake of the NPs was investigated with the Cy5 loaded NPs (red fluorescence) in B16 cell lines by CLSM. DAPI (blue fluorescence) was employed to stain the nucleus of the B16 cells. As shown in Fig. 3, upon incubation of the cells with Cy5-NPs for 2 and 4 h, respectively, the red fluorescence (Cy5) was weak under a confocal microscope. After 8 h incubation, red fluorescence (Cy5) became more visible in the cytoplasm, and more intense red fluorescence was observed after 12 h incubation, suggesting that the Cy5-NPs were efficiently endocytosed by the B16 cells after incubation for approximately 8 h or longer.
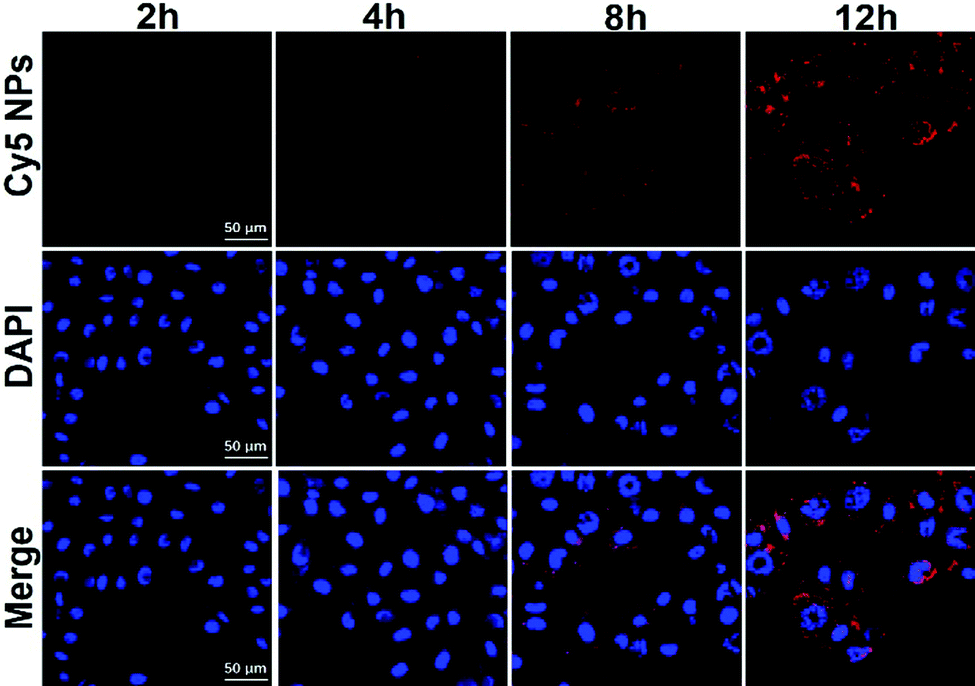 | ||
| Fig. 3 CLSM images showing the intracellular uptake of the Cy5-NPs by the B16 cancer cells after incubation for 2, 4, 8 and 12 h. The scale bars represent 50 μm. | ||
Furthermore, the selective therapeutic efficacy of the PTX-NPs against melanoma cells was examined with RAW 264.7 and B16 cells for comparison, via MTT assays. Both cell lines were incubated with various concentrations of free PTX and the PTX-NPs for 12 h, subject to subsequent incubation for another 2 h with or without UVA irradiation (λ = 365 nm) and were incubated for an additional 24 h for the MTT assays. As shown in Fig. 4, the IC50 (50% of cell growth inhibition concentration) value of the PTX-NPs against B16 with 2 h UVA irradiation was 4.31 μM, significantly more effective than free PTX (IC50 = 10.12 μM), presumably due to the effective internalization of the PTX-NPs into cells and subsequent UVA-triggered PTX release in the presence of high concentration endogenous GSH inside the cancer cells. Without UVA irradiation, the PTX-NPs exhibited very inert activity with a rather high IC50 value of 15.60 μM against the B16 cells, suggesting the importance of UVA (naturally available through natural sunlight exposure on melanoma) irradiation in selective payload release in cancer cells. Conversely, the PTX-NPs exhibited obviously reduced toxicity against the non-cancerous cell line, RAW 264.7, with or without UVA irradiation. The IC50 values of the PTX-NPs against the RAW 264.7 cell line, with and without 2 h UVA irradiation, were 185.90 and 207.00 nM, respectively, approximately 6-fold larger than that of free PTX (31.07 nM). This enhanced safety profile of PTX in non-cancer cells was likely attributed to the relatively low GSH concentration inside non-cancerous cells, which could not effectively trigger the structural disruption of the AO12CB[6] NPs, even under UVA irradiation, attesting the discriminative safety profile of these NPs.
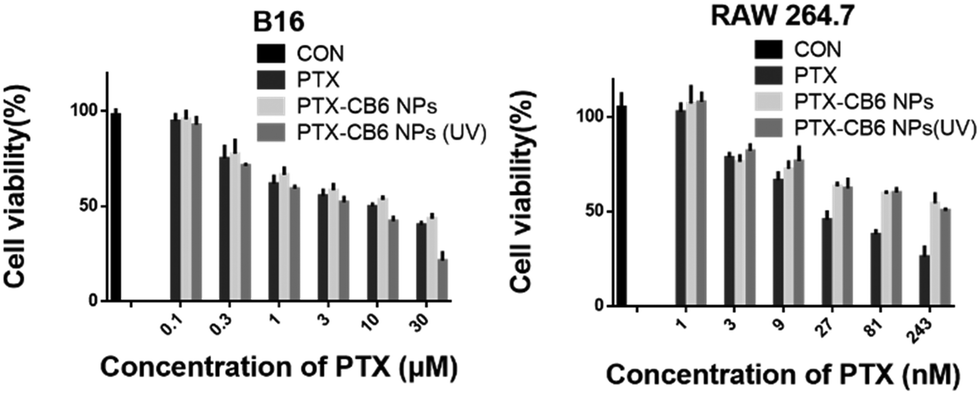 | ||
| Fig. 4 Cytotoxicity of the PTX-loaded AO12CB[6] NPs against B16 (left) and RAW 264.7 (right) cells. The cells were first incubated with PTX or PTX-loaded NPs for 12 h, subjected to subsequent incubation with or without UVA irradiation for 2 h, and incubated for an additional 24 h for the MTT assays. | ||
In addition to the MTT assays, the Annexin V-FITC/propidium iodide (PI) double-staining assay was employed for the examination of the apoptosis rates in the RAW 264.7 and B16 cells treated with PTX and PTX-NPs, respectively, in order to further demonstrate the selective cytotoxicity of the PTX-NPs against cancer cells. As shown in Fig. S3 (ESI†), the apoptosis rate in the B16 cancer cells was enhanced by the PTX-NPs under UVA irradiation for 2 h. Of note, the UVA irradiation of the cells induced negligible apoptosis rate, confirming the safety of UVA light on cells within 2 h irradiation. In the absence of UVA irradiation, the apoptosis rates of the cancer cells induced by the free PTX and PTX-NPs were nearly identical, suggesting that UVA irradiation is a necessary stimulus for endowing the NPs with selective toxicity. Conversely, a significantly decreased apoptosis rate was observed in the RAW 264.7 cells treated with the PTX-NPs without UVA irradiation, in comparison with that of the cells treated with PTX, confirming the safety profile of the PTX-NPs against non-cancerous cells. Even in the presence of UVA irradiation, the apoptosis rate in the RAW 264.7 cells induced by the PTX-NPs was still moderately lower than that of the free PTX, consistent with our MTT results.
In summary, we present a novel, UVA (sunlight)-triggered, GSH-responsive nanoparticle system built upon AO12CB[6] directly, in which a hydrophobic chemotherapeutic drug can be loaded with high loading efficiency and content, for potentially selective therapy of melanoma. The encapsulated payload was selectively released in the presence of 10 mM GSH upon UVA irradiation that is naturally available via the natural sun-bath of the melanoma affected areas. The PTX loaded nanoparticles not only exhibited efficient cellular uptake, but also significantly increased the cytotoxicity and apoptosis rate of cancer cells, with remarkably reduced cytotoxicity against non-cancerous cells under UVA light irradiation. This study provides the first, facile CB[6] derivative based nanomedicine platform for efficient stimuli-responsive release of hydrophobic drugs in melanoma cells in a selective manner. This new discovery will further strengthen the development of other CB[6] derivative based nanoparticles for biomedical applications.
This work was financially supported by the Science and Technology Development Fund, Macao SAR (Grant No. FDCT 030/2017/A1) and the Research Committee at University of Macau (Grant No. MYRG2016-00165-ICMS-QRCM and MYRG2017-00010-ICMS).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621 CrossRef CAS; (b) J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844 CrossRef CAS; (c) S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320 CrossRef CAS; (d) K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394 RSC; (e) Q. Li, J. Sun, J. Zhou, B. Hua, L. Shao and F. Huang, Org. Chem. Front., 2018, 5, 1940 RSC; (f) Q. Li, S.-C. Qiu, J. Zhang, K. Chen, Y. Huang, X. Xiao, Y. Zhang, F. Li, Y.-Q. Zhang, S.-F. Xue, Q.-J. Zhu, Z. Tao, L. F. Lindoy and G. Wei, Org. Lett., 2016, 18, 4020 CrossRef CAS; (g) J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021 RSC.
- (a) D. Shetty, J. K. Khedkar, K. M. Park and K. Kim, Chem. Soc. Rev., 2015, 44, 8747 RSC; (b) Y. Chen, Z. Huang, H. Zhao, J.-F. Xu, Z. Sun and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 8602 CrossRef CAS; (c) X. Yang, S. Li, Q.-W. Zhang, Y. Zheng, D. Bardelang, L.-H. Wang and R. Wang, Nanoscale, 2017, 9, 10606 RSC; (d) Q. Huang, K. I. Kuok, X. Zhang, L. Yue, S. M. Y. Lee, J. Zhang and R. Wang, Nanoscale, 2018, 10, 10333 RSC; (e) H. Chen, Y. Chen, H. Wu, J.-F. Xu, Z. Sun and X. Zhang, Biomaterials, 2018, 178, 697 CrossRef CAS.
- (a) S. Y. Jon, N. Selvapalam, D. H. Oh, J.-K. Kang, S.-Y. Kim, Y. J. Jeon, J. W. Lee and K. Kim, J. Am. Chem. Soc., 2003, 125, 10186 CrossRef CAS; (b) N. Zhao, G. O. Lloyd and O. A. Scherman, Chem. Commun., 2012, 48, 3070 RSC; (c) M. M. Ayhan, H. Karoui, M. Hardy, A. Rockenbauer, L. Charles, R. Rosas, K. Udachin, P. Tordo, D. Bardelang and O. Ouari, J. Am. Chem. Soc., 2015, 137, 10238 CrossRef CAS.
- (a) E. Kim, D. Kim, H. Jung, J. Lee, S. Paul, N. Selvapalam, Y. Yang, N. Lim, C. G. Park and K. Kim, Angew. Chem., Int. Ed., 2010, 49, 4405 CrossRef CAS; (b) K. M. Park, D.-W. Lee, B. Sarkar, H. Jung, J. Kim, Y. H. Ko, K. E. Lee, H. Jeon and K. Kim, Small, 2010, 6, 1430 CrossRef CAS; (c) H. Jung, K. M. Park, J.-A. Yang, E. J. Oh, D.-W. Lee, K. Park, S. H. Ryu, S. K. Hahn and K. Kim, Biomaterials, 2011, 32, 7687 CrossRef CAS; (d) S. Kim, G. Yun, S. Khan, J. Kim, J. Murray, Y. M. Lee, W. J. Kim, G. Lee, S. Kim, D. Shetty, J. H. Kang, J. Y. Kim, K. M. Park and K. Kim, Mater. Horiz., 2017, 4, 450 RSC.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071 CrossRef CAS.
- (a) P. B. Chapman, A. Hauschild, C. Robert, J. B. Haanen, P. Ascierto, J. Larkin, R. Dummer, C. Garbe, A. Testori, M. Maio, D. Hogg, P. Lorigan, C. Lebbe, T. Jouary, D. Schadendorf, A. Ribas, S. J. O'Day, J. A. Sosman, J. M. Kirkwood, A. M. M. Eggermont, B. Dreno, K. Nolop, J. Li, B. Nelson, J. Hou, R. J. Lee, K. T. Flaherty and G. A. McArthur, N. Engl. J. Med., 2011, 364, 2507 CrossRef CAS; (b) A. Hauschild, J.-J. Grob, L. V. Demidov, T. Jouary, R. Gutzmer, M. Millward, P. Rutkowski, C. U. Blank, W. H. Miller, E. Kaempgen, S. Martín-Algarra, B. Karaszewska, C. Mauch, V. Chiarion-Sileni, A.-M. Martin, S. Swann, P. Haney, B. Mirakhur, M. E. Guckert, V. Goodman and P. B. Chapman, Lancet, 2012, 380, 358 CrossRef CAS.
- (a) D. A. Tennant, R. V. Durán and E. Gottlieb, Nat. Rev. Cancer, 2010, 10, 267 CrossRef CAS; (b) R. Mahato, W. Tai and K. Cheng, Adv. Drug Delivery Rev., 2011, 63, 659 CrossRef CAS.
- K. M. Park, K. Suh, H. Jung, D.-W. Lee, Y. Ahn, J. Kim, K. Baek and K. Kim, Chem. Commun., 2009, 71 RSC.
- Y. Shi, R. van der Meel, B. Theek, E. Oude Blenke, E. H. E. Pieters, M. H. A. M. Fens, J. Ehling, R. M. Schiffelers, G. Storm, C. F. van Nostrum, T. Lammers and W. E. Hennink, ACS Nano, 2015, 9, 3740 CrossRef CAS.
- F. Q. Schafer and G. R. Buettner, Free Radical Biol. Med., 2001, 30, 1191 CrossRef CAS.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (b) K. M. Park, K. Baek, Y. H. Ko, A. Shrinidhi, J. Murray, W. H. Jang, K. H. Kim, J.-S. Lee, J. Yoo, S. Kim and K. Kim, Angew. Chem., Int. Ed., 2018, 57, 3132 CrossRef CAS.
- A. de Laat, M. van Tilburg, J. C. von der Leun, W. A. van Vloten and F. R. de Gruijl, Photochem. Photobiol., 1996, 63, 492 CrossRef CAS.
- (a) Y.-w. Hu, Y.-z. Du, N. Liu, X. Liu, T.-t. Meng, B.-l. Cheng, J.-b. He, J. You, H. Yuan and F.-q. Hu, J. Controlled Release, 2015, 206, 91 CrossRef CAS; (b) C. Gao, F. Tang, J. Zhang, S. M. Y. Lee and R. Wang, J. Mater. Chem. B, 2017, 5, 2337 RSC; (c) Q. Cheng, H. Yin, C. Sun, L. Yue, Y. Ding, W. Dehaen and R. Wang, Chem. Commun., 2018, 54, 8128 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experiment details, NP stability data, biocompatibility and apoptosis results. See DOI: 10.1039/c8qm00460a |
| ‡ These two authors contributed to this work equally. |
| This journal is © the Partner Organisations 2019 |