
Unexpected room-temperature phosphorescence from a non-aromatic, low molecular weight, pure organic molecule through the intermolecular hydrogen bond†
Manman
Fang
a,
Jie
Yang
a,
Xueqin
Xiang
b,
Yujun
Xie
a,
Yongqiang
Dong
 b,
Qian
Peng
c,
Qianqian
Li
a and
Zhen
Li
*a
b,
Qian
Peng
c,
Qianqian
Li
a and
Zhen
Li
*a
aDepartment of Chemistry, Hubei Key Laboratory on Organic and Polymeric Opto-Electronic Materials, Wuhan University, Wuhan 430072, P. R. China. E-mail: lizhen@whu.edu.cn; lichemlab@163.com; Fax: +86-27-68756757
bDepartment of Chemistry, Beijing Normal University, Beijing, 100875, P. R. China
cKey Laboratory of Organic Solids, Beijing National Laboratory for Molecular Science (BNLMS), Institute of Chemistry, Chinese Academy of Sciences, 100190 Beijing, China
First published on 18th September 2018
Abstract
Efficient pure organic room temperature phosphorescence (RTP) materials have drawn considerable attention. So far, most pure organic RTP molecules are aromatic compounds, and nonconjugated molecules are really scarce. The only few reported non-aromatic organic phosphorescence materials are polymers without confirmed subtle structures, and there are no reports concerning non-aromatic organic small molecules with persistent RTP. Here, we report an example of a pure non-aromatic organic small molecule, cyanoacetic acid, that shows unexpected persistent RTP behavior with RTP lifetime as long as 862 ms. According to the CAA crystal and theoretical calculations, the presence of strong intermolecular hydrogen bonds is the key factor for its persistent RTP effect. This discovery demonstrates a clear relationship between the molecular structure, packing mode and RTP effect in the non-aromatic system, which will largely extend the current pure organic RTP systems to deeply investigate the origin of light emission.
1 Introduction
Room temperature phosphorescence (RTP) is a fascinating phenomenon, with potential applications such as bioimaging, sensing and displays. However, RTP emission often occurs in inorganic and organometallic materials, which suffer some intrinsic problems, including high cost, toxicity and instability in aqueous environments. Alternatively, regardless of their advantages, efficient pure organic RTP materials are difficult to obtain.1 According to previous reports, there are two key critical requirements for pure organic materials with persistent RTP: (1) the particular functional groups which could favor n–π* transitions and lead to the efficient intersystem crossing (ISC) transition between singlet (S) and triplet (T) states, and (2) a special packing mode or rigid environment in the solid state that could stabilize triplet excitons, either to reduce the radiative transition from T1 to S0 or suppress the non-radiative transition (Fig. 1A).2 In order to obtain efficient organic RTP materials, the host–guest doping system is frequently investigated, regardless of its relative complexity with the very careful control of dosage concentration.3 Fortunately, some pure RTP organic materials with a single component have been developed in recent years.4 Huang et al. obtained ultralong RTP through stabilizing triplet excited states in the presence of intermolecular H-aggregates.4a Tang et al. described the phenomenon of crystallization-induced phosphorescence.4b Chi et al. proposed a new mechanism based on n and π subunits for RTP.4c In contrast to the above cases with aromatic π systems, only scarce examples of nonconjugated pure organic polymers with RTP phenomenon but very short lifetime have been reported, and a few reports have claimed that nonaromatic compounds without the value of lifetime might show delayed fluorescence, rather than room temperature phosphorescence.5 In 2013, Tang et al. observed weak RTP from crystalline natural products of cellulose, rice, starch and bovine serum albumin without any conventional chromospheres.5a Recently, Yuan et al. reported nonconjugated polyacrylonitrile polymer with a short RTP lifetime of 0.2 ms.5b However, polymers are mixtures without exactly accurate structures, thus resulting in the unclear internal mechanism of their RTP effects. If a non-aromatic organic small molecules system could be found, more important information on the relationship between RTP and molecular structure, as well as the molecular packing, could be revealed. According to previous literature and our recent work on pure organic aromatic RTP systems, strong intermolecular interactions play an important role and could even directly determine whether they possess RTP or not.6 Thus, to explore the non-aromatic organic small RTP molecules, the strong intermolecular interactions should be considered seriously. From the basic textbook Organic Chemistry, carboxylic acids could form association complexes due to their strong intermolecular hydrogen bonds.7 From this point, we focused our eyes on different carboxylic acids, considering some other electronic parameters. Excitedly, we observed RTP from cyanoacetic acid (CAA), a non-aromatic organic small molecule with the very low molecular weight of 85 (Fig. 1B). According to the analyses of its crystal and theoretical calculations, the strong intermolecular hydrogen bond is mainly responsible for the RTP effect. To the best of our knowledge, this is the first example of a nonaromatic pure organic small molecule with persistent RTP property.8 This discovery could provide some deep understanding in the inherent mechanism and contribute much to the further development of RTP systems with excellent performance.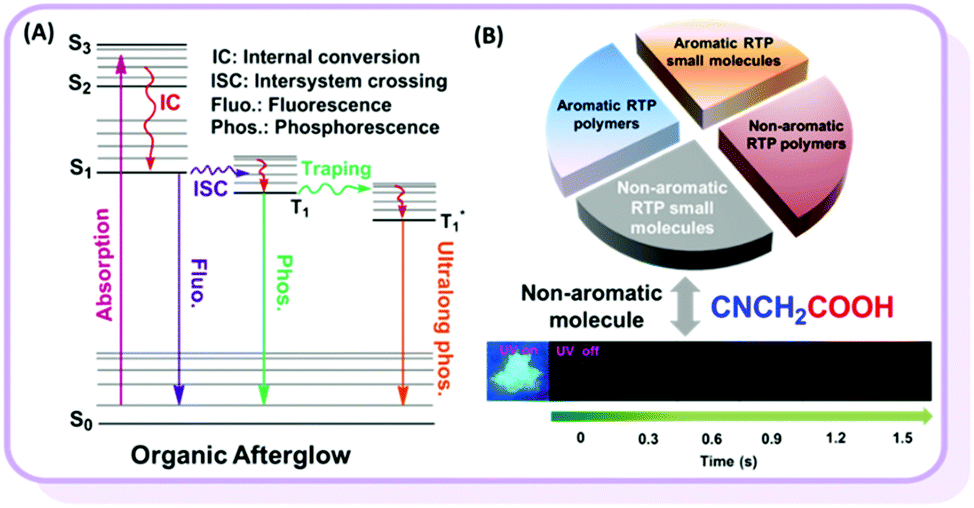 | ||
| Fig. 1 (A) The general diagram for organic afterglow and the category of pure organic room temperature phosphorescence (RTP) materials. (B) The examples for RTP small aromatic molecules, aromatic RTP polymers, and non-aromatic RTP polymers have been reported in recent years, but organic non-aromatic RTP small molecules have never been found. Photographs of CAA crystal taken at room temperature under 365 nm UV light or after ceasing the UV irradiation. | ||
2 Results and discussion
In order to eliminate the possible interference of possible impurities on RTP, commercial CAA was further purified by silica gel chromatography followed by recrystallization (the nuclear magnetic resonance spectrum, elemental analyses, mass spectra, the high-performance liquid chromatogram spectra and gas chromatography-mass spectrometry proved the purity of the CAA sample, as shown in Fig. S1–S4 in the ESI†). When irradiated at 365 nm (Fig. 2A and Fig. S5a, ESI†), both the crystal and powder of CAA exhibited dual fluorescence and phosphorescence at room temperature. The fluorescence peak of CAA crystal centered at 417 nm with a lifetime of 3.44 ns, while a lifetime of 2.68 ns for CAA powder (λmax = 418 nm) was found (Fig. S6 and Table S1, ESI†). No delayed fluorescence (DF) was found, as shown in Fig. S6 (ESI†). The strong fluorescence could be easily observed upon the excitation of a UV lamp. More excitingly, after the UV lamp was turned off, the persistent green RTP emission could be seen by the naked eye due to its ultralong luminescence lifetime of 0.862 s (CAA crystal) (Fig. 2B and Movie S1, ESI†). For CAA powder, the lifetime decreased to 0.674 s (Fig. S5b, ESI†). The absolute photoluminescence quantum efficiencies for CAA crystal and powder were measured to be 8.5% and 7.6% respectively, while their corresponding RTP quantum efficiencies are 2.1% and 1.8%.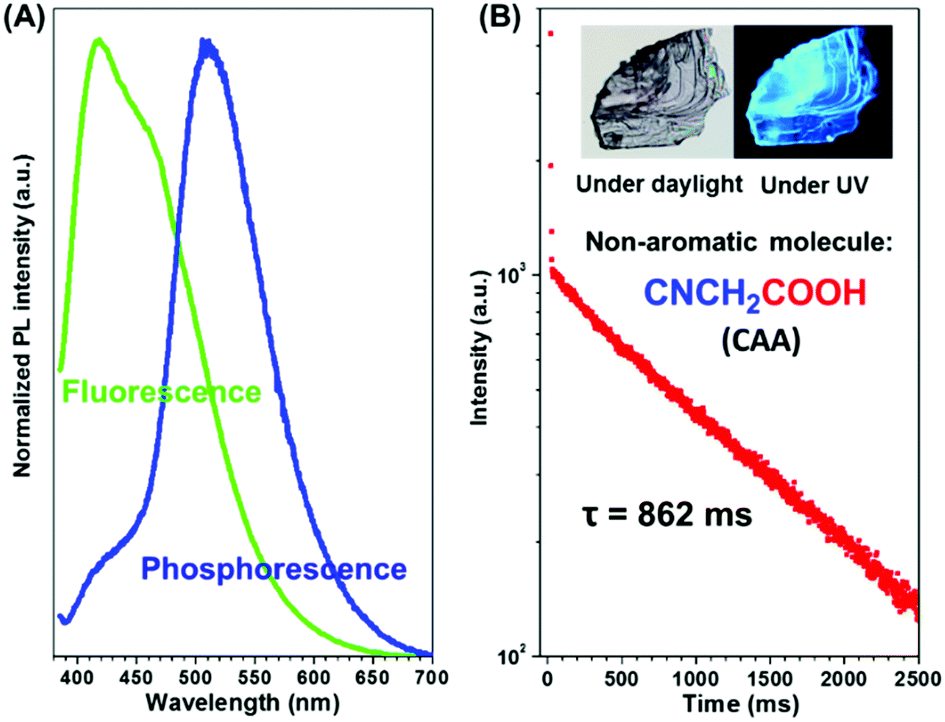 | ||
| Fig. 2 (A) Fluorescence and phosphorescence spectra of CAA crystal at room temperature. (B) Time-resolved measurement of 506 nm emission from CAA crystal. Inset: The crystal image of CAA under daylight and taken upon excitation using the optical microscope Leica M123. Scale bar: 100 μm. | ||
At the low temperature of 77 K, a similar phenomenon was observed, and both the crystal and powder exhibited much longer phosphorescence lifetimes (1.385 s and 1.340 s), as well as much higher PL efficiencies (83.0% and 57.7%), thanks to the rigid molecular conformation at low temperature (Table S1, Fig. S7 and S8, ESI†).
To further investigate the emissive mechanism of CAA, we dissolved CAA in water at different concentrations. As the concentration of CAA increased, the fluorescence intensity increased accordingly (Fig. 3 and Fig. S9, ESI†). Visible emissions were observed with concentrations higher than 1.00 × 10−1 M, but no emission was detected at the concentration lower than 1.00 × 10−2 M (Fig. 3A). This was totally opposite to that of traditional luminogens, which display the concentration quenching effect. Reasonably, as the concentration increased, the absorption was also enhanced, indicating the formation of certain aggregates and energy levels (Fig. 3B). The short PL lifetimes around 5 ns indicated that fluorescence was from the excited singlet state (Fig. S10 and Table S2, ESI†). Similar results were obtained when CAA was dissolved in tetrahydrofuran (THF) (Fig. 3C and D). Also, the excitation spectra of the fluorescence emissions were measured in varying concentrations for both THF and H2O solutions (Fig. S11, ESI†). As easily seen, the excitation spectra were nearly parallel to the X-axis at the concentrations below 0.1 M. Then, enhanced excitation spectra could be observed with the concentrations increasing from 0.1 M to 1 M, which well corresponds to the PL emissions. The solid-state UV-visible spectrum of CAA was also measured (Fig. S12a, ESI†), and the weak absorptions were in the region of 250–500 nm, further confirming the formation of a certain energy level in solid state. Also, the excitation (at 510 nm) spectra of CAA crystal and powder in phosphorescence mode were measured. As shown in Fig. S11a (ESI†), results match well with their corresponding absorption spectra, demonstrating that phosphorescence was not due to artifacts or impurities. The above results also indicated that it was the aggregation, not the single molecule, that gave out the emission, showing the characteristic of aggregation-induced emission.
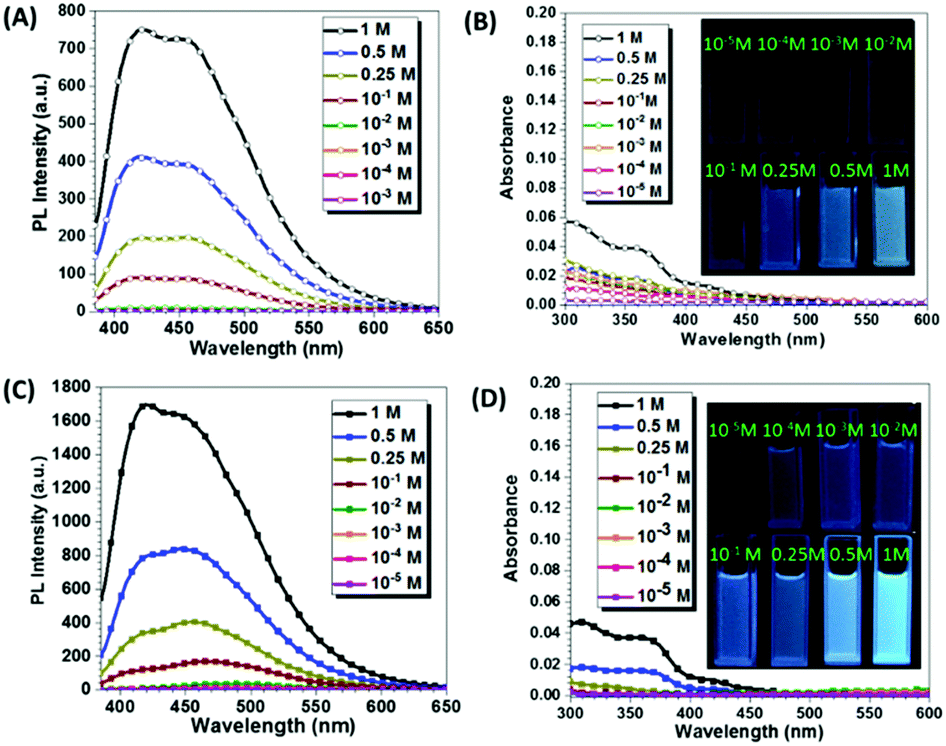 | ||
| Fig. 3 (A) Fluorescence spectra of CAA in H2O solutions at different concentrations (M) (λex = 365 nm). (B) Absorption spectra of CAA in H2O solutions at different concentrations; photographs taken under 365 nm UV light of CAA in H2O. (C) Fluorescence spectra of CAA in THF solutions at varying concentrations (M) (λex = 365 nm). (D) Absorption spectra of CAA in THF solutions at different concentrations; photographs taken under 365 nm UV light of CAA in THF. | ||
As shown in the X-ray diffraction (PXRD) spectra, the crystal of CAA exhibited a high and sharp diffraction peak at 2θ = 29.53° in comparison with that of the CAA powder (Fig. S12b, ESI†). Based on the peak at 2θ = 29.53°, the interlayer spacing was calculated to be 3.0221 Å, according to the Bragg equation (2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ = nλ).
sinθ = nλ).
The fluorescence, phosphorescence and the corresponding phosphorescence lifetimes of CAA solutions at 77 K were also measured. As shown in Fig. S13 (ESI†), the low-temperature fluorescence and phosphorescence both increased with increased concentration (from 10−5 M to 1.00 M), from the invisible emission in dilute solution (0.25 M) to highly bright emission at high concentration, owing to the gradually forming aggregates. Simultaneously, the phosphorescence lifetime became longer, from 22.80 ms to 1.184 ms (Fig. S13 and Table S3, ESI†). These results further confirmed that aggregation was the main reason for the fluorescence and phosphorescence. Besides, they all showed blue-shifted emissions at 77 K compared to those at room temperature, due to the restricted non-radiative motion and small Stokes shifts. However, CAA is a very small non-aromatic molecule with a very low molecular weight of only 85—why could its aggregation induce fluorescence and phosphorescence, but not the dilute solutions? It was reasonable to guess that there should be some influences of the aggregation or packing mode on the emission behaviors.
To gain deep insight on the relationship between the packing and emissive behavior, the single crystal of CAA was carefully investigated, with its structure data presented in Table S4 (ESI†) and Fig. 4, which are different from the previous reports.9 As expected, the CAA molecules have regular packing; the carbonyl group and hydroxyl group between two adjacent CAA molecules were linked by two classic –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯HO– hydrogen bonds, and the strong hydrogen bonds (dO–O = 2.6337(16) Å) extended over the whole CAA crystal.10 This could increase the molecular rigidity and help to lock the molecular conformations. Thus, the intramolecular motions could be restricted, accompanied by the decreased non-radiative transitions and enhanced emission of both fluorescence and phosphorescence. Like rebar, these abundant strong hydrogen bonds brought the CAA molecules into close proximity, leading to the layered structure. The intermolecular electronic couplings between carbonyl and carbonyl units, and cyano and cyano units, were observed in the CAA single crystal (Fig. S14, ESI†). These groups were arranged in the same plane due to the dipole–dipole interactions. The minimum distance between oxygen atom and carbonyl group was 3.5361(22) Å, and the distance decreased to 3.2674(25) Å between the nitrogen atom and cyano unit, which might promote the n–π* transitions. All of them are distributed in a plane, similar to a big π system, which could contribute much to the emission. A short interlayer spacing (3.0225 Å) was measured from the crystal structure of CAA, and the distance between nitrogen atom and carbonyl group was also short [3.6394(16) Å] between the two layers.
O⋯HO– hydrogen bonds, and the strong hydrogen bonds (dO–O = 2.6337(16) Å) extended over the whole CAA crystal.10 This could increase the molecular rigidity and help to lock the molecular conformations. Thus, the intramolecular motions could be restricted, accompanied by the decreased non-radiative transitions and enhanced emission of both fluorescence and phosphorescence. Like rebar, these abundant strong hydrogen bonds brought the CAA molecules into close proximity, leading to the layered structure. The intermolecular electronic couplings between carbonyl and carbonyl units, and cyano and cyano units, were observed in the CAA single crystal (Fig. S14, ESI†). These groups were arranged in the same plane due to the dipole–dipole interactions. The minimum distance between oxygen atom and carbonyl group was 3.5361(22) Å, and the distance decreased to 3.2674(25) Å between the nitrogen atom and cyano unit, which might promote the n–π* transitions. All of them are distributed in a plane, similar to a big π system, which could contribute much to the emission. A short interlayer spacing (3.0225 Å) was measured from the crystal structure of CAA, and the distance between nitrogen atom and carbonyl group was also short [3.6394(16) Å] between the two layers.
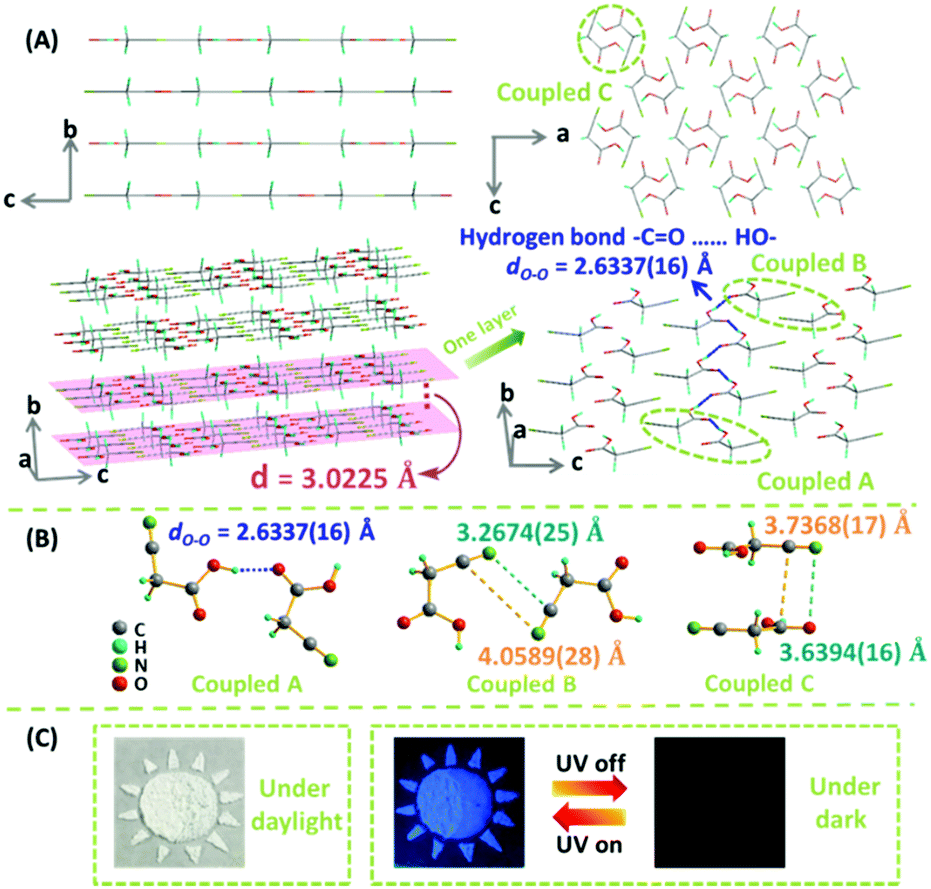 | ||
| Fig. 4 (A) The crystal structure of CAA (CCDC number 1527812†). Molecular packing viewed down the crystallographic a and b (upper) axes. The 3D structure of CAA crystals in which the molecules assemble layer by layer with interlayer (d) spacing of 3.0225 Å and strong –CO⋯HO– bonds [dO–O = 2.6337(16) Å] are widespread in each layer (down). Gray-50%, carbon; red, oxygen; green, nitrogen; sea green, hydrogen. (B) Three kinds of intermolecular coupling states between two molecules obtained from the CAA crystal: coupled A (two molecules coupled together through hydrogen bond), coupled B (two molecules coupled together through the cyano–cyano interaction), and coupled C (two molecules coupled together through the cyano–carbonyl interaction). (C) The time-resolved applications of CAA in the anti-fake application with 4,4′-dibromobenzophenone (no persistent RTP effect). The “Sun”, which was comprised by CAA crystal and 4,4′-dibromobenzophenone, was clearly observed under daylight and under 365 nm ultraviolet irradiation. After turning off the UV lamp, the “Moon,” which was comprised only by CAA crystal, would shine continuously. | ||
Fig. 5 shows the energy level diagrams. The singlet and triplet excited state transition configurations of CAA could be revealed by TD-DFT calculations, in which the red arrows refer to the possible ISC channels. The H and L in the tables (Tables S5–S8, ESI†) refer to the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), respectively. In order to guarantee the possible ISC transition from S1 to Tn, Tn and S1 should contain the same transition orbital compositions, and the difference between their energy level should be less than 0.3 or 0.4 eV.4c In the single crystal of CAA, there were mainly three kinds of intermolecular coupling states between two molecules: coupled A (two molecules coupled together through hydrogen bond), coupled B (two molecules coupled together through the cyano–cyano interaction) and coupled C (two molecules coupled together through the cyano–carbonyl interaction) (Fig. 4B). Their different energy levels might provide some clues to the origin of their RTP effect. For isolated CAA, there were no apparent channels for the ISC transition from S1 to Tn. Although S1 and T1 contained the same transition orbital compositions of H → L, their energy gap was as large as 0.56 eV (Fig. 5A, Table S5 and Fig. S15, ESI†). However, when two CAA molecules interacted through the strong hydrogen bond, two main ISC transition channels were found in coupled A of CAA (S1 → T2 and S1 → T7). Both contained the same transition orbital compositions of H → L, while S1 and T7 also contained H → L+1, with the energy gaps as small as 0.34 and 0.32 eV, respectively (Fig. 5B and Table S6, ESI†). As for coupled B and coupled C of CAA, similar phenomena to the isolated CAA were found. Both gave no apparent ISC transitions (although the S1 to T1 states contained the same orbital compositions; the energy gaps between them were higher than 0.4 eV; see Fig. 5C and D, Tables S7 and S8, ESI†). These calculated results further confirmed that the strong intermolecular hydrogen bonds played the main role in the ISC transition, thus resulting in the abnormal RTP property of CAA. Due to the persistent RTP effect, CAA could be used as an anti-fake material in practical applications. As shown in Fig. 4C, the picture of the “Sun” was clearly observed under daylight and under the ultraviolet irradiation of 365 nm; it was comprised of crystals of CAA and 4,4′-dibromobenzophenone (no persistent RTP effect). In terms of its persistent RTP feature, only the picture of the “Moon” shone continuously after turning off the UV lamp, comprised by the crystal of CAA.
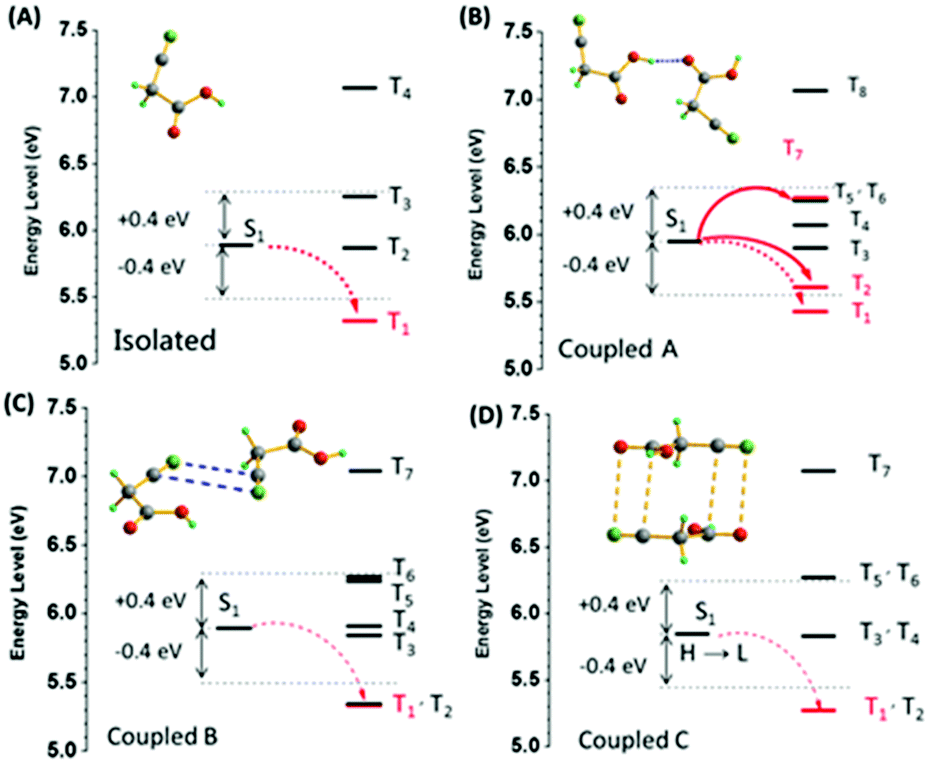 | ||
| Fig. 5 The energy level diagrams of the (A) isolated, (B) coupled A, (C) coupled B, and (D) coupled C for CAA (S0 = 0 eV). | ||
The RTP behavior of CAA prompted us to check the light emission properties of two other analogues, malonic acid (MNA) and malononitril (MNN). As expected, both fluorescence and phosphorescence were obtained in the MNA crystal due to the presence of hydrogen bonds (Fig. S16, ESI†) at room temperature, and fluorescence was observed in the 1 M MNA/H2O solution (Fig. S17, ESI†). However, it exhibited much weaker RTP emission, not visible to the naked eye, and the RTP lifetime and quantum efficiency were just 12 ms and 1.0% (Fig. S18, ESI†), indicating the inefficient ISC transition. This was understandable. As shown in the MNA crystal, similar to most of the previous reports,11 the distances of the –CO⋯HO– hydrogen bonds between two adjacent MNA were different [dN–C = 2.6862(24) and dN–C = 2.6859(17) Å for different hydrogen bonds], much longer than that of CAA [dO–O = 2.6337(16) Å]. The weaker hydrogen bond would lead to the weak suppression of vibrational relaxation as well as the weak electronic interactions, thus resulting in the weak RTP of MNA. Also, the PL behaviors of MNA were studied at 77 K, which restricts non-radiative transition. As shown in Fig. S19 (ESI†), its low-temperature phosphorescence lifetime in crystal was just 343 ms, much shorter than that of CAA (1.385 s). Further on, the corresponding calculation results confirmed that there are no obvious ISC transitions in isolated, coupled H1 [two molecules coupled together through the hydrogen bond with a distance of 2.6862(24) Å] and coupled H2 [two molecules coupled together through the hydrogen bond with a distance of 2.6859(17) Å; see Fig. S20–S22 and Tables S9–S11, ESI†].
When the carboxyl of CAA was replaced by a cyano group to give MNN, only fluorescence was observed, possibly caused by the absence of strong hydrogen bonds [3.2611(48)–3.6777(58) Å for different hydrogen bonds] and the changed electronic structure. Fig. S23 and S24 (ESI†) exhibit the fluorescence spectra of MNN in crystalline state and in the 1 M MNN/H2O solution, with the emission peaks at 464 nm and 457 nm, respectively. The crystal structure of MNN is shown in Fig. 6B, which is different from previous reports.12 Partially similar electronic coupling of intermolecular units between CN and CN units to CAA was observed in the single crystal of MNN, and the electronic interactions between them might lead to the visible fluorescence, while no phosphorescence was observed. Also, its much shorter phosphorescence lifetime at 77 K (143 ms), in comparison with that of CAA (1.385 s), further confirms the inefficient ISC transition for MNN (Fig. S25, ESI†). Furthermore, the calculation of spin orbital coupling constants for MNN was carried out based on the crystal configuration (Table S12 and Fig. S26, ESI†). As shown in Table S12 (ESI†), the spin orbital coupling (SOC) coefficients between S1 and Tn were too small to realize the spin orbital coupling as well as the ISC transition.
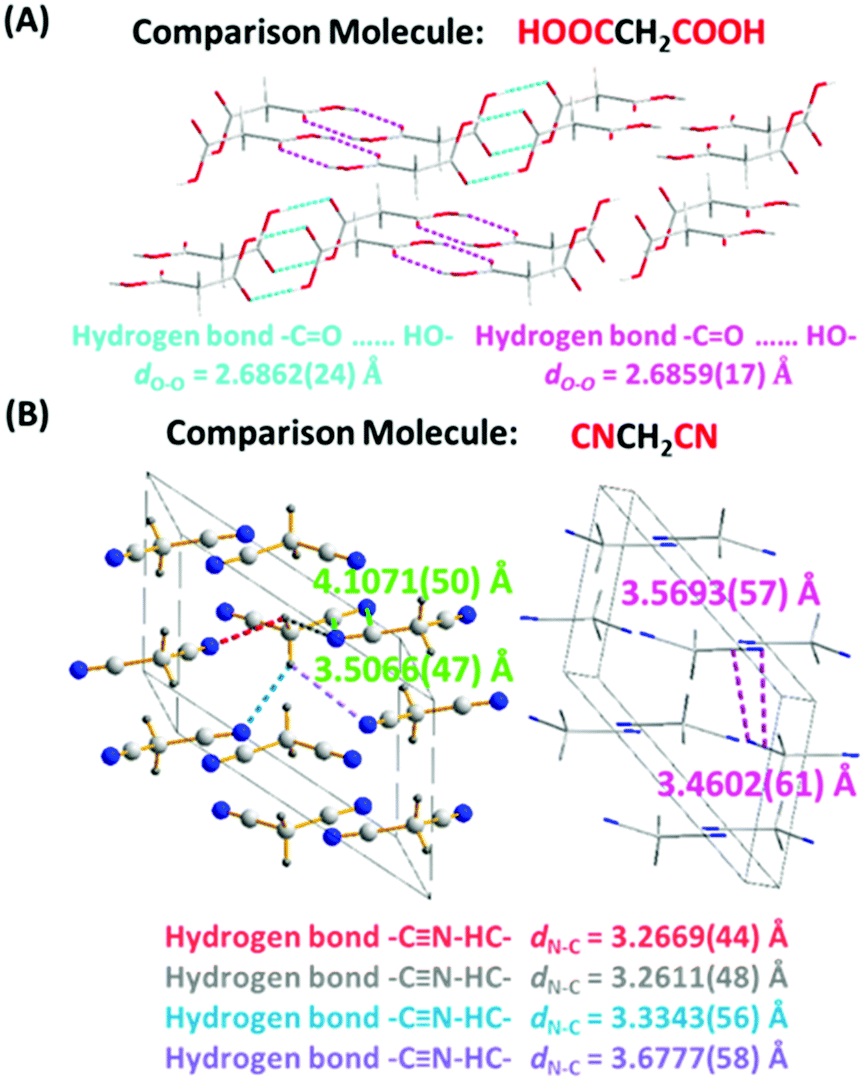 | ||
| Fig. 6 (A) The crystal structure of MNA (CCDC number 1528578†). The hydrogen bonds are indicated by dashed lines of different colors. The distances of hydrogen bonds between two adjacent malonic acid molecules are 2.6862(24) and 2.6859(17) Å, respectively. Gray-50%, carbon; red, oxygen; gray-25%, hydrogen. (B) The crystal structure of MNN (CCDC number 1829539†). The minimum distance from nitrogen atom to cyano unit is 3.4602(61) Å. The hydrogen bonds are indicated by dashed lines of different colors. The distances of hydrogen bonds between two adjacent malononitril molecules are 3.2611(48)–3.6777(58) Å. Gray-50%, carbon; red, oxygen; gray-25%, hydrogen; blue, nitrogen. | ||
Further on, another two compounds of methyl cyanoacetate and ethyl cyanoacetate were investigated to check the effect of the CO group on the RTP phenomenon. Since the COOH group was replaced by COOCH3 or COOC2H5 in these two molecules, no strong –CO⋯HO– bond could be formed. Thus, they were both liquid, without RTP effect, at room temperature. Even at 77 K, they only showed phosphorescence with lifetimes of about 224 and 996 ms for the restricted non-radiative motion at low temperature, which are much shorter than those of CAA (>1.3 s) (Fig. S27 and S28, ESI†). These results further confirm that the hydrogen bond formed between the COOH groups, rather than the CO groups, should be mainly responsible for the RTP effect.
Based on the above experimental and calculated results, it is clear that intermolecular hydrogen bonds play an important role in RTP. For CAA crystal, the strong hydrogen bonds [2.6337(16) Å], with the aid of the electron-withdrawing ability of the cyano-group, extend all over the crystal, directly leading to a long-range order arrangement and promoting phosphorescence emission. When the cyano-group of CAA is replaced by a carboxyl group in MNA, the intermolecular hydrogen bonds become weaker, with the bond lengths of 2.6862(24) and 2.6859(17) Å. Although the amount of hydrogen bonds increases, these are not strong enough to ensure the efficient phosphorescence as presented above, in the discussions on the data of single crystals and calculations. As for the MNN crystal, no strong hydrogen bonds exist [3.2611(48)–3.6777(58) Å], thus the non-phosphorescence. Also, the IR spectra of CAA, MNA and MNN in crystal and solution states demonstrate their different hydrogen bond strengths, in good accordance with results from the single crystals (Fig. S33, ESI†). Thus, in order to obtain strong intermolecular interaction and realize the RTP effect in non-aromatic systems, the molecular structure and electronic property should both be taken into consideration, as they could heavily affect the status of molecular packing in the aggregated state.
3 Conclusions
In summary, room-temperature phosphorescence was observed in the non-aromatic organic small molecule CAA, with a τ value as long as 862 ms. The strong intermolecular interactions through hydrogen bonds are the main driving force for its phosphorescence emission. The obtained experimental results confirmed that without the presence of classic aromatic groups, RTP is observed in nonaromatic pure organic compounds, opening a new angle for the consideration of organic luminogens. Also, RTP presents for the CAA crystal but not in solution, which demonstrates the importance of molecular packing and presence of possible molecular interactions. This phenomenon could be termed as a Molecular Uniting Set Identified Characteristic (MUSIC).13 Perhaps, the traditional thoughts on the light emitting materials should be further expanded from the aromatic to nonaromatic ones, with much deeper understanding of the inherent mechanism. Surely, more examples should be explored to achieve this change of thought, just like the transition of S1 to Tn required for RTP performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Science Foundation of China (no. 51573140, 21734007), the Fundamental Research Funds for the Central Universities (2042017kf0247), and Hubei Province (2017CFA002) for financial support. Many thanks for the help with crystallographic structural analysis from Prof. Jianrong Li and Bin Wang in Beijing University of Technology.Notes and references
- (a) S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 26, 9920 CrossRef PubMed; (b) S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988 RSC; (c) S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386 CrossRef CAS; (d) J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Diasb and M. R. Bryce, Chem. Commun., 2016, 52, 2612 RSC; (e) C. Li, X. Tang, L. Zhang, C. Li, Z. Liu, Z. Bo, Y. Don, Y. Tian, Y. Dong and B. Tang, Adv. Opt. Mater., 2015, 3, 1184 CrossRef CAS; (f) Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef PubMed; (g) L. Paul, S. Chakrabarti and K. Ruud, J. Phys. Chem. Lett., 2017, 8, 1253 CrossRef CAS PubMed; (h) G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747 CrossRef CAS PubMed; (i) O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205 CrossRef CAS PubMed; (j) M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081 RSC.
- (a) D. Lee, X. Ma, J. Jung, E. Jeong, H. Hashemi, A. Bregman, J. Kieffer and J. Kim, Phys. Chem. Chem. Phys., 2015, 17, 19096 RSC; (b) J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC; (c) S. Kuno, H. Akeno, H. Ohtani and H. Yuasa, Phys. Chem. Chem. Phys., 2015, 17, 15989 RSC; (d) M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. Youk, J. Gierschner and J. Kim, Nat. Commun., 2015, 6, 8947 CrossRef PubMed.
- (a) K. Totani, Y. Okada, S. Hirata, M. Vacha and T. Watanabe, Adv. Opt. Mater., 2013, 1, 283 CrossRef; (b) S. Hirata, K. Totani, H. Kaji, M. Vacha, T. Watanabe and C. Adachi, Adv. Opt. Mater., 2013, 1, 438 CrossRef; (c) J. Wei, B. Liang, R. Duan, Z. Cheng, C. Li, T. Zhou, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 15589 CrossRef CAS PubMed; (d) S. Hirata, K. Totani, T. Yamashita, C. Adachi and M. Vacha, Nat. Mater., 2014, 13, 938 CrossRef CAS PubMed.
- (a) Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685 CrossRef CAS PubMed; (b) Y. Gong, G. Chen, Q. Peng, W. Yuan, Y. Xie, S. Li, Y. Zhang and B. Tang, Adv. Mater., 2015, 27, 6195 CrossRef CAS PubMed; (c) Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y. Wu, P. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181 CrossRef CAS PubMed; (d) W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Tang, Chem, 2016, 1, 592 CrossRef CAS; (e) S. Hirata and M. Vacha, J. Phys. Chem. Lett., 2016, 7, 1539 CrossRef CAS PubMed; (f) E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Previtali, S. Righetto and E. Cariati, J. Phys. Chem. Lett., 2017, 8, 1894 CrossRef CAS PubMed; (g) Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed; (h) M. Shimizu, T. Kinoshita, R. Shigitani, Y. Miyake and K. Tajima, Mater. Chem. Front., 2018, 2, 347–354 RSC; (i) H. Liu, Y. Gao, J. Cao, T. Li, Y. Wen, Y. Ge, L. Zhang, G. Pan, T. Zhou and B. Yang, Mater. Chem. Front., 2018, 2, 1853 RSC; (j) Y. Wang, X. Bin, X. Chen, S. Zheng, Y. Zhang and W. Yuan, Macromol. Rapid Commun., 2018, 1800528 CrossRef PubMed.
- (a) Y. Gong, Y. Tan, J. Mei, Y. Zhang, W. Yuan, Y. Zhang, J. Sun and B. Tang, Sci. China: Chem., 2013, 56, 1178 CrossRef CAS; (b) Q. Zhou, B. Cao, C. Zhu, S. Xu, Y. Gong, W. Yuan and Y. Zhang, Small, 2016, 12, 6586 CrossRef CAS PubMed; (c) A. Pfister, G. Zhang, J. Zareno, A. F. Horwitz and C. L. Fraser, ACS Nano, 2008, 2, 1252 CrossRef CAS PubMed; (d) M. Palner, K. Pu, S. Shao and J. Rao, Angew. Chem., Int. Ed., 2015, 54, 11477 CrossRef CAS PubMed; (e) X. H. Chen, W. J. Luo, H. L. Ma, Q. Peng, W. Z. Yuan and Y. M. Zhang, Sci. China: Chem., 2017, 60, DOI:10.1007/s11426-017-9114-4.
- (a) W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090 CrossRef CAS; (b) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed; (c) Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 6195 CrossRef CAS PubMed; (d) J. Yang, Z. Ren, Z. Xie, Y. Liu, C. Wang, Y. Xie, Q. Peng, B. Xu, W. Tian, F. Zhang, Z. Chi, Q. Li and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 880 CrossRef CAS PubMed; (e) Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Li and Z. Li, Adv. Mater., 2017, 29, 1606829 CrossRef PubMed.
- J. Mcmurry, Organic Chemistry, Thomson Learning, Inc., USA, 2008 Search PubMed.
- (a) Z. Li, M. Fang and Q. Li, CN Pat., 201710214462.7, 2017 Search PubMed; (b) C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174–2194 RSC.
- (a) J. A. Kanters, G. Roelofsen and L. H. Straver, Acta Crystallogr., 1978, 34, 1393 CrossRef; (b) J. A. Kanters, G. Roelofsen and L. H. Straver, Acta Crystallogr., 1978, 34, 1396 CrossRef.
- (a) G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond, Oxford University Press, New York, 1999 Search PubMed; (b) Y. M. Legrand, A. Lee and M. Barboiu, Science, 2010, 329, 299 CrossRef CAS PubMed; (c) K. Saito, A. W. Rutherford and H. Ishikita, Nat. Commun., 2015, 6, 8488 CrossRef CAS PubMed.
- (a) V. R. Thalladi, M. Nusse and R. Boese, J. Am. Chem. Soc., 2000, 122, 9227 CrossRef CAS; (b) M. K. Mishra, U. Ramamurty and G. R. Desiraju, Chem. – Asian J., 2015, 10, 2176 CrossRef CAS PubMed; (c) S. Bhattacharya, V. G. Saraswatula and B. K. Saha, Cryst. Growth Des., 2013, 13, 3651 CrossRef CAS; (d) J. A. Goedkoop and C. H. MacGillavry, Acta Crystallogr., 1975, 10, 125 CrossRef; (e) N. R. Jagannathan, S. S. Rajan and E. Subramanian, J. Chem. Crystallogr., 1994, 24, 75 CrossRef CAS; (f) R. S. Gopalan, P. Kumaradhas, G. U. Kulkarni and C. N. R. Rao, J. Mol. Struct., 2000, 521, 97 CrossRef.
- (a) M. T. Dove and A. I. M. Rae, Faraday Discuss. Chem. Soc., 1980, 69, 98 RSC; (b) N. Nakamura, S. Tanisaki and K. Obatake, Phys. Lett. A, 1971, 34, 372 CrossRef CAS; (c) K. Obatake and S. Tanisaki, Phys. Lett. A., 1973, 44, 341 CrossRef CAS.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structure information of the compound; Chart S1, Fig. S1–S33 and Tables S1–S14. CCDC 1527812, 1829539 and 1528578. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00396c |
| This journal is © the Partner Organisations 2018 |